

Abstract
Interferon (IFN) consensus sequence-binding protein (ICSBP) is a transcription factor playing a critical role in the regulation of lineage commitment, especially in myeloid cell differentiation. In this study, we have characterized the phenotype and activation pattern of subsets of dendritic cells (DCs) in ICSBP−/− mice. Remarkably, the recently identified mouse IFN-producing cells (mIPCs) were absent in all lymphoid organs from ICSBP−/− mice, as revealed by lack of CD11clowB220+Ly6C+CD11b− cells. In parallel, CD11c+ cells isolated from ICSBP−/− spleens were unable to produce type I IFNs in response to viral stimulation. ICSBP−/− mice also displayed a marked reduction of the DC subset expressing the CD8α marker (CD8α+ DCs) in spleen, lymph nodes, and thymus. Moreover, ICSBP−/− CD8α+ DCs exhibited a markedly impaired phenotype when compared with WT DCs. They expressed very low levels of costimulatory molecules (intercellular adhesion molecule [ICAM]-1, CD40, CD80, CD86) and of the T cell area-homing chemokine receptor CCR7, whereas they showed higher levels of CCR2 and CCR6, as revealed by reverse transcription PCR. In addition, these cells were unable to undergo full phenotypic activation upon in vitro culture in presence of maturation stimuli such as lipopolysaccharide or poly (I:C), which paralleled with lack of Toll-like receptor (TLR)3 mRNA expression. Finally, cytokine expression pattern was also altered in ICSBP−/− DCs, as they did not express interleukin (IL)-12p40 or IL-15, but they displayed detectable IL-4 mRNA levels. On the whole, these results indicate that ICSBP is a crucial factor in the regulation of two possibly linked processes: (a) the development and activity of mIPCs, whose lack in ICSBP−/− mice may explain their high susceptibility to virus infections; (b) the generation and activation of CD8α+ DCs, whose impairment in ICSBP−/− mice can be responsible for the defective generation of a Th1 type of immune response.
Keywords: transcription factor, dendritic cell subsets, interferon, differentiation, maturation
Introduction
IFN consensus sequence-binding protein (ICSBP)* is a component of the IFN regulatory factor (IRF) transcription factor family, acting as an important regulator of IFN-inducible genes (1). ICSBP binds with other members of the IRF family and with the hematopoietic-specific member of the Ets family PU.1 to form transcriptional complexes apparently critical for the regulation of the immune system (2). The expression of ICSBP is restricted to myeloid and lymphoid cell lineages, including cells of monocyte/macrophage lineage, B lymphocytes, and activated T lymphocytes (3). Some reports have provided evidence indicating that ICSBP plays a critical role in modulating the immune response by influencing the differentiation and maturation of immune cells and by affecting cytokine expression (3, 4). Much of our knowledge on the in vivo role of ICSBP has stemmed from studies in mice lacking the expression of this transcription factor. Of interest, knockout (ICSBP−/−) mice were found to be highly susceptible to infection with various pathogens including vaccinia virus and LCMV, bacteria as Listeria monocytogenes and Yersinia enterocolitica, and parasites such as Leishmania major and Toxoplasma gondii, whose effective host control is associated with a Th1 immune response in normal mice (3, 5). Of note, several studies indicate that ICSBP has a role in regulating pathways affecting lineage commitment and myeloid cell differentiation (2, 6). In this regard, ICSBP−/− mice are characterized by altered hematopoiesis and develop a myeloproliferative disease resembling to chronic myelogenous leukemia (CML) in humans (7). The development and the response to cytokines of myeloid progenitors were also found to be altered in these mice (2, 4). Moreover, myeloid cells from ICSBP−/− mice have been reported to exhibit defective apoptosis, indicating that ICSBP plays a role in the control of cell growth and differentiation of myeloid cells at different developmental stages (8). Recently, it has been reported that ICSBP can affect the proliferative potential of myeloid cells at the progenitor cell level, playing a role in promoting macrophage differentiation, thus inhibiting the development of granulocytes (9). In the present study, we have studied the role of ICSBP in dendritic cell (DC) development and maturation by characterizing different DC subsets in ICSBP−/− mice as compared with control animals.
DCs are the most powerful APCs, playing a key role in triggering the immune system against infectious agents and cancer. In tuning the immune response of the host to pathogens, DCs undergo several stages of maturation by differentiating from immature DCs, specialized in the capture of antigens at the various portals of microbe entry, to mature APCs, with potent ability to prime naive helper and cytotoxic T cells in secondary lymphoid organs (10).
DC maturation can be triggered by products of bacterial or viral pathogens, through direct interaction with pattern-recognition receptors (PRRs), such as Toll-like receptors (TLRs) (11). Alternatively, DCs can be indirectly activated by cytokines produced by infected cells (12). One important family of infection-induced cytokines are type I IFNs (IFN-α/β). IFN-α/β not only represent one of the most important line of innate defense against pathogens, but also possess a variety of immunomodulatory activities (13–15), including promoting effects on the differentiation/activation of DCs (16, 17).
Several subpopulations of DCs have been described both in humans and in mice, based on phenotype, functional potential and microenvironmental localization, which are capable of inducing distinct types of responses (18, 19). Recently, a rare DC subset with plasmacytoid characteristics has been identified as the major IFN-producing cells (IPCs) first in humans (20, 21) and subsequently in mice (22–24). These cells represent a crucial cell type in the regulation of the immune response, as upon interaction with various types of pathogens, they trigger the innate immunity by producing large amounts of IFN-α/β, which may subsequently initiate the adaptive immune response by promoting differentiation of DCs and a Th1 type of immune response (25, 26). Mouse IPCs express markers associated with lymphoid lineage, but lack expression of myeloid markers and express low levels of the DC marker CD11c (23, 24).
In mice, mature CD11c+ DCs can be classified in two major DC subsets, on the basis of the expression of CD8, as an αα homodimer (27, 28). These CD8α− and CD8α+ DCs require different cytokines for their development (29) and exhibit distinct biological functions, such as the ability to induce Th1 and Th2 responses, respectively (30). Furthermore, they display different anatomical distribution in lymphoid organs, as CD8α+ DCs are located in the T cell areas, while CD8α− DCs reside in the marginal zone (31). Whether this heterogeneity reflects the existence of distinct lineages of DCs or different maturation stages, or both, remains controversial.
In this study, we found that IPCs are almost undetectable in ICSBP−/− mice and that this profound defect is associated with a marked impairment in the numbers and activation properties of CD8α+ DCs in all secondary lymphoid organs. These results unravel a previously unrecognized important role of ICSBP in the regulation of the development and activation of DCs.
Materials and Methods
Mice.
ICSBP-deficient mice were generated as described (7). Homozygous deficient (−/−) and WT mice (+/+) on a (C57BL/6 × 129/Sv) F2 background were bred and maintained under specific pathogen-free conditions.
DC Isolation and Culture.
DCs were isolated from lymphoid organs using a method similar to that described by Vremec et al. (27). In brief, spleens, thymuses, mesenteric and skin-draining (mandibular, axillary, inguinal, and popliteal) lymph nodes from 3–5 mice were pooled and cut into small fragments. Fragments were digested in RPMI 1640 (Bio-Whittaker) containing 10% FCS (SEBAM), 1 mg/ml type III collagenase (Worthington Biochemicals), and 325 K units/ml DNase I (Sigma-Aldrich), with periodic pipetting to break up fragments, for 25 min at room temperature. EDTA (0.1 M, pH 7.2; Sigma-Aldrich) was added for an additional 5 min, to allow disruption of DC–T cell complexes. Cells were washed, resuspended in Nycodenz (1.077 g/ml; Life Technologies), overlaid on an additional layer of Nycodenz, and centrifuged at 1,700 g for 20 min. The low-density fraction was collected, washed, and either directly used for phenotypic analysis, or further incubated on ice with anti–CD11c-FITC (Becton Dickinson) followed by anti–FITC-Microbeads (Miltenyi Biotech). The positive fraction was recovered using a MACS separation column and checked on a FACSort™ (Becton Dickinson) for purity. The cells obtained were routinely >95% CD11c+. In some experiments, DCs were overnight cultured in IMDM medium (Bio-Whittaker) supplemented with 10% heat-inactivated FCS, 100 U/ml penicillin, 100 μg/ml streptomycin (both from Bio-Whittaker), with or without added LPS, 0.5 μg/ml, or poly (I:C), 100 μg/ml (all from Sigma-Aldrich).
mAbs and Flow Cytometry.
The following mAbs (all from BD PharMingen) were used: anti-CD8a (53–6.7) either PE- or FITC-labeled, anti–CD11b-PE (M1/70), anti-CD45R/B220 FITC (RA3–6B2), anti–CD54-biotin (3E2), anti–CD40-biotin (HM40–3), anti–CD80-biotin (16–10A1), anti–CD86-biotin (GL1), anti–H2Db-biotin (28–14–8), anti–I-Ad/I-Ed-biotin (2G9), anti–Ly6C-biotin (AL-21), anti–CD45RB-biotin (16A), and anti-CD11c (HL3), which was used in either PE-, FITC-, or biotin-conjugated form. Biotinylated mAbs were detected with streptavidin-Red670 (Life Technologies). Stained cells were analyzed on a FACSort™ flow cytometer (Becton Dickinson). Viable cells were selected for analysis based on forward- and side-scatter properties.
RT-PCR and Analysis of Amplified Products.
Total RNA was extracted from 0.5–2 × 106 magnetically-purified CD11c+ splenic DCs by using the miniprep total RNA purification kit (QIAGEN). 500 ng of RNA was incubated at 25°C for 10 min with Oligo-p(dT)15 (Boehringer) in the presence of 50U RNase inhibitors (Boehringer) and reverse-transcribed using 20 U of AMV reverse transcriptase (Boehringer) for 1 h at 42°C in a final volume of 20 μl (10 mM Tris, 50 mM KCl, 5 mM MgCl2, 1 mM dNTPs; pH 8.3). PCR was performed on 2 μl of each cDNA sample using 1.25 U of Thermoprime Plus DNA polymerase (Advanced Biotechnologies) in a final volume of 50 μl containing 75 mM Tris, 20 mM Ammonium Persulphate, 0.1% Tween 20, 1.5 mM MgCl2, 0.2 mM dNTPs, 10 pmol of sense primer, and 10 pmol of antisense primer at pH 8.8. The specific primer pairs used were as follows: TLR3: 5′-TCGGATTCTTGGTTTCAAGG-3′ (sense) and 5′-CTTGCTGAACTGCGTGAT GT-3′ (antisense); TLR4: 5′-AGTGGGTCAAGGAACAGA AGCA-3′ (sense) and 5′-CTTTACCAGCTCATTTCTCACC-3′ (antisense); CCR2: 5′-GGGCTCACTATGCTGCAAAT-3′ (sense) and 5′-CGAAACAGGGTGTGGAGAAT-3′ (antisense); CC R6: 5′-ACTCTTTGTCCTCACCCTACCG-3′ (sense) and 5′-AT CCTGCAGCTCGTATTTCTTG-3′ (antisense); CCR7: 5′-AC AGCGGCCTCCAGAAGAACAGCGG-3′ (sense) and 5′-TGAC GTCATAGGCAATGTTGAGCTG-3′ (antisense); IL-4: 5′-AT GGGTCTCAACCCCCAGCTA-3′ (sense) and 5′-CGAGTAAT CCATTTGCATGAT-3′ (antisense); IL-12p40: 5′-AACTGGCG TTGGAAGCACGG-3′ (sense) and 5′-GAACACATGCCCACT TGCTG-3′ (antisense); IL-15: 5′-CATATGGAATCCAACTGGA TAGATGTAAGATA-3′ (sense) and 5′-CATATGCTCGAGGGA CGTGTTGATGAACAT-3′ (antisense); β-actin: 5′-TGACGGG GTCACCCACACTGTGCCCATCTA-3′ (sense) and 5′-CTAG AAGCATTGCGGTGGAGCATGGAGGG-3′ (antisense). All pri- mers were obtained from Invitrogen. The samples were amplified for 30–40 cycles at different annealing temperatures, optimal to each primer combination Tm. Amplified products (10 μl) were separated by agarose gel electrophoresis on a 1.2% TAE gel and visualized by ethidium bromide staining and UV transillumination. β-actin RT-PCR was run in parallel to normalize the levels of mRNA in the samples. The relative density of amplified bands was determined by LKB XL Ultroscan densitometer (Amersham Biosciences).
In Vitro Stimulation of DCs and IFN Bioassay.
3 × 105 magnetically sorted splenic DCs (>97% CD11c+) were infected with Newcastle disease virus (NDV; 576 UE/ml) for 1 h at 37°C, 5% CO2, then washed and cultured in 300 μl/well of a 48 well-plate for 18 h, after which the supernatant was harvested. 50 μl of each sample was assayed for IFN-α/β biological activity by measuring its ability to confer resistance to vesicular stomatitis virus (VSV) infection upon L929 cells as described elsewhere (32). Each IFN unit, as expressed in the text, represents 4 IU.
Results
ICSBP−/− Mice Lack mIPCs and Show Impaired IFN-α/β Production during Viral Infection.
Murine IPCs display a plasmacytoid morphology and have been phenotypically characterized as CD11clowCD11b−B220+Ly6C+CD 45 RB+MHC-IIlow (22–24). To identify mIPCs in ICSBP−/− and WT mice, DC-enriched cell populations were obtained from spleen, thymus, skin-draining and mesenteric lymph nodes by Nycodenz density-gradient centrifugation and the low-density fraction was subsequently stained with a panel of monoclonal antibodies. The staining for CD11c marker allowed us to gate on a population expressing this molecule at high and low levels, as indicated in Fig. 1 A. DCs were further stained for CD11b and Ly6C, as the expression of these surface markers allowed to identify mIPCs, which have been characterized as CD11clowCD11b−Ly6C+ cells (23, 24). As shown in Fig. 1 B, the flow cytometric analysis revealed a well-defined CD11b−Ly6C+ DC population (gate R3) in all organs from control mice. Strikingly, ICSBP−/− mice lacked these cells in all tissues. To confirm these results, we stained the cells with anti-CD11b coupled with anti-CD45RB, and analyzed the number of mIPCs, as CD11b−CD45RB+ in the CD11c+ gate (23). As shown in Fig. 1 C, a region of CD11b−CD45RB+ cells (gate R4) could be identified in control mice, while these cells were almost undetectable in all organs from ICSBP−/− mice.
Figure 1.

ICSBP−/− mice lack IPCs in all lymphoid organs. DCs were isolated from spleen, thymus, skin-draining and mesenteric lymph nodes from ICSBP−/− or WT mice. Nycodenz-enriched DCs were triple-stained for CD11c, CD11b, and alternatively for Ly6C, CD45RB, CD40, I-A, or B220 markers and then analyzed by flow cytometry for detection of mouse IPCs. (A) A region (R2 gate) was drawn on DC populations expressing CD11c at both low and high levels. (B and C) R2-gated DCs from the indicated lymphoid organs were analyzed for the expression of CD11b and Ly6C (B), or CD45RB (C). Regions (R3 and R4) identify IPCs (see text). (D) Surface phenotype of IPCs in spleens from ICSBP−/− and WT mice. Filled histograms show specific staining for the indicated markers in the CD11b−CD11clow cell population (R2 gate). Open lines represent isotype-matched control. Data are representative of one experiment of three.
To better characterize the surface phenotype of mIPCs, splenic plasmacytoid cells were further analyzed by using three-color analysis for the expression of CD11c, CD11b, and alternately B220, MHC-II, CD40, Ly6C, and CD45RB. As shown in Fig. 1 D, CD11clowCD11b− cells from control mice (gate R2) were found positive for B220 and CD45RB, expressed low levels of MHC-II and considerable levels of Ly6C, but were negative for the activation marker CD40, in accordance to what has been recently reported by Asselin-Paturel et al. (23). In contrast, the mIPCs (gate R2) were almost undetectable in spleens from ICSBP−/− mice, as revealed by the absence of staining for B220, MHC-II, CD40, and Ly6C markers.
To check whether the absence of mIPCs in lymphoid tissues of ICSBP−/− mice was indeed associated with a defective production of IFN-α/β upon virus stimulation, we purified DCs from spleens of ICSBP−/− or WT mice and infected them with NDV. The IFN-α/β production was then determined in the culture supernatants by biological assay. As expected, NDV infection did not induce significant release of IFN-α/β in DCs from ICSBP−/− mice, whereas large amounts of this cytokine were found in control-DC cultures (Fig. 2) .
Figure 2.

Defective production of IFN-α/β in DCs from ICSBP−/− mice after in vitro infection with NDV. Magnetically-purified DCs (>97% CD11c+), obtained from spleens of ICSBP−/− and WT mice, were infected with NDV for 1 h. Virus was then washed out, and cells were incubated for 18 h, at 37°C. The supernatant was harvested and assayed for IFN-α/β bioactivity, as described in Materials and Methods. Data are representative of two independent experiments.
ICSBP−/− Mice Exhibit Severe Reductions in and Altered Phenotype of CD8α+ DCs.
Two major DC subsets have been identified in the mouse lymphoid organs on the basis of their differential CD8α expression, named CD8α− and CD8α+ DCs (27). Additional reports have further indicated that the CD8α− DCs can be in turn distinguished on the basis of CD4 expression (33). This complex heterogeneity could reflect a different state of activation, maturation, mobilization (34–36), or divergent ontogeny of DCs (37).
We therefore examined the distribution of CD8α− and CD8α+ DC subsets in spleens, thymus, mesenteric and skin-draining lymph nodes from ICSBP−/− or WT mice. Gradient-enriched DCs were double-stained for CD11c and CD8α markers and analyzed by flow cytometry. As illustrated in Fig. 3 A, showing the expression of CD8α in spleen DCs gated by CD11c positivity and forward side scatter properties, 32% of splenic DCs were CD8α+ in WT mice, similarly to values previously found in other mouse strains (19). Surprisingly, spleen CD8α+ detected in ICSBP−/− mice only represented 3.6% of the total DC population. To further analyze the maturation and activation phenotype of CD8α− and CD8α+ subsets of splenic DCs from ICSBP−/− and control mice, we performed three-color flow cytometric analysis combining the CD11c and CD8α markers alternatively with the costimulatory antigens CD40, CD80, CD86, intercellular adhesion molecule (ICAM)-1, and the MHC class I and class II molecules. As shown in Fig. 3 B, the CD8α+ DC subpopulation from ICSBP−/− mice expressed significantly lower levels of the costimulatory antigens CD40, CD80, CD86, and ICAM-1 with respect to the WT counterparts. In contrast, in the CD8α− DC-subset all the considered costimulatory antigens and activation markers were expressed at comparable levels in ICSBP−/− and WT mice. The altered phenotype of CD8α+ DCs in ICSBP−/− mice was further confirmed by the morphologic analysis of the forward scatter profile (Fig. 3 C), showing that, consistently with previous reports (36), splenic CD8α+ DCs were bigger than CD8α− DCs in WT mice. Conversely, CD8α+ DCs recovered from the spleens of ICSBP−/− mice proved to be significantly smaller than CD8α− DCs.
Figure 3.

Impaired number and phenotype of CD8α+ DCs in spleens from ICSBP−/− mice. Spleens from ICSBP−/− or WT mice were pooled and enriched for DCs by Nycodenz density-gradient centrifugation, as described in Materials and Methods. (A) The low-density cell fraction was double-stained for CD11c and CD8α expression. The histograms show the percentage of CD8α+ DCs in ICSBP−/− and WT mice, gated for CD11c positivity and by forward side scatter properties. (B) The CD8α− and CD8α+ CD11c+ DC-subsets were additionally stained for ICAM-1, CD40, CD80, CD86, MHC class I, or class II molecules. Histograms show specific staining for the indicated antigens in CD8α+ CD11c+ (left) and CD8α- CD11c+ (right) gated populations, in ICSBP−/− (filled) and WT (open) mice. The broken profiles represent the background fluorescence for control isotype-matched antibodies. (C) Forward scatter profiles of CD8α− (filled histograms) and CD8α+ (open histograms) CD11c+ DCs from ICSBP−/− and WT mice. (D) Density plot analysis showing CD4 and CD8α expression in CD11c-gated DCs from ICSBP−/− and WT mice. Results are representative of at least five independent experiments.
CD8α− DCs can also express the CD4 marker and CD4+CD8α− and CD4−CD8α− DCs have been described to be present in the mouse spleen in a 3:1 ratio (19). To determine whether the CD4 molecule was differentially expressed in CD8α− splenic DCs from ICSBP−/− mice, we performed immunofluorescent staining of gradient-enriched CD11c+ DCs for CD4 and CD8α expression. As shown in Fig. 3 D, the percentage of CD4+ DCs was significantly higher in ICSBP−/− than in control mice. In fact, the representation of the CD4+CD8α− versus CD4−CD8α− DC subsets was in a 5:1 ratio in knockout mice, which contrasts with the 2.5:1 ratio found in WT animals.
The analysis of the other lymphoid organs, where the frequencies of CD8α+ DCs are generally more consistent, revealed that this DC subset was markedly reduced in all of them in ICSBP−/− mice (Fig. 4 A). Moreover, three-color analysis showed that the expression of ICAM-1, CD40, CD80, and CD86 in the CD8α+ DC-subset from skin-draining and mesenteric lymph nodes was also impaired (Fig. 4 B). In fact, CD8α+ DCs from control mice could be clearly separated in two populations, one expressing intermediate to high levels of each activation marker (gate R3), and one showing low or no expression of these molecules (gate R4). Remarkably, the highly activated CD8α+ DC population (gate R3) was barely detectable in lymph nodes from ICSBP−/− mice, as indicated by the absence of ICAM-1hi, CD40+, and CD86+ cells, even though considerable levels of CD80 expression were retained. Overall, these results indicate that ICSBP is essential for the development and maturation of mouse CD8α+ DCs.
Figure 4.
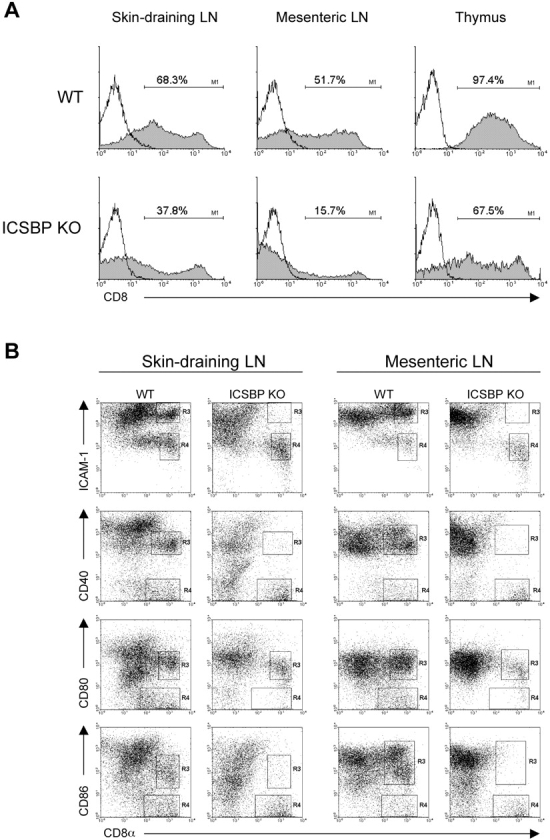
Highly reduced percentage of CD8α+ DCs in all lymphoid organs from ICSBP−/− mice. Density gradient-enriched DCs were obtained from thymus, skin-draining and mesenteric lymph nodes of ICSBP−/− or WT mice. (A) DC preparations were double stained for CD11c and CD8α expression and then analyzed by flow cytometry. Filled histograms show CD8α expression in CD11c-gated populations of the indicated organs. Open lines represent isotype-matched controls. (B) Dot plot analysis of CD8α+ and CD8α−, CD11c-gated DCs, alternatively labeled with anti–ICAM-1, CD40, CD80, or CD86 specific antibodies. Regions were drawn (R3 and R4) to identify CD8α+ subpopulations expressing the indicated molecules at different levels, in lymph nodes from ICSBP−/− or WT mice. Representative data of one experiment out of four are shown.
Altered Response of CD8α+ DCs from ICSBP−/− Mice to Activation Signals.
Because DCs are extremely tuned to respond to activation stimuli, even manipulation and short-time culture has been described to activate DCs (38, 39). We therefore wanted to address whether the unbalance of CD8α+ and CD8α− DC subsets might affect the ability of ICSBP−/− DCs to undergo phenotypic maturation upon in vitro culture. To this end, CD11c+ DCs were magnetically sorted from spleens of ICSBP−/− and WT mice, cultured for 18 h and then stained for surface markers expression. Fig. 5 A shows the mean fluorescence intensity values of the indicated markers in CD8α+ -gated and CD8α−-gated populations. CD8α+ DCs from ICSBP−/− mice exhibited a clear-cut reduction in the expression of the costimulatory molecules CD40, CD80, and CD86, the adhesion antigen ICAM-1 and the activation markers MHC class I and class II, as compared with the control counterparts. In contrast, CD8α− DCs from ICSBP−/− and WT mice exhibited similar levels of CD40, CD80, CD86, and ICAM-1. Interestingly, the expression of MHC class I and class II was significantly lower in ICSBP−/− CD8α− DCs, indicating that optimal activation was compromised also in this subset.
Figure 5.

Reduced activation of CD8α+ splenic DCs from ICSBP−/− mice, after short-time culture. CD11c+ DCs were purified from spleens by magnetic cell-sorting (see Materials and Methods). Cells obtained from both ICSBP−/− and WT mice were cultured in complete medium for 24 h, at 37°C. (A) Overnight-cultured DCs were stained for CD8α and alternatively for ICAM-1, CD40, CD80, CD86, or the molecules MHC class I and class II. The mean fluorescence distributions for the indicated antigens in CD8α+ and CD8α− subsets are represented by black bars for DCs isolated from ICSBP−/− mice or white bars for DCs from WT mice. (B) CD8α+ DCs from cultures, treated or not with LPS or poly (I:C), were stained for the costimulatory molecules CD40, CD80, or CD86. The staining for the indicated antigens is represented by filled histograms for CD8α+ DCs from ICSBP−/− mice and open histograms for CD8α+ DC from WT mice. The broken profiles show isotype-matched controls. (C) Total RNA was extracted from freshly isolated magnetically sorted CD11c+ splenic DCs from ICSBP−/− and control mice. TLR3 and TLR4 mRNA levels were detected by RT-PCR. Representative data of one experiment out of four are shown.
Next, we examined whether in vitro treatment with different maturation stimuli, such as LPS or poly (I:C) could activate DCs from ICSBP−/− mice. As shown in Fig. 5 B, addition of LPS or poly (I:C) to the DC cultures, increased the expression of the costimulatory markers CD40, CD80, and CD86 in DCs from both genotypes. Nevertheless, the intensity of expression of these molecules was significantly lower in CD8α+ DCs from ICSBP−/− mice, indicating that full activation did not occur in ICSBP−/− DCs. To assess whether the defective responsiveness of ICSBP−/− DCs to these two stimuli was due to altered expression of PRRs, we analyzed the expression of TLR4 and TLR3, which are known to recognize LPS (40) and poly (I:C; reference 41), respectively. Fig. 5 C shows TLR3 and TLR4 mRNA expression, as revealed by RT-PCR, in freshly isolated, magnetically sorted splenic DCs from ICSBP−/− and WT mice. Interestingly, ICSBP−/− DCs did not express detectable TLR3 mRNA, whereas they expressed higher levels of TLR4 mRNA than control DCs.
Chemokine Receptor and Cytokine Expression in DCs from ICSBP−/− Mice Reflects the Prevalence of CD8α− Subsets.
The differential expression of chemokine receptors identifies distinct DC maturation stages and ensures a correct trafficking of these cells to lymphoid organs (42). We then performed RT-PCR for the detection of chemokine receptors implicated in the anatomical localization and maturation stage of CD8α+ and CD8α− DC subsets in lymphoid tissues (43). Notably, freshly isolated splenic DCs from ICSBP−/− mice expressed significantly higher levels of CCR2 (twofold) and CCR6 (threefold) as compared with the WT counterparts, while they showed lower levels (2.5-fold) of CCR7, whose expression is associated with maturating DCs (44; Fig. 6 A).
Figure 6.

Expression of cytokines and chemokine receptors in DCs from ICSBP−/− mice. DCs were isolated from pooled spleens from ICSBP−/− or WT mice. CD11c+ DCs were magnetically sorted, as indicated in Fig. 5. Total RNA was extracted from freshly isolated DC preparations and assayed for the expression of the indicated chemokine receptors (A) or cytokines (B) by RT-PCR. Representative data of one experiment out of three are shown.
Although CD8α+ and CD8α− DC subsets appear to be equally competent at presenting antigen to T cells in vivo, they are known to possess distinct cytokine profiles, which, under certain experimental conditions, can drive either the Th1 or Th2 response (45, 46). Therefore, the imbalance between CD8α+ and CD8α− DC subsets in ICSBP−/− mice, could result in altered expression of cytokines, which might in turn affect some DC functions. To evaluate the expression pattern of cytokines, RNA was extracted from freshly-isolated magnetically sorted CD11c+ DCs from ICSBP−/− or WT mice, and IL-12p40, IL-15, or IL-4 mRNA expression was evaluated by RT-PCR. As illustrated in Fig. 6 B, ICSBP−/− DCs did not express any detectable level of IL-12p40 and showed very low levels of IL-15, but they displayed some IL-4 mRNA expression. In contrast, DCs from WT mice showed a clear-cut expression of both IL-12p40 and IL-15 mRNA, while they did not apparently express IL-4 mRNA.
Discussion
This study provides the first evidence that ICSBP, a transcriptional factor acting in the IFN signaling, plays a key role in the development of mIPCs and in the generation and activation of CD8α+ DCs. The most striking observation reported in this paper is the lack of mIPCs in ICSBP−/− mice. This recently identified cell subset, which is responsible for the production of large amounts of type I IFN after virus infection, is characterized by a plasmacytoid morphology and by the expression of specific markers. In particular, mouse IPCs exhibit low levels of the DC marker CD11c, lack of the myeloid marker CD11b and are characterized by the coexpression of the neutrophil marker Ly6C and the B cell marker B220 (22–24). Moreover, these cells express significant levels of CD45RB and MHC class II, but lack of CD40 marker (23, 24). The lack of IPCs in ICSBP−/− mice was revealed not only by the absence of a defined population of CD11clowCD11b−Ly6C+B220+CD45RB+MHC-II+CD40− cells in spleen, thymus, skin-draining and mesenteric lymph nodes, but also by the marked defect in the production of type I IFN by DC cultures from these mice. The lack of IFN-α/β production may explain the susceptibility of ICSBP-deficient mice to some viral infections such as vaccinia virus (VV; reference 7). Of note, even though ICSBP−/− mice can control VSV infection, they develop an impaired CTL response to this virus, which has been reported to activate IPC localized in the marginal zone (47). Taken together, our data suggest a key role of this transcription factor in the development of mIPC lineage with consequent profound deficits in immune functions. Although the precise stage of the mIPC developmental program controlled by ICSBP remain to be clarified, we favor the hypothesis that the activity of this transcriptional factor becomes a critical determinant in an IPC-committed precursor, as ICSBP−/− mice are still competent at producing other DC subsets.
Another important finding reported in this study is the marked impairment in the number and activation stage of CD8α+ DCs observed in various tissues from ICSBP−/− mice. CD8α+ and CD8α− DCs were originally reported as distinct lineages, lymphoid- and myeloid-derived, respectively, endowed with distinct cytokine requirements and development regulation mechanisms (48), as mice deficient for RelB, a transcription factor of the nuclear factor (NF)-kB family, normally generate CD8α+ DCs but lack of CD8α− DC subtype (49). However, recent studies have challenged this concept by demonstrating that both CD8α− and CD8α+ DCs can be generated from either lymphoid or myeloid progenitors (50) and a common precursor population, yielding CD8α+ and CD8α− as well as B220+ DCs, has been characterized (51). The marked impairment in the levels of CD8α+ DCs in lymphoid tissues from ICSBP−/− mice and the poorly activated phenotype of these cells reported in the present study might account for the impaired Th1 response previously observed in these mice (5, 52). In addition, CD8α+ DCs from ICSBP−/− mice displayed low levels of the surface markers CD40, CD80, CD86 ICAM-1, and MHC class I and II along with a significantly small size as evaluated by morphologic analysis. Consistent with our data, Aliberti et al., (2002) have recently reported that ICSBP is preferentially expressed by the CD8α+ DC subset and that ICSBP−/− mice display a selective reduction of CD8α+DEC205+ DCs, which is ascribed to an intrinsic defect of bone marrow–derived progenitors (53). Of interest, we also found that, even though the absence of ICSBP did not affect the expression of costimulatory molecules in CD8α− DCs, it influenced the frequencies of CD4+ and CD4−. At present, the lineage relationship between CD4+ and CD4− DCs relative to the CD8α− subtype is unknown (54); however, as these subtypes could reflect different maturation stages (33), the prevalence of CD4+CD8α− DCs in ICSBP−/− mice is likely to be correlated to a specific role of this factor in controlling DC maturation. The concept that ICSBP can play a role in DC maturation is also supported by our findings that ICSBP−/− DCs expressed high levels of CCR6 and CCR2, receptors both present on immature DCs (55, 56), as well as low levels of CCR7, whose expression is up-regulated in mature DCs, driving their migration into T cell areas of secondary lymphoid organs (44). The control by ICSBP on maturation of CD8α+ DCs, and to a lower extent of CD8α− DCs, was also evident upon activation by LPS or poly (I:C) treatment (57), as these agents failed to induce full phenotypic activation in DCs from ICSBP−/− mice. Notably, the unresponsiveness of ICSBP−/− DCs to poly (I:C) correlated with lack of expression of TLR3, which has been recently reported to recognize dsRNA (41). In contrast, these cells were found to exhibit high levels of TLR4, indicating that the inability of CD8α+ DCs to be activated by LPS is likely due to an intrinsic defect, rather than to a lack of this specific PRR. Notably, ICSBP−/− DCs showed a qualitative difference in cytokine expression with respect to DCs from control mice, as IL-12 and IL-15 mRNAs were undetectable, while some expression of IL-4 was observed. This cytokine profile appears to be consistent with the Th2 bias of ICSBP deficient mice (5) as well as with the preferential presence of CD8α− DCs in lymphoid organs (58).
On the whole, our findings demonstrate that ICSBP is essential for the development of mIPC and plays an important role in the generation of CD8α+ DCs. This transcription factor may also affect the terminal stages of CD8α− DC maturation. We suggest that the activity of ICSBP becomes relevant at different stages in the distinct DC lineages. It may control the development of a mIPC-committed precursor and, in parallel, may delay the differentiation program of a progenitor of CD8α+ DCs (51, 54). Alternatively, if CD8α+ DCs and mIPC represent different maturation stages of the same DC subpopulation, a question raised by some authors (23, 58), we can argue that ICSBP may act as a key factor in controlling the developmental maturation program of these cells.
Acknowledgments
We are grateful to Cinzia Gasparrini for secretarial assistance.
This work was supported by grants from the Italian Association for Cancer Research (Project AIRC no. F89) and Italian Ministry of Health (Project on Cytokines no. 98/JB/T).
Footnotes
Abbreviations used in this paper: DC, dendritic cell; ICAM, intercellular adhesion molecule; ICSBP, IFN consensus sequence-binding protein; IPC, IFN-producing cell; NDV, Newcastle disease virus; PRR, pattern-recognition receptor; TLR, Toll-like receptor.
References
- 1.Nguyen, H., J. Hiscott, and P.M. Pitha. 1997. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 8:293–312. [DOI] [PubMed] [Google Scholar]
- 2.Nagamura-Inoue, T., T. Tamura, and K. Ozato. 2001. Transcription factors that regulate growth and differentiation of myeloid cells. Int. Rev. Immunol. 20:83–105. [DOI] [PubMed] [Google Scholar]
- 3.Tamura, T., and K. Ozato. 2002. ICSBP/IRF-8: its regulatory roles in the development of myeloid cells. J. Interferon Cytokine Res. 22:145–152. [DOI] [PubMed] [Google Scholar]
- 4.Scheller, M., J. Foerster, C.M. Heyworth, J.F. Waring, J. Lohler, G.L. Gilmore, R.K. Shadduck, T.M. Dexter, and I. Horak. 1999. Altered development and cytokine responses of myeloid progenitors in the absence of transcription factor, interferon consensus sequence binding protein. Blood. 94:3764–3771. [PubMed] [Google Scholar]
- 5.Giese, N.A., L. Gabriele, T.M. Doherty, D.M. Klinman, L. Tadesse-Heath, C. Contursi, S.L. Epstein, and H.C. Morse III. 1997. Interferon (IFN) consensus sequence-binding protein, a transcription factor of the IFN regulatory factor family, regulates immune responses in vivo through control of interleukin 12 expression. J. Exp. Med. 186:1535–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedman, A.D. 2002. Transcriptional regulation of granulocyte and monocyte development. Oncogene. 21:3377–3390. [DOI] [PubMed] [Google Scholar]
- 7.Holtschke, T., J. Lohler, Y. Kanno, T. Fehr, N. Giese, F. Rosenbauer, J. Lou, K.P. Knobeloch, L. Gabriele, J.F. Waring, et al. 1996. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 87:307–317. [DOI] [PubMed] [Google Scholar]
- 8.Gabriele, L., J. Phung, J. Fukumoto, D. Segal, I.M. Wang, P. Giannakakou, N.A. Giese, K. Ozato, and H.C. Morse III. 1999. Regulation of apoptosis in myeloid cells by interferon consensus sequence-binding protein. J. Exp. Med. 190:411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tamura, T., T. Nagamura-Inoue, Z. Shmeltzer, T. Kuwata, and K. Ozato. 2000. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 13:155–165. [DOI] [PubMed] [Google Scholar]
- 10.Banchereau, J., F. Briere, C. Caux, J. Davoust, S. Lebeque, Y.-J. Liu, B. Pulendran, and K. Palucka. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811. [DOI] [PubMed] [Google Scholar]
- 11.Underhill, D.M., and A. Ozinsky. 2002. Toll-like receptors: key mediators of microbe detection. Curr. Opin. Immunol. 14:103–110. [DOI] [PubMed] [Google Scholar]
- 12.Reis e Sousa, C. 2001. Dendritic cells as sensors of infection. Immunity. 14:495–498. [DOI] [PubMed] [Google Scholar]
- 13.Belardelli, F., and I. Gresser. 1996. The neglected role of type I interferon in the T cell-response: implications for its clinical use. Immunol. Today. 17:369–372. [DOI] [PubMed] [Google Scholar]
- 14.Belardelli, F., and M. Ferrantini. 2002. Cytokines as a link between innate and adaptive antitumor immunity. Trends Immunol. 23:201–208. [DOI] [PubMed] [Google Scholar]
- 15.Le Bon, A., and D.F. Tough. 2002. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 14:432–436. [DOI] [PubMed] [Google Scholar]
- 16.Montoya, M., G. Schiavoni, F. Mattei, I. Gresser, F. Belardelli, P. Borrow, and D.F. Tough. 2002. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 99:3263–3271. [DOI] [PubMed] [Google Scholar]
- 17.Santini, S.M., C. Lapenta, M. Logozzi, S. Parlato, M. Spada, T. Di Pucchio, and F. Belardelli. 2000. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL. J. Exp. Med. 191:1777–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu, Y.J. 2001. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell. 106:259–262. [DOI] [PubMed] [Google Scholar]
- 19.Shortman, K., and Y.-J. Liu. 2002. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2:151–161. [DOI] [PubMed] [Google Scholar]
- 20.Cella, M., D. Jarrossay, F. Facchetti, O. Alebardi, H. Nakajima, A. Lanzavecchia, and M. Colonna. 1999. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5:919–923. [DOI] [PubMed] [Google Scholar]
- 21.Siegal, F.P., N. Kadowaki, M. Shodell, P.A. Fitzgerald-Bocarsly, K. Shah, S. Ho, S. Antonenko, and Y.-J. Liu. 1999. The nature of the principal type 1 interferon-producing cells in human blood. Science. 284:1835–1837. [DOI] [PubMed] [Google Scholar]
- 22.Bjorck, P. 2001. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 98:3520–3526. [DOI] [PubMed] [Google Scholar]
- 23.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 24.Nakano, H., M. Yanagita, and M.D. Gunn. 2001. CD11c+ B220+Gr-1+ cells in mouse lymph nodes and spleens display characteristics of plasmacytoid dendritic cells. J. Exp. Med. 194:1171–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kadowaki, N., S. Antonenko, J.Y.-N. Lau, and Y.-J. Liu. 2000. Natural interferon α/β-producing cells link innate and adaptive immunity. J. Exp. Med. 192:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent Th1 polarization. Nat. Immunol. 1:305–310. [DOI] [PubMed] [Google Scholar]
- 27.Vremec, D., M. Zorbas, R. Scollay, D.J. Saunders, C.F. Ardavin, L. Wu, and K. Shortman. 1992. The surface phenotype of dendritic cells purified from mouse thymus and spleen: investigation of the CD8 expression by a subpopulation of dendritic cells. J. Exp. Med. 176:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anjuère, F., P. Martín, I. Ferrero, M. López Fraga, G. Martínez del Hoyo, N. Wright, and C. Ardavín. 1999. Definition of dendritic cell subpopulations present in the spleen, Peyer's patches, lymph nodes, and skin of the mouse. Blood. 93:590–598. [PubMed] [Google Scholar]
- 29.O'Keeffe, M., H. Hochrein, D. Vremec, J. Pooley, R. Evans, S. Woulfe, and K. Shortman. 2002. Effects of administration of progenipoietin 1, Flt-3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood. 99:2122–2130. [DOI] [PubMed] [Google Scholar]
- 30.Moser, M., and K.M. Murphy. 2000. Dendritic cell regulation of Th1-Th2 development. Nat. Immunol. 1:199–205. [DOI] [PubMed] [Google Scholar]
- 31.McLellan, A.D., M. Kapp, A. Eggert, C. Linden, U. Bommhardt, E.B. Brocker, U. Kammerer, and E. Kampgen. 2002. Anatomic location and T-cell stimulatory functions of mouse dendritic cell subsets defined by CD4 and CD8 expression. Blood. 99:2084–2093. [DOI] [PubMed] [Google Scholar]
- 32.Tovey, M.G., J. Begon-Lours, and I. Gresser. 1974. A method for the large scale production of potent interferon preparations. Proc. Soc. Exp. Biol. Med. 146:809–815. [DOI] [PubMed] [Google Scholar]
- 33.Vremec, D., J. Pooley, H. Hochrein, L. Wu, and K. Shortman. 2000. CD4 and CD8 expression by dendritic cell subtypes in mouse thymus and spleen. J. Immunol. 164:2978–2986. [DOI] [PubMed] [Google Scholar]
- 34.Merad, M., L. Fong, J. Bogenberger, and E.G. Engleman. 2000. Differentiation of myeloid dendritic cells into CD8a-positive dendritic cells in vivo. Blood. 96:1865–1872. [PubMed] [Google Scholar]
- 35.Wu, L., A. D'Amico, H. Hochrein, M. O'Keeffe, K. Shortman, and K. Lucas. 2001. Development of thymic and splenic dendritic cell populations from different hemopoietic precursors. Blood. 98:3376–3382. [DOI] [PubMed] [Google Scholar]
- 36.Del Hoyo, G.M., P. Martin, C.F. Arias, A.R. Marin, and C. Ardavin. 2002. CD8α(+) dendritic cells originate from the CD8α(−) dendritic cell subset by a maturation process involving CD8α, DEC-205, and CD24 up-regulation. Blood. 99:999–1004. [DOI] [PubMed] [Google Scholar]
- 37.Kamath, A.T., J. Pooley, M.A. O'Keeffe, D. Vremec, Y. Zhan, A.M. Lew, A. D'Amico, L. Wu, D.F. Tough, and K. Shortman. 2000. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 165:6762–6770. [DOI] [PubMed] [Google Scholar]
- 38.Gallucci, S., M. Lolkema, and P. Matzinger. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255. [DOI] [PubMed] [Google Scholar]
- 39.De Smedt, T., E. Butz, J. Smith, R. Maldonado-Lopez, B. Pajak, M. Moser, and C. Maliszewski. 2001. CD8α(−) and CD8α(+) subsets of dendritic cells undergo phenotypic and functional maturation in vitro and in vivo. J. Leukoc. Biol. 69:951–958. [PubMed] [Google Scholar]
- 40.Chow, J.C., D.W. Young, D.T. Golenbock, W.J. Christ, and F. Gusovsky. 1999. Toll-like receptor-4 mediates lipopolysaccharide-induced signal transduction. J. Biol. Chem. 274:10689–10692. [DOI] [PubMed] [Google Scholar]
- 41.Alexopoulou, L., A.C. Holt, R. Medzhitov, and R.A. Flavell. 2001. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 413:732–738. [DOI] [PubMed] [Google Scholar]
- 42.Yoshie, O. 2000. Role of chemokines in trafficking of lymphocytes and dendritic cells. Int. J. Hematol. 72:399–407. [PubMed] [Google Scholar]
- 43.Caux, C., S. Ait-Yahia, K. Chemin, O. de Bouteiller, M.C. Dieu-Nosjean, B. Homey, C. Massacrier, B. Vanbervliet, A. Zlotnik, and A. Vicari. 2000. Dendritic cell biology and regulation of dendritic cell trafficking by chemokines. Springer Semin. Immunopathol. 22:345–369. [DOI] [PubMed] [Google Scholar]
- 44.Saeki, H., A.M. Moore, M.J. Brown, and S.T. Hwang. 1999. Cutting edge: secondary lymphoid-tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in the emigration pathway of mature dendritic cells from the skin to regional lymph nodes. J. Immunol. 162:2472–2475. [PubMed] [Google Scholar]
- 45.Maldonado-Lopez, R., C. Maliszewski, J. Urbain, and M. Moser. 2001. Cytokines regulate the capacity of CD8α(+) and CD8α(−) dendritic cells to prime Th1/Th2 cells in vivo. J. Immunol. 167:4345–4350. [DOI] [PubMed] [Google Scholar]
- 46.Maldonado-Lopez, R., and M. Moser. 2001. Dendritic cell subsets and the regulation of Th1/Th2 responses. Semin. Immunol. 13:275–282. [DOI] [PubMed] [Google Scholar]
- 47.Barchet, W., M. Cella, B. Odermatt, C. Asselin-Paturel, M. Colonna, and U. Kalinke. 2002. Virus-induced interferon α production by a dendritic cell subset in the absence of feedback signaling in vivo. J. Exp. Med. 195:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shortman, K. 2000. Burnet oration: dendritic cells: multiple subtypes, multiple origins, multiple functions. Immunol. Cell Biol. 78:161–165. [DOI] [PubMed] [Google Scholar]
- 49.Wu, L., A. D'Amico, K.D. Winkel, M. Suter, D. Lo, and K. Shortman. 1998. RelB is essential for the development of myeloid-related CD8α− dendritic cells but not of lymphoid-related CD8α1 dendritic cells. Immunity. 9:839–847. [DOI] [PubMed] [Google Scholar]
- 50.Traver, D., K. Akashi, M. Manz, M. Merad, T. Miyamoto, E.G. Engleman, and I.L. Weissman. 2000. Development of CD8α-positive dendritic cells from a common myeloid progenitor. Science. 290:2152–2154. [DOI] [PubMed] [Google Scholar]
- 51.Del Hoyo, G.M., P. Martin, H.H. Vargas, S. Ruix, C.F. Arias, and C. Ardavin. 2002. Characterization of a common precursor population for dendritic cells. Nature. 415:1043–1047. [DOI] [PubMed] [Google Scholar]
- 52.Wu, C.Y., H. Maeda, C. Contursi, K. Ozato, and R.A. Seder. 1999. Differential requirement of IFN consensus sequence binding protein for the production of IL-12 and induction of Th1-type cells in response to IFN-gamma. J. Immunol. 162:807–812. [PubMed] [Google Scholar]
- 53.Aliberti, J., O. Schulz, D.J. Pennington, H. Tsujimura, C. Reis e Sousa, K. Ozato, and A. Sher. 2002. Essential role for ICSBP in the in vivo development of murine CD8+ dendritic cells. Blood. 10.1182/blood-2002-04-1088. [DOI] [PubMed]
- 54.Ardavin, C., G. Martinez del Hoyo, P. Martin, F. Anjuere, C.F. Arias, A.R. Marin, S. Ruiz, V. Parrillas, and H. Hernandez. 2001. Origin and differentiation of dendritic cells. Trends Immunol. 22:691–700. [DOI] [PubMed] [Google Scholar]
- 55.Vanbervliet, B., B. Homey, I. Durand, C. Massacrier, S. Ait-Yahia, O. de Bouteiller, A. Vicari, and C. Caux. 2002. Sequential involvement of CCR2 and CCR6 ligands for immature dendritic cell recruitment: possible role at inflamed epithelial surfaces. Eur. J. Immunol. 32:231–242. [DOI] [PubMed] [Google Scholar]
- 56.Kucharzik, T., J.T. Hudsafon, III, R.L. Waikel, W.D. Martin, and I.R. Williams. 2002. CCR6 expression distinguishes mouse myeloid and lymphoid dendritic cell subsets: demonstration using a CCR6 EGFP knock-in mouse. Eur. J. Immunol. 32:104–112. [DOI] [PubMed] [Google Scholar]
- 57.Mattei, F., G. Schiavoni, F. Belardelli, and D.F. Tough. 2001. IL-15 is expressed by dendritic cells in response to type I IFN, double-stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J. Immunol. 167:1179–1187. [DOI] [PubMed] [Google Scholar]
- 58.Hochrein, H., K. Shortman, D. Vremec, B. Scott, P. Hertzog, and M. O'Keeffe. 2001. Differential production of IL-12, IFN-α, and IFN-gamma by mouse dendritic cell subsets. J. Immunol. 166:5448–5455. [DOI] [PubMed] [Google Scholar]