

The mechanisms that regulate NK cell function as a first line of defense against infection and transformation have exceeded our expectations in terms of their sophistication and complexity. Quiescent, circulating NK cells can be activated by either soluble mediators, such as interferons and cytokines, or by direct cell–cell contact. Activation occurs during contact not only when NK cells sense the loss of MHC class I expression on the surface of other cells but also with cells that have undergone other alterations induced by infection or cellular stress. Notably, expression of ligands for receptor NKG2D induced by stress or transformation results in activation signals that can overcome the inhibition mediated by MHC class I–specific receptors (1–4). Yet another mode of NK cell activation upon cellular stress is described in this issue by Michaëlsson et al. (5). Replacement of a peptide on an MHC class I molecule by a peptide released from a heat shock protein prevents recognition by an NK cell inhibitory receptor. Therefore, processing of a heat shock protein, which can occur during the cellular stress that accompanies infection or inflammation, into a peptide that modifies an MHC class I molecule represents another mechanism for the immunosurveillance of stressed cells by NK cells.
Both the innate and adaptive arms of the immune system are galvanized upon infection or transformation of healthy cells. Antigen-specific receptors of the adaptive immune system are selected to make the important distinction between normal structures and those that are either foreign or aberrant. In contrast, the innate immune system uses receptors that recognize structurally conserved molecular patterns on microorganisms, such as polysaccharides and CpG nucleotide sequences. NK cells are a subpopulation of lymphocytes that bridge these two arms of the immune response. While they do not use V(D)J recombination to generate antigen-specific receptors, they nevertheless use a complex arsenal of receptors that either activate or inhibit their effector functions (cytotoxicity and/or cytokine release). There are multiple NK cell activation receptors on a given NK cell, which recognize ligands expressed on other cells, and an array of inhibitory receptors that monitor the integrity of MHC class I expression on other cells.
Missing Self.
The ‘missing self’ hypothesis was the first breakthrough in understanding specific recognition of target cells by NK cells, when it was appreciated that loss of MHC class I expression leads to sensitivity to lysis by NK cells (6). The hypothesis was validated by the discovery of three different families of inhibitory receptors specific for MHC class I on human and mouse NK cells that regulate self-tolerance of NK cells. Human NK cells express the family of killer cell Ig-like receptors (KIRs), and mouse NK cells express the Ly-49 family of lectin-like receptors, that bind classical class I molecules (7, 8). In addition, a heterodimer of the lectin-like CD94 and NKG2A in humans and mice binds the nonclassical class I molecule HLA-E and Qa1, respectively (8). The clonal distribution of these inhibitory receptors is such that some human NK cells are inhibited by recognition of HLA-B or HLA-C by a KIR, while others are inhibited by recognition of HLA-E by CD94/NKG2A (9; Fig. 1) . The down-regulation of MHC molecules frequently observed in tumors and in virus-infected cells can lead to sensitivity to NK cells. The loss of inhibitory signals due to a reduction in HLA class I on target cells usually results in NK cell activation because the ligands of the many activation receptors (e.g., NKp46, NKp44, NKp30, 2B4, etc.) are widely expressed, particularly on hematopoietic cells (10). However, NK cells can also be activated by cells that still express MHC class I.
Figure 1.
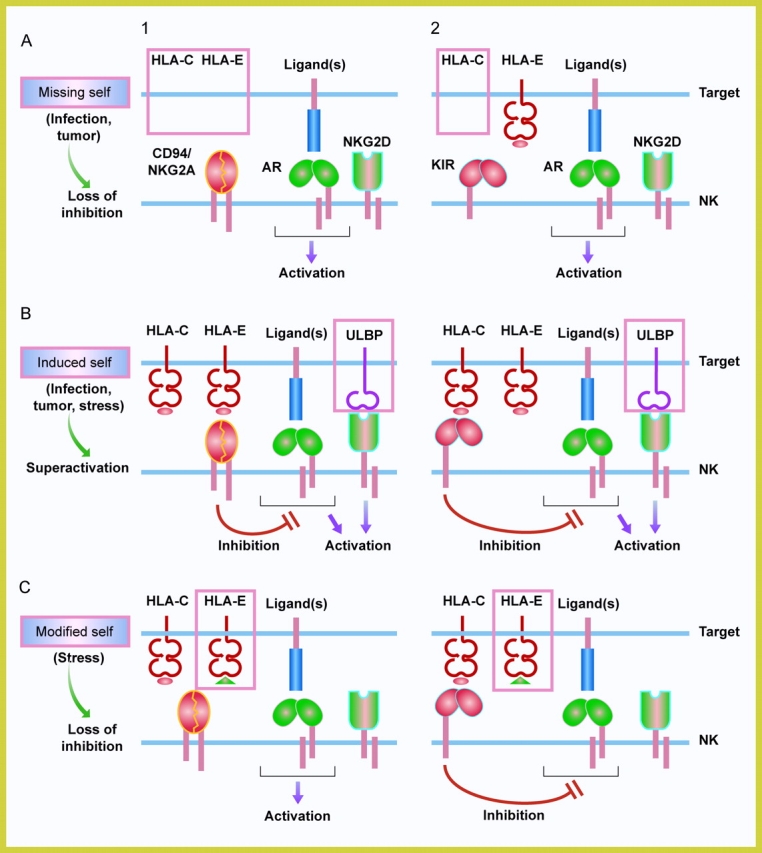
Outcomes of surveillance by NK cells. NK cells survey cells for changes in self, represented here by MHC class I and MHC class I–related molecules (e.g., ULBP, MICA). (1) Depicts an NK cell that is inhibited by the HLA-E-specific receptor CD94/NKG2A, and (2) depicts one that is inhibited by an HLA-C–specific KIR. The KIR family also includes inhibitory receptors with three Ig domains that bind to HLA-A and HLA-B. A representative activation receptor (AR) of the natural cytotoxicity receptor family is shown in association with an immunoreceptor tyrosine-based activation motif (ITAM)-containing subunit, such as DAP12. NK cells express multiple activation receptors on their cell surface. NKG2D is a lectin-like receptor that is associated with the subunit DAP10. (A) “Missing self”: a global down-regulation of the expression of MHC class I molecules, as seen in certain tumors or during viral infections, results in the loss of surface HLA-E. The lack of engagement of the inhibitory CD94/NKG2A leads to activation of NK cells (A1). The loss of a single class I allotype, such as HLA-C, may activate NK cells that express an HLA-C-specific inhibitory KIR (A2). (B) “Induced self”: expression of ligands of NKG2D is induced on target cells due to cellular stress, transformation, or viral infections and results in an activation of NK cells despite the presence of inhibitory receptors (Superactivation). It is not known if NKG2D engagement bypasses inhibition or if it overrides inhibition by enhancing activation signals from other receptors. ULBP is a family of molecules with α1-α2 domains. NKG2D binds also to MICA and MICB, which have α1-α2-α3 domains. ULBP and MICA/B proteins are not associated with β2-microglobulin and do not bind peptides. (C) “Modified self”: stress-induced molecule hsp60 provides a signal sequence-derived peptide that binds to HLA-E and prevents recognition by inhibitory CD94/NKG2A. This results in activation of NK cells expressing CD94NKG2A (C1) but not of NK cells expressing inhibitory KIR (C2).
Stress-inducible Ligands.
It has become evident recently that factors other than MHC class I expression are determinants of sensitivity to NK cells. Binding of activation receptor NKG2D to inducible ligands on target cells leads to elimination by NK cells, despite normal expression of MHC class I (1–4). NKG2D binds with high affinity to a number of different ligands, which are distantly related to MHC class I, such as MICA/MICB and ULBP proteins in humans, and Rae-1 and H60 proteins in mice (11). These proteins are generally not expressed on normal cells but can be induced by stress and up-regulated on tumors and virally infected cells (12–14). Engagement of NKG2D by its ligands on target cells either bypasses or overcomes the inhibition provided by MHC class I–specific receptors (Fig. 1). NKG2D is also expressed by activated CD8+ T cells and is present on some γ/δ T cells and NK1.1+ T cells. NKG2D provides potent costimulation to CD28− virus-specific CD8 T cells (12). In addition, NKG2D expression is inducible in macrophages by treatment with lipopolysaccharides, interferon-α/β, and interferon-γ (15). The ubiquitous expression of NKG2D on NK cells and the inducible expression of its ligands on other cells provides an efficient mechanism for the rapid activation of NK cells which is not tied to down-regulation of MHC class I.
Stress-induced HLA-E Modification.
Another strategy for detection of stressed cells has now been revealed by the work of Michaëlsson et al. (5). HLA-E is the relatively nonpolymorphic ligand of CD94/NKG2A and CD94/NKG2C. It has a very selective preference for nonameric peptides, derived from the signal sequence of other class I molecules, that contain conserved anchor residues at position 2 and 9 (16). However, the amino acid sequence of the peptide in HLA-E is known to affect binding by CD94/NKG2A (17, 18). Similarly, the mouse functional homologue of HLA-E, Qa1, binds mouse CD94/NKG2A only if certain amino acids are present at position 5 (19). In fact, Kraft et al. have suggested that competitive peptide replacement in Qa1 may provide a mechanism for activation of NK cells (19). In the new study reported in this issue (5), a peptide from the signal sequence of stress protein hsp60 is shown to load onto HLA-E and to compete effectively with an MHC class I–derived peptide. Furthermore, HLA-E loaded with the hsp60-derived peptide does not bind CD94/NKG2A or CD94/NKG2C. The side chain at position 5 of that peptide contributes to the lack of binding. The authors further show that coexpression of hsp60 with HLA-E in transfected cells up-regulates surface expression of HLA-E. Enhanced surface expression of HLA-E was also observed in stressed cells and did not result in increased protection from NK cells. Although not shown experimentally, it seems feasible that peptides derived from stress-induced hsp60 could achieve competitive peptide replacement in HLA-E in a physiological situation because peptides derived from signal sequences of MHC class I are apparently in limited supply. Indeed, expression of several HLA class I alleles is necessary to obtain maximal protection from NK cells that are inhibited through HLA-E recognition by CD94/NKG2A (9).
A competing strategy to avoid recognition by NK cells is used by CMV. A peptide derived from the signal sequence of the CMV glycoprotein UL40 binds to HLA-E and up-regulates its expression (20). The UL40-derived peptide is loaded onto HLA-E independently of the peptide transporter TAP and is recognized by CD94/NKG2A, which makes escape possible even in cells where TAP has been disabled. The specific nature of the peptide bound to HLA-E can also modulate NK cell responses in other ways. Some NK cells express the activating receptor CD94/NKG2C. Surprisingly, HLA-E loaded with a peptide derived from the signal sequence of the nonclassical class I molecule HLA-G, whose expression is limited to a few cell types, favored recognition by NKG2C over that by the inhibitory NKG2A (21). Even though binding of tetrameric HLA-E loaded with the hsp60 signal peptide to cells expressing CD94/NKG2C was undetectable, it would be interesting to test if there is functional recognition of stress-modified HLA-E by this activating receptor, as it would enhance NK cell responses even further. In addition to its role in inhibition of NK cells, HLA-E loaded with self and viral peptides can activate a subset of CD8+ T cells (22). It remains to be seen whether the hsp60-derived peptide will suppress or activate T cell responses, by either competing with peptides recognized by those T cells or by inducing hsp60-specific T cell responses.
Peptide Matters.
The influence of MHC class I–bound peptide on recognition by inhibitory receptors had previously been reported for the KIR3DL and KIR2DL receptors specific for HLA-B and HLA-C, respectively (23, 24). Although many different peptides are compatible with recognition by KIR, certain side chains at position 7 and 8 of the nonamer peptide interfere with KIR binding. Crystal structures of two KIR2DL:HLA-C complexes confirmed why these receptors bind only when certain peptide side chains are present. Direct contacts are made by KIR with the peptide backbone, and certain peptide side chains cannot be accommodated in the tight space (25, 26).
The reason for the peptide selectivity in the recognition of HLA-B and HLA-C by inhibitory KIR has remained elusive. It is clear that such selectivity is not the basis for self/nonself discrimination by NK cells since there is no correlation between the protective effect of a peptide and its origin. In other words, self-peptides and viral peptides that have been eluted from HLA class I come in both flavors, those that are compatible with KIR binding and those that are not. Regardless, a strategy similar to that used by HLA-E through a stress-induced peptide is not feasible in the case of KIR recognition because of the very high allelic polymorphism of its HLA-B and HLA-C ligands. Each inhibitory KIR recognizes a large group of HLA ‘allotypes’ that share specific structural determinants but otherwise bind completely different sets of peptides. Therefore, it is not possible for a nonpolymorphic self protein to carry all possible combinations of anchor residues for binding to multiple HLA allotypes and to include an amino acid side chain at the right position in all cases to block KIR binding.
Unlike inhibitory KIR which bind MHC class I on top of the peptide binding groove, the lectin-like Ly-49A inhibitory receptor in mice has a completely different binding site, far removed from the peptide binding site (27, 28). Thus, there is no apparent peptide specificity for MHC class I recognition by Ly49A, and the contribution of peptides bound to H-2Dd is to ensure proper folding and cell surface expression (29).
Conclusions.
In addition to revealing a novel strategy used by stressed cells to initiate NK cell activation, the new findings of Michaëlsson et al. also illustrate how useful the dual system of inhibition by KIR and CD94/NKG2A can be. These two receptor systems are not simply redundant. For example, loss of a single HLA-B or HLA-C allele, as occurs frequently in tumor cells, could not be detectable by NK cells expressing CD94/NKG2A, whereas NK cells inhibited by a KIR specific for the product of the lost allele will get activated (Fig. 1). Conversely, stress-induced modification of HLA-E would not prevent inhibition of NK cells expressing inhibitory KIR but would activate CD94/NKG2A-expressing NK cells due to loss of inhibition (Fig. 1). It will be important to test the role of HLA-E in alerting NK cells to cellular stress in physiological settings such as infections or inflammation. Furthermore, given the widespread expression of CD94/NKG2A on cytotoxic T cells, it remains to be seen whether this stress-induced immunosurveillance mechanism can also switch the inhibition of T cell effector functions to activation, thereby influencing the adaptive arm of the immune response as well.
References
- 1.Bauer, S., V. Groh, J. Wu, A. Steinle, J.H. Phillips, L.L. Lanier, and T. Spies. 1999. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 285:727–729. [DOI] [PubMed] [Google Scholar]
- 2.Cosman, D., J. Müllberg, C.L. Sutherland, W. Chin, R. Armitage, W. Fanslow, M. Kubin, and N.J. Chalupny. 2001. ULBPs, novel MHC class I-related molecules bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity. 14:123–133. [DOI] [PubMed] [Google Scholar]
- 3.Diefenbach, A., E.R. Jensen, A.M. Jamieson, and D.H. Raulet. 2001. Rae1 and H60 ligands of the NKG2D receptor stimulate tumour immunity. Nature. 413:165–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cerwenka, A., J.L. Baron, and L.L. Lanier. 2001. Ectopic expression of retinoic acid early inducible-1 gene (RAE-1) permits natural killer cell-mediated rejection of a MHC class I-bearing tumor in vivo. Proc. Natl. Acad. Sci. USA. 98:11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Michaëlsson, J., C. Teixeira de Matos, A. Achour, L.L. Lanier, K. Kärre, and K. Söderström. 2002. HLA-E–mediated presentation of an hsp60 signal peptide prevents natural killer (NK) cell inhibitory receptor engagement: a novel mechanism for NK cell detection of stressed cells. J. Exp. Med. 196:1403–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kärre, K., H.G. Ljunggren, G. Piontek, and R. Kiessling. 1986. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature. 319:675–678. [DOI] [PubMed] [Google Scholar]
- 7.Long, E.O., and S. Rajagopalan. 2000. HLA class I recognition by killer cell Ig-like receptors. Semin. Immunol. 12:101–108. [DOI] [PubMed] [Google Scholar]
- 8.Raulet, D.H., R.E. Vance, and C.W. McMahon. 2001. Regulation of the natural killer cell receptor repertoire. Annu. Rev. Immunol. 19:291–330. [DOI] [PubMed] [Google Scholar]
- 9.Valiante, N.M., M. Uhrberg, H.G. Shilling, K. Lienert-Weidenbach, K.L. Arnett, A. D'Andrea, J.H. Phillips, L.L. Lanier, and P. Parham. 1997. Functionally and structurally distinct NK cell receptor repertoires in the peripheral blood of two human donors. Immunity. 7:739–751. [DOI] [PubMed] [Google Scholar]
- 10.Moretta, A., C. Bottino, M. Vitale, D. Pende, C. Cantoni, M.C. Mingari, R. Biassoni, and L. Moretta. 2001. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu. Rev. Immunol. 19:197–223. [DOI] [PubMed] [Google Scholar]
- 11.Cerwenka, A., and L.L. Lanier. 2001. Natural killer cells, viruses, and cancer. Nat. Rev. Immunol. 1:41–49. [DOI] [PubMed] [Google Scholar]
- 12.Groh, V., R. Rhinehart, J. Randolph-Habecker, M.S. Topp, S.R. Riddell, and T. Spies. 2001. Costimulation of CD8alphabeta T cells by NKG2D via engagement by MIC induced on virus-infected cells. Nat. Immunol. 2:255–260. [DOI] [PubMed] [Google Scholar]
- 13.Das, H., V. Groh, C. Kuijl, M. Sugita, C.T. Morita, T. Spies, and J.F. Bukowski. 2001. MICA engagement by human Vgamma2Vdelta2 T cells enhances their antigen-dependent effector function. Immunity. 15:83–93. [DOI] [PubMed] [Google Scholar]
- 14.Tieng, V., C. Le Bouguénec, L. Du Merle, P. Bertheau, P. Desreumaux, A. Janin, D. Charron, and A. Toubert. 2002. Binding of Escherichia coli adhesin AfaE to CD55 triggers cell-surface expression of the MHC class I-related molecule MICA. Proc. Natl. Acad. Sci. USA. 99:2977–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jamieson, A.M., A. Diefenbach, C.W. McMahon, N. Xiong, J.R. Carlyle, and D.H. Raulet. 2002. The role of the NKG2D immunoreceptor in immune cell activation and natural killing. Immunity. 17:19–29. [DOI] [PubMed] [Google Scholar]
- 16.O'Callaghan, C.A., J. Tormo, B.E. Willcox, V.M. Braud, B.K. Jakobsen, D.I. Stuart, A.J. McMichael, J.I. Bell, and E.Y. Jones. 1998. Structural features impose tight peptide binding specificity in the nonclassical MHC molecule HLA-E. Mol. Cell. 1:531–541. [DOI] [PubMed] [Google Scholar]
- 17.Brooks, A.G., F. Borrego, P.E. Posch, A. Patamawenu, C.J. Scorzelli, M. Ulbrecht, E.H. Weiss, and J.E. Coligan. 1999. Specific recognition of HLA-E, but not classical, HLA class I molecules by soluble CD94/NKG2A and NK cells. J. Immunol. 162:305–313. [PubMed] [Google Scholar]
- 18.Valés-Gómez, M., H.T. Reyburn, R.A. Erskine, M. López-Botet, and J.L. Strominger. 1999. Kinetics and peptide dependency of the binding of the inhibitory NK receptor CD94/NKG2-A and the activating receptor CD94/NKG2-C to HLA-E. EMBO J. 18:4250–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kraft, J.R., R.E. Vance, J. Pohl, A.M. Martin, D.H. Raulet, and P.E. Jensen. 2000. Analysis of Qa-1b peptide binding specificity and the capacity of CD94/NKG2A to discriminate between Qa-1-peptide complexes. J. Exp. Med. 192:613–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomasec, P., V.M. Braud, C. Rickards, M.B. Powell, B.P. McSharry, S. Gadola, V. Cerundolo, L.K. Borysiewicz, A.J. McMichael, and G.W.G. Wilkinson. 2000. Surface expression of HLA-E, an inhibitor of natural killer cells, enhanced by human cytomegalovirus gpUL40. Science. 287:1031–1033. [DOI] [PubMed] [Google Scholar]
- 21.Llano, M., N. Lee, F. Navarro, P. García, J.P. Albar, D.E. Geraghty, and M. López-Botet. 1998. HLA-E-bound peptides influence recognition by inhibitory and triggering CD94/NKG2 receptors: preferential response to an HLA-G-derived nonAm. Eur. J. Immunol. 28:2854–2863. [DOI] [PubMed] [Google Scholar]
- 22.Romagnani, C., G. Pietra, M. Falco, E. Millo, P. Mazzarino, R. Biassoni, A. Moretta, L. Moretta, and M.C. Mingari. 2002. Identification of HLA-E-specific alloreactive T lymphocytes: A cell subset that undergoes preferential expansion in mixed lymphocyte culture and displays a broad cytolytic activity against allogeneic cells. Proc. Natl. Acad. Sci. USA. 99:11328–11333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malnati, M.S., M. Peruzzi, K.C. Parker, W.E. Biddison, E. Ciccone, A. Moretta, and E.O. Long. 1995. Peptide specificity in the recognition of MHC class I by natural killer cell clones. Science. 267:1016–1018. [DOI] [PubMed] [Google Scholar]
- 24.Rajagopalan, S., and E.O. Long. 1997. The direct binding of a p58 killer cell inhibitory receptor to human histocompatibility leukocyte antigen (HLA)-Cw4 exhibits peptide selectivity. J. Exp. Med. 185:1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyington, J.C., S.A. Motyka, P. Schuck, A.G. Brooks, and P.D. Sun. 2000. Crystal structure of an NK cell immunoglobulin-like receptor in complex with its class I MHC ligand. Nature. 405:537–543. [DOI] [PubMed] [Google Scholar]
- 26.Fan, Q.R., E.O. Long, and D.C. Wiley. 2001. Crystal structure of the human natural killer cell inhibitory receptor KIR2DL1-HLA-Cw4 complex. Nat. Immunol. 2:452–460. [DOI] [PubMed] [Google Scholar]
- 27.Tormo, J., K. Natarajan, D.H. Margulies, and R.A. Mariuzza. 1999. Crystal structure of a lectin-like natural killer cell receptor bound to its MHC class I ligand. Nature. 402:623–631. [DOI] [PubMed] [Google Scholar]
- 28.Matsumoto, N., M. Mitsuki, K. Tajima, W.M. Yokoyama, and K. Yamamoto. 2001. The functional binding site for the C-type lectin-like natural killer cell receptor Ly49A spans three domains of its major histocompatibility complex class I ligand. J. Exp. Med. 193:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Correa, I., and D.H. Raulet. 1995. Binding of diverse peptides to MHC class I molecules inhibits target cell lysis by activated natural killer cells. Immunity. 2:61–71. [DOI] [PubMed] [Google Scholar]