

Summary
Mre11/Rad50 complexes in all organisms function in the repair of DNA double-strand breaks. In budding yeast, genetic evidence suggests that the Sae2 protein is essential for the processing of hairpin DNA intermediates and meiotic double-strand breaks by Mre11/Rad50 complexes, but the biochemical basis of this functional relationship is not known. Here we demonstrate that recombinant Sae2 binds DNA and exhibits endonuclease activity on single-stranded DNA independently of Mre11/Rad50 complexes, but hairpin DNA structures are cleaved cooperatively in the presence of Mre11/Rad50 or Mre11/Rad50/Xrs2. Hairpin structures are not processed at the tip by Sae2 but rather at single-stranded DNA regions adjacent to the hairpin. Truncation and missense mutants of Sae2 inactivate this endonuclease activity in vitro, and fail to complement Δsae2 strains in vivo for meiosis and recombination involving hairpin intermediates, suggesting that the catalytic activities of Sae2 are important for its biological functions.
Introduction
DNA double-strand breaks (DSBs) in chromosomal DNA are recombinogenic in that they can lead to non-homologous or homologous recombination between the DNA ends and with other parts of the genome. Numerous factors have been identified in eukaryotic cells that mediate and regulate recombination, including the Mre11/Rad50 complex which is conserved in all organisms and functions in the repair of DNA double-strand breaks as well as in the signaling of DNA damage. In budding yeast, Mre11 and Rad50 also associate with the Xrs2 protein which mediates the signaling functions of the complex and is the functional equivalent of Nbs1 (Nibrin) in mammalian cells.
Null strains of any of the components of the Mre11/Rad50/Xrs2 (MRX) complex in S. cerevisiae confer extreme sensitivity to ionizing radiation and other DNA damaging agents, defects in telomere maintenance, non-homologous end joining (NHEJ), and meiotic DSB repair (Krogh and Symington, 2004). Genetic studies have revealed that mre11, rad50 and xrs2 strains exhibit hyper-recombination in vegetatively growing cells and also accumulate gross chromosomal rearrangements (Chen and Kolodner, 1999; Symington, 2002). Deletion of any of the MRX components delays resection of DSB ends; however, the deletion strains still complete homologous recombination with nearly wild-type efficiency (Ivanov et al., 1994). NHEJ is also defective in Δmre11, Δrad50, and Δxrs2 strains, and end joining between linear DNA ends is reduced as severely as in strains lacking Ku70 and Ku80 proteins (Boulton and Jackson, 1998; Moore and Haber, 1996).
Several rad50 hypomorphic mutants were isolated by Kleckner and colleagues in which meiotic recombination was initiated but blocked by covalent Spo11-DNA intermediates (Alani et al., 1990; Keeney et al., 1997). These rad50S mutants are only mildly sensitive to MMS when compared to Δrad50 strains during vegetative growth, and therefore are considered separation of function mutants. mre11S mutants were also identified and exhibit phenotypes similar to the rad50S mutants (Nairz and Klein, 1997; Tsubouchi and Ogawa, 1998), as do strains expressing Mre11 nuclease-deficient mutants (Moreau et al., 1999). In contrast, Spo11-dependent breaks do not occur in Δrad50, Δmre11, or Δxrs2 deletion strains. Taken together, this evidence suggests that MRX is involved in the removal of protein adducts from DSBs during meiosis and may also be involved in the 5′ to 3′ resection of ends that is required for 3′ overhang formation and the initiation of meiotic recombination.
The SAE2/COM1 gene was also shown to be required for meiotic DSB repair (McKee and Kleckner, 1997; Prinz et al., 1997). The Δsae2 sporulation phenotype is identical to rad50S mutants in that Spo11-conjugated DNA breaks accumulate in these strains, the first suggestion that the role of Sae2 may intersect with that of the MRX complex. More recently, Sae2 was also found to play important roles in DNA recombination in vegetative cells. Rattray et al. isolated mutant alleles of Sae2 in a genetic screen for intrachromosomal recombination between inverted repeats (Rattray et al., 2001). In this assay, Δsae2 strains, as well as rad50S mutants and mre11 nuclease-deficient mutants, accumulate aberrant recombination products that included a large amplification of part of the genomic DNA between the repeats. In further analysis of these products, Rattray et al. suggest that Δsae2/rad50S strains may fail to process a hairpin fold-back structure that arises during the course of break-induced replication, thus leading to chromosomal amplification events (Rattray et al., 2005).
Lobachev et al. also described a role for Sae2 and the MRX complex in processing of hairpin-capped ends using a different genetic assay in S. cerevisiae (Lobachev et al., 2002). Closely-spaced inverted repeats were inserted into the LYS2 gene on chromosome II while a 5′ truncated lys2 allele was present on chromosome III. The presence of the inverted repeats was shown to increase homologous recombination between the lys2 alleles by over 1000-fold and DSBs were seen at the site of the inverted repeats, suggesting that the repeats extruded into cruciform structures in vivo which are highly susceptible to breakage. Δsae2, Δmrx, rad50S, and strains expressing Mre11 nuclease-deficient mutants failed to promote homologous recombination at the site of the inverted repeats. Moreover, these strains accumulated chromosomes with hairpin-capped ends at the sites of the inverted repeats, as well as large amplifications of the entire chromosome arm adjacent to the repeats. Taken together, these results suggest that Sae2 and the MRX complex play a unique role in S. cerevisiae in the processing of DNA structures containing hairpins, and that Mre11 nuclease activity also is essential for this process.
Hairpin processing has previously been linked to the function of the Mre11/Rad50 complex in E. coli (Gibson et al., 1992). Leach and colleagues showed that closely-spaced inverted repeats that are likely to form cruciform structures are unstable in wild-type E. coli strains but are stabilized in strains lacking the SbcC/D complex, which are homologs of Rad50 and Mre11, respectively. Consistent with this observation, SbcC/D and MRN(X) homologs from humans and from yeast were shown to exhibit manganese-dependent cleavage of hairpin structures in vitro, in addition to 3′ to 5′ exonuclease activity (Connelly et al., 1998; Paull and Gellert, 1998; Trujillo and Sung, 2001).
In this study we address the role of the Sae2 protein in hairpin processing with the S. cerevisiae MRX complex, using purified proteins in vitro. Surprisingly, we found that Sae2 itself exhibits endonuclease activity on single-stranded DNA and single-strand/double-strand transitions and shows a strong preference for hairpin structures. MRX appears to stimulate Sae2 endonuclease activity on single-stranded DNA adjacent to hairpins, and also facilitates Sae2 endonucleolytic cutting through its 3′ to 5′ exonucleolytic activity. These results provide a biochemical explanation for the observed cooperativity of MRX and Sae2 in vivo on hairpin structures.
RESULTS
Recombinant Sae2 expression and mutant analysis
To determine the biochemical basis of the role of Sae2 in DNA repair we created an expression construct for recombinant S. cerevisiae Sae2 protein that includes his6 and maltose-binding protein (MBP) affinity tags to facilitate purification and to maximize solubility. To determine essential domains necessary for Sae2 activities we also created several mutant forms of the gene based on sequence homology, genetic data, and published literature describing Sae2 phosphorylation (Fig. 1A). The C-terminus of Sae2 shows the highest degree of conservation among Sae2 homologs in other fungi (Supp. Fig. 1), thus a C-terminal truncation was made that removes amino acids 251–345 (Sae2(ΔC)). In addition, internal SacI restriction sites were used to remove amino acids 21–172, leaving the 20 amino acids of the N-terminus in frame with 173–345 at the C-terminus (Sae2(ΔN)). We also made a specific missense mutant, Sae2(G270D), which was discovered in a genetic screen for mutants deficient in intrachromosomal recombination in vivo (Rattray et al., 2001)(data not shown). Lastly, the Sae2(5A) and Sae2(5D) were based on the putative Tel1/Mec1 phosphorylation sites, S73, T90, S249, T279, and S289 identified by the Longhese group (Baroni et al., 2004). In the Sae2(5A) mutant each serine or threonine was changed to an alanine. Baroni et al. showed that yeast strains expressing the Sae2(5A) mutant were nearly as sensitive to methyl-methane-sulfonate (MMS) as Δsae2 strains. In the Sae2(5D) mutant the five phosphorylation sites were changed to aspartates to mimic phosphorylation.
Figure 1.

Expression and DNA binding activity of recombinant Sae2. (A) Schematic representation of full-length wild-type Sae2, and mutants G270D, 5D (residues indicated changed to aspartate), 5A (residues indicated changed to alanine), ΔN (Δ a.a. 21 to 173) and ΔC (Δ a.a. 251 to 345). (B) SDS-PAGE of purified Sae2 wild-type and mutant proteins, approximately 100 ng total protein each. (C) Wild-type (wt) and mutant Sae2 proteins were incubated with a 249 bp double-stranded DNA substrate and analyzed in a 8% native polyacrylamide gel in the presence of wild-type (wt) or R20M (RM) MRX complex. Protein-DNA complex 1 and complex 2 are indicated (described in text).
The wild-type and mutant Sae2 proteins were expressed in E. coli and purified using multiple affinity, ion-exchange, and gel filtration chromatographic steps (see Experimental Procedures). This purification strategy yielded recombinant Sae2 proteins at nearly 100% homogeneity (Fig. 1B). The last step of the purification procedure involves separation on a Superdex200 gel filtration column, in which the wild-type Sae2 protein and all of the mutants except for the Sae2(ΔN) eluted as three distinct multimeric forms (Supp. Fig. 2). Sedimentation analysis of these forms by analytical ultracentrifugation suggests that recombinant wild-type Sae2 forms oligomeric complexes, primarily dimeric, consistent with the reported self-interaction of Sae2 in 2-hybrid assays (Uetz et al., 2000). In contrast to the wild-type protein, the Sae2(ΔN) mutant protein lacks the dimeric form and shows a much more prominent peak later in the elution profile which was determined to be a monomer (Supp. Fig. 4). Thus, the Sae2(ΔN) mutant does not exhibit the peak corresponding to the wild-type dimer and therefore may lack a self-association domain. Analysis of a functional YFP-Sae2 fusion protein expressed from the endogenous locus (Lisby et al., 2004) showed a predominant species that eluted from gel filtration similar to the dimer peak from the recombinant wild-type MBP-Sae2 protein (data not shown), suggesting that Sae2 is also present as a multimer in vivo.
Sae2 binds to DNA
Sae2 has been shown to form foci at sites of DSBs, both in the presence and absence of MRX (Lisby et al., 2004). To test whether Sae2 is itself a DNA-binding protein we performed gel mobility shift assays. Sae2 was incubated with a [32P]-labeled 249 bp blunt double-stranded DNA substrate and analyzed in a 8% native acrylamide gel, as shown in Fig. 1C. We found that Sae2 forms a stable complex with DNA, evident by the mobility shift of the substrate (Fig. 1C, lane 9, “complex 1”). A comparison of protein from each of the gel filtration peaks showed that the aggregate (peak #1) did not bind DNA, but the dimer form (peak #2) and the putative monomer form produced stable complexes with DNA. (Fractions in the range of the putative monomeric peak were used for all of the experiments shown here, for wild-type as well as mutant proteins, because the binding efficiency was highest with this fraction and also because it facilitated a direct comparison with the Sae2(ΔN) mutant.) The Sae2(5D), Sae2(5A) and Sae2(ΔC) mutants all bound DNA similarly compared to wildtype Sae2 (Fig. 1C, lanes 11, 12, and 14) while the Sae2(G270D) and the Sae2(ΔN) mutant proteins showed a nearly complete loss of DNA-binding activity (Figure 1C, lanes 10 and 13).
Since in vivo data suggest that MRX and Sae2 cooperate in DNA end processing, we also investigated whether MRX affects Sae2 DNA binding. To do this, recombinant MRX was expressed in a baculovirus system similar to the coexpression of human MRN complexes (Paull and Gellert, 1998, 1999) (Supp. Fig. 5). MRX alone binds DNA but at these concentrations (11 nM) does not form discrete higher order complexes in this assay (Fig. 1C, lane 2). Interestingly, in the presence of both MRX and Sae2, the majority of the labeled DNA migrated more slowly in the gel, forming a complex that was not observed with either protein preparation alone (Fig. 1C, lane 3, “complex 2”). A larger complex was also formed, similar to the protein-DNA shift with Sae2 alone (“complex 1”), but the complex in the presence of MRX migrated slightly higher in the gel compared to the complex in the absence of MRX (Fig. 1C, compare lanes 16 and 17).
The Sae2 mutants were also tested in combination with wild-type MRX, which showed that neither the Sae2(G270D) nor the Sae2(ΔN) mutant formed the cooperative protein-DNA complex #2. We also tested for protein-protein interactions between MRX and wild-type Sae2 in the absence of DNA in vitro but no significant interaction was observed (data not shown), suggesting that if these proteins do directly interact with each other it must be in the context of a protein-DNA complex.
Effects of Sae2 on MRX exo- and endonuclease activities
Sae2 has been shown to act cooperatively with MRX in vivo (Lobachev et al., 2002), therefore we tested whether Sae2 affects MRX nuclease activity in vitro. Purified recombinant MRX complex exhibited 3′Π5′ exonuclease activity on a 3′ recessed double strand DNA substrate in manganese but not in magnesium, as was reported previously for MRX (Trujillo and Sung, 2001) and similar to our previous findings with human MRN (Paull and Gellert, 1998, 1999)(Fig. 2A, lanes 2–5). The addition of the Sae2 protein stimulated the exonuclease activity of the MRX complex 2 to 5-fold when compared to MRX alone, and Sae2 did not exhibit any exonuclease activity by itself on this substrate (Fig. 2A, lanes 6–8 and 9–10, respectively). Wildtype MR was also tested and showed similar exonuclease activity to the full complex, while complexes containing Mre11 nuclease domain mutants, M(D16A)R and Mre11-3/R, did not exhibit exonuclease activity (Fig. 2C), as previously predicted (Bressan et al., 1998; Furuse et al., 1998; Lewis et al., 2004).
Figure 2.
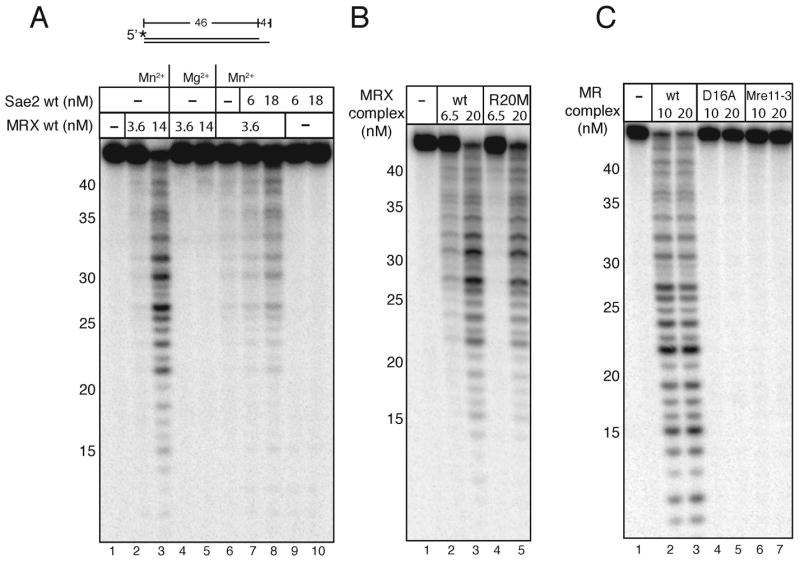
Nuclease activities of MR and MRX complexes. (A) MRX was incubated with a 5′ [32P]-labeled 46 bp DNA substrate containing a 4 nt recessed 3′ end in 1 mM MnCl2 or 5 mM MgCl2 with wild-type Sae2 protein as indicated. (B) The Rad50S MR(R20M)X complex was assayed in comparison to the wild-type complex as in (A) in 1 mM MnCl2. (C) The nuclease-deficient complexes M(D16A)R and M(Mre11-3)R were assayed in comparison to the wild-type MR complex as in (A) in 1 mM MnCl2.
rad50S strains exhibit a phenotype similar to that of Δsae2 strains in meiosis, as well as in cruciform recombination assays in vegetative cells (Alani et al., 1990; Lobachev et al., 2002). The specific defect in Rad50S MRX complexes is not understood, but a recent study showed that rad50S strains could be partially suppressed for defects in single-strand annealing by the overexpression of Sae2 (Clerici et al., 2005). To test the idea that Rad50S mutants exhibit a defect in Sae2 association or functional cooperativity, we expressed and purified one of the Rad50S mutants described by Alani et al., MR(R20M)X (Alani et al., 1990). We found that this mutant complex is active in forming the unique DNA-protein complex that requires both Sae2 and MRX (Fig. 1C, lane 18) and shows wild-type levels of 3′ to 5′ exonuclease activity in manganese (Fig. 2B, lane 5).
Lobachev et al. demonstrated that MRX and Sae2 are required for the resolution of hairpin-capped DSBs at sites of cruciform formation in vivo (Lobachev et al., 2002). Considering this evidence, we tested our MR and MRX complexes for hairpin endonuclease activity on an oligonucleotide hairpin substrate in vitro and found that both forms of the complex exhibit relatively inefficient but detectable cutting of the hairpin at the tip (Fig. 3A, lane 5; here shown with MR). A previous study showed that recombinant yeast MR and MRX cut DNA hairpin structures at the tip (Trujillo et al., 2003; Trujillo and Sung, 2001), similar to results with E. coli SbcC/D (Connelly et al., 1998) and with human MRN (Paull and Gellert, 1999). In contrast to our expectation, the addition of Sae2 did not increase MR hairpin cutting activity (Fig. 3A, lanes 3–4 and 6–7).
Figure 3.

Sae2 cleaves hairpin DNA. (A) Wild-type MR and Sae2 were incubated with a 3′ [32P]-labeled hairpin DNA substrate with 1 mM MnCl2 and 0.5 mM ATP as indicated and separated in a denaturing polyacrylamide gel. Substrate was also incubated with Mung Bean nuclease as a control to show the location of hairpin cut at the tip (MB). Numbers in the “M” lane indicate the positions of DNA standards run in the same gel. (B) Reactions were performed as in (A) with Sae2 only, 5 mM MgCl2, and internally-labeled hairpin substrates as shown. (C) Reactions were performed with substrates identical to those in (B) except lacking the hairpin loops.
Sae2 endonuclease activity
During the course of these experiments, we noticed that the addition of recombinant Sae2 (35 nM) to the hairpin substrate resulted in the production of several novel products (Fig. 3A, lanes 4 and 9) that were not seen with the MR complex alone. The size of the products suggested that these were fragments of the 3′ overhang adjacent to the hairpin, not cleavage products from the hairpin loop. This was a striking observation, as Sae2 was not expected to be a nuclease.
In addition to the MRX-independent products observed with Sae2 alone, we also found that the presence of MRX stimulated the production of 3′ overhang products by lower levels of Sae2 (3 nM), as seen in Fig. 3A, comparing lanes 2, 3, and 8. With this level of Sae2, no cleavage products were observed in the absence of MRX, but significant cutting was seen in the presence of both Sae2 and MRX (lane 3).
To investigate the MRX-independent DNA cleavage by Sae2 in greater detail, we generated a similar hairpin substrate but with an internal [32P] label, as shown in Fig. 3B (substrate 1). Addition of high levels of wild-type Sae2 to this substrate generated products consistent with cleavage of the substrate within the single-stranded overhang (Fig. 3B, lanes 2–4). Unlike MRX nuclease activity which is manganese-specific, this activity was seen in both magnesium and manganese, and the assays shown here were performed in magnesium. To test the hypothesis that Sae2 cuts single-stranded DNA adjacent to hairpin structures, variants of this substrate were generated that included complementary strands annealed to the overhang to block potential cut sites but retain either a 5 or 11 nucleotide gap. When compared to the hairpin without a complementary strand, Sae2 was able to cut the variant including an 11-nucleotide gap but with lower efficiency (Fig. 3B, lanes 10–12). Reduction of the gap to 5 nucleotides resulted in nearly complete inhibition of Sae2 cutting activity (Fig. 3B, lanes 6–8). These results demonstrated that Sae2 itself is an endonuclease that can cleave single-stranded DNA at ends or in gaps.
To investigate the requirement for DNA hairpin substrates in Sae2 endonuclease activity, duplex DNA substrates were created that are identical to the hairpin structures in Fig. 3B but lacking the hairpin loops. Sae2 showed minimal endonuclease activity on the substrate containing the 3′ overhang (Fig. 3C, lanes 2–4), 10-fold lower compared to the equivalent hairpin-containing substrate. The cut sites were also not as strongly localized to the single-stranded region adjacent to the single-strand/double-strand transition as observed with the hairpin substrate. Sae2 also exhibited even lower activity on the substrates containing complementary strands (Fig. 3C, lanes 6–8 and 10–12). The endonuclease activity of Sae2 on single-stranded DNA therefore appears to be strongly stimulated by the presence of an adjacent hairpin structure.
Hairpin removal by Sae2 and MRX
The ability of Sae2 to cleave DNA adjacent to hairpin structures suggested the possibility that MRX might stimulate Sae2, in contrast to our expectation that Sae2 would stimulate MRX activity. To test this hypothesis, an internally labeled hairpin (as in Fig. 3B, substrate 1) was prepared and incubated with recombinant MRX and Sae2 in the presence of magnesium, which supports Sae2 but not MRX endonuclease activity. Although Sae2 can cleave a single-stranded overhang adjacent to the hairpin without MRX (Fig. 3B), the concentrations of Sae2 used here in combination with MRX were 3 to 12-fold lower than the levels required for Sae2 endonuclease activity in the absence of MRX. Under these conditions (1.5 to 3 nM Sae2), minimal cleavage of the substrate with Sae2 alone was observed (Fig. 4A, lanes 5 and 6). However, in the presence of both Sae2 and MRX, significantly more cleavage of the hairpin was seen (Fig. 4A, lanes 3 to 4), 10-fold higher levels of cleavage compared with Sae2 alone.
Figure 4.

Sae2 and MRX cooperatively cleave hairpin structures. (A) Wild-type MRX and Sae2 were incubated in 5 mM MgCl2 with an internally [32P]-labeled hairpin DNA substrate as in Fig. 3B. The primary cleavage products (arrow) are not cut at the tip of the hairpin but in the single-stranded overhang as shown in the diagram. MB control as in Fig. 3. (B) Reactions were performed as in (A) but with substrates lacking the hairpin loop. (C) Wild-type MRX and Sae2 were incubated in 5 mM MgCl2 with an internally [32P]-labeled hairpin DNA substrate with the opposite polarity compared to the substrate shown in (A). (D) Wild-type MRX and wild-type and mutant Sae2 proteins were incubated in 5 mM MgCl2 with an internally [32P]-labeled hairpin DNA substrate as in (B). (E) Wild-type Sae2 and wild-type MRX and Rad50S MR(R20M)X complexes were assayed as in (A).
It is important to note also that the levels of the MRX complex required for this cooperative activity with Sae2 (10 nM) were 16-fold lower than the levels required for hairpin cleavage at the tip by MRX alone (Fig. 3A). The stimulatory effect of MRX on Sae2 on this substrate was specific to hairpin structures, because no stimulation or cooperativity was observed with an identical DNA substrate that lacked the hairpin structure (Fig. 4B, lanes 3–4). MRX and Sae2 thus act cooperatively on a hairpin substrate to cleave the single-stranded region adjacent to the hairpin stem. The fact that this activity occurs in magnesium indicates that Mre11 nuclease activity is not absolutely required for product formation. We also created an internally labeled hairpin substrate with the opposite polarity and found that this substrate was also cut by MRX and Sae2 (Fig. 4C, lanes 3 and 4), indicating that the polarity of the single-stranded DNA adjacent to the hairpin does not affect the cooperative activity of MRX and Sae2.
The Sae2 mutants described above were tested in comparison to wild-type Sae2 with MRX on the hairpin substrate, as shown in Fig. 4D. None of the Sae2 proteins showed any activity in the absence of MRX (Fig. 4D, lanes 9 to 14). The Sae2(5D) mutant was active in cooperative cleavage of the hairpin with MRX in this assay, (Fig. 4D, lane 5), while all of the other mutants were inactive. This demonstrates that both the N and C termini of Sae2 are required for hairpin substrate cleavage, and that cooperative DNA binding with MRX is not sufficient for cooperative cleavage activity, since the Sae2(ΔC) and the Sae2(5A) mutants bind DNA with MRX in gel mobility shift assays yet hairpin cleavage by these mutants is not stimulated by MRX. The inability of the Sae2(5A) mutant to function with MRX is not likely to be because of loss of phosphorylation since the wild-type protein does not appear to be phosphorylated and is not affected by addition of a phosphatase (data not shown).
rad50S strains accumulate hairpin-capped DSBs at sites of inverted repeats in vivo (Lobachev et al., 2002). Consistent with this deficiency, the MR(R20M)X complex exhibited 2-fold lower levels of Sae2 stimulation, in comparison to wild-type MRX, using the internally-labeled hairpin substrate in magnesium (Fig. 4E, lanes 9 – 11). This suggests that MR(R20M)X can act cooperatively with Sae2 but exhibits a subtle defect in this functional interaction.
MRX exonuclease activity can facilitate hairpin removal by Sae2
The hairpin cleavage assay above suggested that MRX stimulates Sae2 cutting of single-stranded DNA adjacent to a hairpin structure, independently of MRX nuclease activity. Yet Mre11 nuclease activity is clearly required in vivo for the processing of hairpin-capped DSBs (Lobachev et al., 2002). To resolve this contradiction, we considered the substrate specificity of Sae2 as shown in Fig. 3B above, where we found that a single-stranded DNA gap of 11 nucleotides allowed Sae2 cutting but a gap of 5 nucleotides did not. We hypothesized that one role of Mre11 exonuclease activity may be to widen gaps adjacent to hairpins to allow Sae2 endonuclease activity. To test this idea, an internally labeled hairpin was constructed containing an 8-nucleotide loop hairpin and fully paired hairpin at either end, with a 5-nucleotide gap between the 3′ and 5′ ends. When MRX was incubated with this substrate in manganese, the gap was widened due to MRX 3′Π5′ exonuclease digestion (Fig. 5A). When Sae2 was added in a second step with magnesium to inhibit exonuclease activity by MRX, cleavage in the ssDNA region was observed which was dependent on both MRX and Sae2 (Figure 5A, lanes 2–7). Therefore, on hairpin substrates that contain small gaps, MRX stimulates Sae2 endonuclease cutting by resecting the 3′ strand and enlarging the single-stranded region. To confirm that exonuclease activity is important for this cooperative effect, we tested the nuclease-deficient M(D16A)R and Mre11-3/R complexes with this hairpin structure. Neither the M(D16A)R nor the Mre11-3/R complexes facilitated efficient Sae2 cutting of the hairpin substrate (Figure 5B, lanes 6–7 and 9–10), consistent with the lack of exonuclease activity seen with these mutants (Fig. 2C). In budding yeast, cruciform structures are processed into hairpin-containing DNA ends by an unknown nuclease (Lobachev et al., 2002). Since it is possible that this processing event may leave only nicks instead of gaps, we also tested the activity of MRX and Sae2 on a double-ended hairpin substrate containing only a nick on one strand. This substrate was processed efficiently by MRX and Sae2 (Fig. 5C), similar to the gapped substrate, thus nicks are functional as entry points for MRX and Sae2.
Figure 5.

Mre11 exonuclease activity facilitates Sae2 hairpin removal. (A) An internally [32P]-labeled double hairpin DNA substrate was incubated first with wild-type MRX in 1 mM MnCl2 for 20 minutes; Sae2 was then added with 5 mM MgCl2 and the reactions were continued for an additional 30 minutes before separation on a denaturing polyacrylamide gel. MB control as in Fig. 3. (B) Reactions were performed as in (A) but with wild-type MR and Mre11 nuclease-deficient complexes M(D16A)R and M(Mre11-3)R as indicated. (C) Hairpin cleavage assays as in (A) except that the substrate has a nick instead of a gap. (D) Model of MRX/Sae2 hairpin processing. Dotted arrow represents predicted MRX/Sae2-independent cleavage of the cruciform; Pacman represents MRX 3′ to 5′ exonuclease activity; bold arrow represents Sae2 cleavage. The location of the cruciform cleavage site is assumed to be at the base of the cruciform as shown but this has not been demonstrated in vivo.
Sae2 nuclease activity on branched DNA structures
As shown above with the hairpin DNA substrates, Sae2 itself exhibits endonuclease activity on DNA duplexes containing single-stranded DNA. To characterize this activity further, we tested wildtype Sae2 with a branched DNA structure as shown in Fig. 6A (substrate 1). We observed that wildtype Sae2 generated several endonucleolytic products from this 5′-labeled substrate, nearly all within the 15nt 5′ flap region (Fig. 6B, lanes 2–3). In addition, Sae2 also removed the radiolabeled nucleotide at the 5′ end of the 5′ flap, and cleaved the DNA within the duplex region at several points, although the cutting within the duplex region was much less efficient compared to the cutting within the single-stranded DNA region.
Figure 6.

Sae2 cleaves single-stranded DNA in branched DNA structures. (A) Diagram of DNA substrates 1 to 4 with locations of [32P] labels (asterisks) and summary of predominant cleavage sites (arrows) as shown. (B) Wild-type and mutant Sae2 proteins were incubated with Substrate 1 in 5 mM MgCl2 as indicated. (C) Reactions with wild-type Sae2 on Substrates 1, 2, and 3 as in (B). (D) Wild-type Sae2 protein was incubated as in (B) with Substrate 4. (E) Wild-type Sae2 protein incubated as in (B) with substrates 1, 5, and 6.
The Sae2 mutants were also tested on the branched substrate, which showed that the Sae2(5D) mutant showed similar activity as wild-type Sae2 (Fig. 6B, lanes 6–7), while the Sae2(5A) and Sae2(ΔC) mutants both showed intermediate levels of endonuclease activity (Fig. 6B, lanes 8–9 and 12–13). In contrast, the Sae2(G270D) and Sae2(ΔN) mutants exhibited essentially no endonuclease activity on this substrate (Fig. 6B, lanes 4–5 and 10–11), consistent with the lack of DNA binding activity observed with these mutants (Fig. 1C). The fact that the Sae2(5A) and Sae2(ΔC) mutants were able to cleave the single-stranded DNA in the branched substrate yet did not show any nuclease activity with MRX on the hairpin substrate (Fig. 4C) suggests that these mutants may be specifically defective in hairpin-specific interactions with the substrate. Alternatively, the Sae2(5A) and Sae2(ΔC) mutants may have a more subtle defect in MRX/DNA interactions despite the fact that they do form the MRX-dependent protein-DNA complex in the gel mobility shift assay and are not grossly defective in DNA binding (Fig. 1C).
To test the DNA structure requirements for Sae2 endonuclease activity, complementary strands were annealed to either the 5′ or 3′ flap of the branched substrate (Fig. 6A). When the branched DNA included a complementary strand that fully paired with the 5′ flap (Substrate 2), Sae2 no longer cut along the 5′ flap or removed the 5′ label but showed enhanced cutting within the duplex region (Fig. 6C, lane 4). In contrast, Sae2 did show 5′ flap-cutting activity on Substrate 3, which contains a complementary strand annealed to the 3′ flap (Fig. 6C, lane 6). Labeling of the top strand of the branched substrate (Substrate 4) showed that Sae2 also cut the top strand exclusively within the 3′ flap region (Fig. 6D, lanes 2–3), but not at the base of the 3′ flap. Taken together, these data demonstrate that Sae2 is an endonuclease with a strong preference for single-stranded DNA and single-strand/double-strand junctions but is also able to cut duplex DNA at a low level when it is present adjacent to a region of single-stranded DNA. A side-by-side comparison of fully base-paired DNA with branched DNA showed that Sae2 exhibits no detectable activity on DNA lacking single-stranded regions (Fig. 6E).
Activity of Sae2 mutants in vivo
To determine the in vivo effects of the Sae2 mutant proteins we characterized, we expressed each mutant in S. cerevisiae from a low-copy CEN plasmid as a C-terminal Calmodulin-Binding-Protein (CBP) fusion under the control of the endogenous SAE2 promoter. Δsae2 strains are deficient in sporulation, and we found that none of the mutants characterized here complemented the null strain in meiosis (Table 1).
Table 1.
Summary of in vitro and in vivo phenotypes of Sae2 mutants
| DNA binding | MRX-DNA binding | ssDNA endonuclease activity | hairpin endonuclease activity | sporulation (% of wt) | LYS2 Conversion (median frequency ± SD × 107) | |
|---|---|---|---|---|---|---|
| Sae2 wt | + | + | +++ | +++ | 81.4 ± 9 | 1950 ± 440 |
| Sae2(G270D) | − | − | −/+ | − | 0.2 ± 0.4 | 23 ± 2 |
| Sae2(5D) | + | + | +++ | +++ | 0.2 ± 0.4 | 170 ± 160 |
| Sae2(5A) | + | + | ++ | − | < 0.2 | 40 ± 21 |
| Sae2(ΔN) | − | − | − | − | < 0.2 | 31 ± 17 |
| Sae2(ΔC) | + | + | ++ | − | < 0.2 | 21 ± 18 |
|
| ||||||
| vector only | < 0.2 | 20 ± 5.5 | ||||
We also tested the functions of the mutants in a strain background containing inverted Alu repeats inserted in a LYS2 allele on chromosome II, and a truncated lys2 allele on chromosome III, as described previously (Lobachev et al., 2002). Extrusion of the cruciform, processing, and recombination between the two lys2 alleles leads to the generation of LYS2 colonies, which was shown to be ~65-fold reduced in Δsae2, rad50S, and mre11 nuclease-deficient strains (Lobachev et al., 2002). We found that expression of wild-type Sae2 on the plasmid complemented Δsae2 similar to previous reports for wildtype strains (Baroni et al., 2004; Lobachev et al., 2002) (Table 1). In contrast, strains expressing the sae2(G270D), sae2(5A), sae2(ΔN), and sae2(ΔC) mutant strains all showed similar deficiencies in hairpin-capped DSB resolution as Δsae2 strains containing the vector only. Interestingly, the sae2(5D) mutant shows partial complementation of the Δsae2 strain (8.5-fold higher frequency of recombination compared to vector only), but still 11.5-fold lower than the wild-type strain, indicating that the aspartate residues do not fully substitute for the dynamic phosphorylation of Sae2 that occurs in vivo (Baroni et al., 2004). Overall, however, the phenotypes observed with these mutants in cruciform-dependent recombination in vivo are consistent with the activities of the mutants in hairpin removal assays in vitro.
Discussion
In this study we demonstrate that the Sae2 protein from S. cerevisiae is an endonuclease and that it cooperates with the MRX complex in processing of hairpin structures in vitro. These results are consistent with observations in yeast showing similar phenotypes of Δsae2, rad50S, and mre11 nuclease-deficient strains in assays that involve the processing of hairpin-containing DNA intermediates. Mutations in Sae2 that block cooperative cleavage activity with MRX in vitro also are inactive in recombination assays in vivo, suggesting that the biochemical activities described here form the basis of Sae2-MRX functional cooperativity in cells.
Hairpin cleavage vs. hairpin removal
Mre11/Rad50 complexes from several species have been shown to cleave hairpin structures through Mre11-mediated endonuclease activity at the tip (or in the loop) of a hairpin structure (Connelly et al., 1998; Paull and Gellert, 1999; Trujillo and Sung, 2001). In E. coli this activity was postulated to be the basis of the instability of closely spaced inverted repeats, which can extrude into hairpin-containing cruciform structures in vivo (Connelly et al., 1998). In sbcC or sbcD mutant strains (Sharples and Leach, 1995), inverted repeats are stabilized (Connelly and Leach, 1996). A similar observation was made in S. cerevisiae with inverted repeats in the genome, which exhibit 60 to 70-fold lower rates of repeat-induced recombination in strains lacking components of the MRX complex (Lobachev et al., 2002).
In our experiments, however, we found that hairpin cleavage at the tip by recombinant MRX is extremely inefficient, and concentrations over 150 nM were required to visualize hairpin cleavage by yeast MR or MRX in vitro. Consistent with this observation, a previous characterization of MR and MRX hairpin cutting in vitro utilized 400 nM or higher concentrations of the complexes (Trujillo et al., 2003; Trujillo and Sung, 2001). In the study by Trujillo et al., ATP was shown to be required for hairpin cleavage activity (Trujillo and Sung, 2001), and this was the case in our assays as well (data not shown). In contrast, only ~50 nM human MRN is sufficient for high levels of hairpin cleavage in vitro and this cutting is ATP-independent (Paull and Gellert, 1999). It is not clear why there is such a difference between the human and the yeast complexes, but it may explain why there is a strong dependence on another nuclease for hairpin processing in yeast.
In S. cerevisiae, deletion of the SAE2 gene dramatically reduces the rate of recombination initiated at sites of inverted repeats, similar to the results in strains lacking MRX, rad50S strains, or mre11 nuclease-deficient strains (Lobachev et al., 2002). This suggests that Sae2 contributes to MRX-dependent processing and/or recombination at these sites, consistent with the observation that Sae2 regulates the association of MRX with sites of double-strand breaks (Lisby et al., 2004). Δsae2 strains also fail to facilitate intrachromosomal recombination in yeast using a genetic assay for break-induced recombination, similar to rad50S and mre11 nuclease-deficient strains (Rattray et al., 2001). In this assay, similar to the assay with inverted repeats, the Δsae2, rad50S, and mre11 nuclease-deficient strains accumulate large palindromic gene amplifications that are postulated to result from replication initiated from a hairpin intermediate (Rattray et al., 2005).
To determine the biochemical role of Sae2 in these processes we purified the recombinant protein and show here that Sae2 exhibits endonuclease activity on single-stranded DNA and increased nuclease activity on single-stranded DNA adjacent to hairpin structures. In contrast to the high levels required for hairpin cutting at the tip, as little as 10 nM MRX was sufficient to promote Sae2 cleavage of single-stranded DNA adjacent to hairpins in vitro. This was observed in the presence of 1.5 to 3 nM Sae2, and under these conditions no cutting was seen with either protein alone. Based on this data, we conclude that MRX and Sae2 act cooperatively to remove hairpin structures in vivo through the cleavage of single-stranded DNA adjacent to the hairpins.
Processing of hairpin intermediates
The cruciform structures generated at sites of inverted repeats in vivo were shown to undergo multiple processing steps. In both wild-type and Δmre11 strains, double-strand breaks were formed at inverted repeat sites at a low level (Lobachev et al., 2002), indicating that MRX does not make (or at least is not solely responsible for) this first processing event. In Δmre11 cells, these double-strand breaks were shown to be covalently closed by a hairpin structure. It is not known what the structure of the ends is immediately after the formation of the double-strand break since the breaks are spontaneous and the intermediates are transient. It is possible that the cruciform is broken at the highly constrained junction point, as suggested by Lobachev et al, and that this produces hairpin-capped ends with an adjacent nick or gap. Resection of the discontinuous strand at this nick or gap by Mre11 exonuclease activity and stimulation of Sae2 endonuclease activity in this single-stranded region would result in complete removal of the hairpin structure. Failure to perform this processing would result in increased stability of the hairpin-capped double-strand break intermediate which could become a fully sealed hairpin after fill-in synthesis and ligation, as observed in the Δmre11 strain (Lobachev et al., 2002). Replication of this chromosomal fragment generates a large amplification of the entire chromosomal arm, which was observed in Δmrx, Δsae2, rad50S, and mre11 nuclease-deficient strains.
In this study we show that Sae2 endonuclease activity requires a single-stranded region greater than 5 nt in length. One possibility for the role of Mre11 exonuclease activity in vivo is to resect DNA at sites of nicks and gaps to increase the length of single-stranded DNA, as shown in Fig. 5. If this is the case, then MRX could act in at least two ways to facilitate Sae2 activity on hairpin structures: first to resect the discontinuous strand from 3′ to 5′ to enlarge a nick or a gap, and second to stimulate Sae2 cleavage (see model in Fig. 5C).
MRX-Sae2 interactions
What is the biochemical basis of MRX-Sae2 cooperativity on hairpin DNA substrates? One possibility is that MRX directly associates with Sae2 and stimulates its catalytic activity. We have looked for direct protein-protein interactions between MRX and Sae2 in the absence of DNA in vitro and did not find any convincing evidence of this. Consistent with this observation, no 2-hybrid interactions were reported between Sae2 and MRX components in a global interaction screen even though Sae2-Sae2 interactions was observed (Uetz et al., 2000). However, our data does show cooperative DNA binding with MRX and Sae2, suggesting that the proteins may interact on DNA.
The R20M Rad50 mutant is one of several rad50S alleles isolated genetically on the basis of meiosis-specific defects in recombination and repair (Alani et al., 1990). rad50S strains were shown to be deficient in hairpin processing (Lobachev et al., 2002) and generated large palindromic amplifications at sites of inverted repeats, similar to a Δsae2 strain (Rattray et al., 2001). In vitro, an MRX complex containing the Rad50 R20M mutant exhibited a partial deficiency in Sae2 cooperative nuclease activity. Increasing the ionic strength of the reaction buffer decreased the ability of MRX(R20M) to stimulate Sae2 but did not affect the activity of the wild-type MRX protein (data not shown). This is consistent with the suggestion that MRX complexes in rad50S strains are impaired in Sae2 association, and the observation that overexpression of Sae2 can partially rescue a rad50S strain for defects in DNA processing and single-strand annealing (Clerici et al., 2005). It is also possible that other rad50s alleles may have stronger effects on the cooperative activities of MRX and Sae2.
Sae2 phosphorylation
Sae2 was shown by Longhese and colleagues to be phosphorylated during S phase and after DNA damage in a Tel1/Mec1-dependent manner (Baroni et al., 2004). Mutation of the serine or threonine in the five Sae2 SQ/TQ sites to alanine blocked phosphorylation and caused hypersensitivity to DNA damaging agents and reduced rates of cruciform-induced recombination, similar to a Δsae2 strain. Here we investigated the role of Sae2 phosphorylation by mutating all of the sites to alanine (5A) as in the “2, 5, 6, 8, 9” mutant used by Baroni et al., and also mutated these residues to aspartate to mimic phosphorylation (5D). The Sae2(5A) mutant bound to DNA by itself in a gel mobility shift assay and also formed higher order complexes with MRX on DNA similar to the wild-type protein (Fig. 1). In assays of MRX-independent flap cleavage, the Sae2(5A) mutant did exhibit partial activity (Fig. 6) yet failed to cleave hairpin structures cooperatively with MRX (Fig. 4). This defect in functional activity is likely the basis of the inactivity of the 5A mutant in vivo (Table 1) (Baroni et al., 2004).
The wild-type recombinant Sae2 protein used in this study was expressed in E. coli, where it is unlikely to be phosphorylated. The fact that the wild-type and 5D mutant proteins act identically in all of the in vitro assays presented in this study suggests that Sae2 phosphorylation is not essential for catalytic activity. In addition, we found that preincubation of wild-type Sae2 with either a phosphatase to remove phosphorylation or purified ATM protein to phosphorylate Sae2 had no apparent effect on its catalytic activity (data not shown). Taken together, this data suggest that the phenotype of the 5A mutant in vivo is likely due to the effects of the amino acid substitutions on Sae2 activity, rather than the absence of phosphorylation.
The Sae2(5D) mutant showed wild-type levels of hairpin endonuclease, flap endonuclease, and DNA binding in vitro, yet only partially complements a Δsae2 strain for inverted repeat recombination in vivo (Table 1). We do not know the reason for this, but it is clear that the aspartate residues do not fully recapitulate the effects of phosphorylation in vivo. Phosphorylation of Sae2 does not appear to be essential for Sae2 catalytic activities, either MRX-dependent or independent, but could play a role in protein-protein interactions with other factors. The fact that Sae2 is phosphorylated during a normal S phase as well as in response to DNA damage (Baroni et al., 2004) suggests that the role of phosphorylation involves S-phase-specific interactions. Preliminary data from another laboratory shows that the Sae2(5D) mutant does function similarly to the wild-type enzyme in other DNA repair assays (Sang Eun Lee, personal communication), thus there may be differences in the requirement for Sae2 phosphorylation depending on the type of damage and the assay used.
Sae2 functional domains
The recombinant his6-MBP-Sae2 protein we produced in this study formed several distinct multimeric complexes (Supp. Fig. 2, 3, 4). The dimeric and the smallest form, likely monomeric, showed the highest specific activity (data not shown) so this was used for the functional analysis. Interestingly, deletion of the N-terminus changed the distribution of multimeric forms such that the majority of this protein separated on a gel filtration column in the monomeric region. This Sae2(ΔN) mutant protein was confirmed to be a monomer by analytical ultracentrifugation. The N-terminus of Sae2 thus appears to contain a domain that controls Sae2 homotypic interactions. This mutant exhibited no activity in either MRX-dependent or MRX-independent cleavage events, similar to the Sae2(G270D) mutant which was isolated in a genetic screen for sae2 mutants deficient in intrachromosomal recombination. Neither of these mutants exhibited significant DNA binding activity, which is likely the cause of the loss of catalytic activity. In contrast, the Sae2(ΔC) mutant lacking the C-terminus did show DNA binding and MRX cooperative DNA binding, as well as partial endonuclease activity on flap DNA structures, yet failed to exhibit cooperative endonuclease activity with MRX on hairpin DNA substrates. The C-terminal domain may thus be responsible for recognizing or responding to hairpin structures with MRX. The Sae2(ΔC) and the Sae2(5A) mutants exhibit identical properties in DNA binding and nuclease activities, perhaps indicating that one or more of the mutations in the C-terminus of the Sae2(5A) mutant (S249A, T279A, or S289A) is responsible for the defects observed with this protein in vitro and in vivo.
Sae2 and sequence conservation
There are no recognizable domains or functional motifs in Sae2, and homology searches with the S. cerevisiae protein yield potential homologs only within other fungi. Amino acid sequence alignment of these fungal proteins shows several conserved regions, mostly in the C-terminal half of the protein (Supp. Fig. 1). The active site for Sae2 endonuclease activity is clearly not located between residues 250 and 345 because the Sae2(ΔC) mutant still exhibits at least partial nuclease activity. There are several conserved residues between amino acids 190 and 215 that are suggestive of a conserved active site region, and further mutagenesis is underway to determine which residues are responsible if this is the case.
The N-terminal domain of Sae2 mediates multimeric complex formation by Sae2 and is essential for nuclease activity. This region of Sae2, specifically the first 33 amino acids, was also reported to exhibit one of the highest rates of positive selection in the S. cerevisiae genome (Sawyer and Malik, 2006). In contrast to sequence conservation, positive selection occurs where there are more non-synonymous changes (alterations of the amino acid sequence) compared to synonymous changes (silent mutations). Signatures of positive selection were also found in the XRS2, POL4, and NEJ1 genes in a comparison of 7 Saccharomyces species. These 4 genes were among only 72 open reading frames showing positive selection in this analysis. One possible explanation for this phenomenon is that organisms are constantly in evolutionary conflict with mobile genetic elements, which can transpose and invade genomes to the detriment of their hosts. These 4 genes are all involved in the regulation or catalysis of NHEJ, including Sae2 which was recently found to be required for a pathway of microhomology-mediated end joining but to negatively regulate Ku-mediated NHEJ (Lee and Lee, 2007). Successful end joining is important for the success of retrotransposition in yeast (Downs and Jackson, 1999; Yu et al., 2004) and Sae2 has also been identified as a negative regulator of Ty1 retrotransposition (Scholes et al., 2001). The surprising lack of sequence conservation in the N-terminus of Sae2 may thus reflect a rapid and ongoing evolutionary selection for changes in Sae2 that effectively control mobile genetic elements.
Experimental Procedures
Plasmid construction (see Supplemental Data)
Protein Purification (see Supplemental Data)
DNA substrates
The 249 bp fragment used in the gel mobility shift assays in Fig. 1 was internally labeled by PCR amplification. The PCR product was purified on a 1% agarose gel and extracted with a Gel Extraction kit (Qiagen).
All 5′ [32P]-labeled oligonucleotide substrates were labeled with T4 polynucleotide kinase (PNK) (NEB). 3′ [32P]-labeled oligonucleotide substrates were labeled with cordycepin (NEN) using terminal deoxytransferase (TdT) (Boehringer). For oligonucleotide sequences and description of the preparation of internally labeled substrates, see Supplementary Data.
DNA Binding Assays
Gel mobility shift assays were performed in a volume of 28μL for Sae2 only and 20μL for Sae2/MRX. Reactions contained 0.014 nM DNA substrate, 25 mM 3-(N-morpholino) propanesulfonic acid, pH 7.0 (MOPS), 2 mM DTT, 10 mM ethylenediaminetetraacetic acid (EDTA), 100 mM NaCl. The reactions were incubated on ice for 20 minutes before separation in a 8% 37.5:1 acrylamide/bisacryalmide (EMD) native gel (Sae2 only) containing 89 mM Bis-Tris, pH 7.0, 8.9 mM boric acid, and 2 mM EDTA. Gels were dried and analyzed by phosphorimager (GE).
Nuclease Assays
Exonuclease reactions were performed in 10μL with 1 nM DNA substrate, 25 mM MOPS, 2 mM DTT, 50 mM NaCl, 1mM MnCl2 or 5mM MgCl2 as indicated at 37°C for 20 minutes. Endonuclease reactions were performed similarly but for 45 minutes. Reactions shown in Fig. 5 were performed in two stages as described in the figure legend.
Yeast Strains (see Supplemental Data)
Supplementary Material
Acknowledgments
We are grateful to Sang Eun Lee for sharing unpublished data, to Kirill Lobachev, Lorraine Symington, and Rodney Rothstein for yeast strains, to Maria Pia Longhese for plasmids, and to members of the Paull lab for critical comments about this project. This work was supported by NIH grant R01 CA094008 to T.T.P. A.R. and R.G. were supported by the Intramural Research Program of the NIH, National Cancer Institute (A.R.) and National Institute of Diabetes and Digestive and Kidney Diseases (R.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alani E, Padmore R, Kleckner N. Analysis of wild-type and rad50 mutants of yeast suggests an intimate relationship between meiotic chromosome synapsis and recombination. Cell. 1990;61:419–436. doi: 10.1016/0092-8674(90)90524-i. [DOI] [PubMed] [Google Scholar]
- Baroni E, Viscardi V, Cartagena-Lirola H, Lucchini G, Longhese MP. The functions of budding yeast Sae2 in the DNA damage response require Mec1- and Tel1-dependent phosphorylation. Mol Cell Biol. 2004;24:4151–4165. doi: 10.1128/MCB.24.10.4151-4165.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskara V, Dupre A, Lengsfeld B, Hopkins BB, Chan A, Lee JH, Zhang X, Gautier J, Zakian VA, Paull TT. Rad50 Adenylate Kinase Activity Regulates DNA Tethering by Mre11/Rad50 complexes. Mol Cell. 2007;25:647–661. doi: 10.1016/j.molcel.2007.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Jackson SP. Components of the Ku-dependent non-homologous end-joining pathway are involved in telomeric length maintenance and telomeric silencing. EMBO J. 1998;17:1819–1828. doi: 10.1093/emboj/17.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressan DA, Olivares HA, Nelms BE, Petrini JH. Alteration of N-terminal phosphoesterase signature motifs inactivates saccharomyces cerevisiae mre11. Genetics. 1998;150:591–600. doi: 10.1093/genetics/150.2.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–85. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- Clerici M, Mantiero D, Lucchini G, Longhese MP. The Saccharomyces cerevisiae Sae2 protein promotes resection and bridging of double strand break ends. J Biol Chem. 2005;280:38631–38638. doi: 10.1074/jbc.M508339200. [DOI] [PubMed] [Google Scholar]
- Connelly JC, Kirkham LA, Leach DR. The SbcCD nuclease of Escherichia coli is a structural maintenance of chromosomes (SMC) family protein that cleaves hairpin DNA. Proc Natl Acad Sci U S A. 1998;95:7969–7974. doi: 10.1073/pnas.95.14.7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connelly JC, Leach DR. The sbcC and sbcD genes of Escherichia coli encode a nuclease involved in palindrome inviability and genetic recombination. Genes Cells. 1996;1:285–291. doi: 10.1046/j.1365-2443.1996.23024.x. [DOI] [PubMed] [Google Scholar]
- Downs JA, Jackson SP. Involvement of DNA end-binding protein Ku in Ty element retrotransposition. Mol Cell Biol. 1999;19:6260–6268. doi: 10.1128/mcb.19.9.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuse M, Nagase Y, Tsubouchi H, Murakami-Murofushi K, Shibata T, Ohta K. Distinct roles of two separable in vitro activities of yeast Mre11 in mitotic and meiotic recombination. EMBO J. 1998;17:6412–6425. doi: 10.1093/emboj/17.21.6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson FP, Leach DR, Lloyd RG. Identification of sbcD mutations as cosuppressors of recBC that allow propagation of DNA palindromes in Escherichia coli K-12. J Bacteriol. 1992;174:1222–1228. doi: 10.1128/jb.174.4.1222-1228.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov EL, Sugawara N, White CI, Fabre F, Haber JE. Mutations in XRS2 and RAD50 delay but do not prevent mating-type switching in Saccharomyces cerevisiae. Mol Cell Biol. 1994;14:3414–3425. doi: 10.1128/mcb.14.5.3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell. 1997;88:375–384. doi: 10.1016/s0092-8674(00)81876-0. [DOI] [PubMed] [Google Scholar]
- Klar AJ. Mating-Type Functions for Meiosis and Sporulation in Yeast Act through Cytoplasm. Genetics. 1980;94:597–605. doi: 10.1093/genetics/94.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Lee K, Lee SE. Saccharomyces cerevisiae Sae2- and Tel1-Dependent Single Strand DNA Formation at DNA Break Promotes Microhomology-Mediated End Joining. Genetics. 2007 doi: 10.1534/genetics.107.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis LK, Storici F, Van Komen S, Calero S, Sung P, Resnick MA. Role of the nuclease activity of Saccharomyces cerevisiae Mre11 in repair of DNA double-strand breaks in mitotic cells. Genetics. 2004;166:1701–1713. doi: 10.1534/genetics.166.4.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Lobachev KS, Gordenin DA, Resnick MA. The Mre11 complex is required for repair of hairpin-capped double-strand breaks and prevention of chromosome rearrangements. Cell. 2002;108:183–193. doi: 10.1016/s0092-8674(02)00614-1. [DOI] [PubMed] [Google Scholar]
- McKee AH, Kleckner N. A general method for identifying recessive diploid-specific mutations in Saccharomyces cerevisiae, its application to the isolation of mutants blocked at intermediate stages of meiotic prophase and characterization of a new gene SAE2. Genetics. 1997;146:797–816. doi: 10.1093/genetics/146.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz K, Klein F. mre11S--a yeast mutation that blocks double-strand-break processing and permits nonhomologous synapsis in meiosis. Genes & Dev. 1997;11:2272–2290. doi: 10.1101/gad.11.17.2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Gellert M. The 3′ to 5′ exonuclease activity of Mre 11 facilitates repair of DNA double-strand breaks. Mol Cell. 1998;1:969–979. doi: 10.1016/s1097-2765(00)80097-0. [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M. Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes & Dev. 1999;13:1276–1288. doi: 10.1101/gad.13.10.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prinz S, Amon A, Klein F. Isolation of COM1, a new gene required to complete meiotic double-strand break-induced recombination in Saccharomyces cerevisiae. Genetics. 1997;146:781–795. doi: 10.1093/genetics/146.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, McGill CB, Shafer BK, Strathern JN. Fidelity of mitotic double-strand-break repair in Saccharomyces cerevisiae: a role for SAE2/COM1. Genetics. 2001;158:109–122. doi: 10.1093/genetics/158.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattray AJ, Shafer BK, Neelam B, Strathern JN. A mechanism of palindromic gene amplification in Saccharomyces cerevisiae. Genes Dev. 2005;19:1390–1399. doi: 10.1101/gad.1315805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Malik HS. Positive selection of yeast nonhomologous end-joining genes and a retrotransposon conflict hypothesis. Proc Natl Acad Sci U S A. 2006;103:17614–17619. doi: 10.1073/pnas.0605468103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholes DT, Banerjee M, Bowen B, Curcio MJ. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics. 2001;159:1449–1465. doi: 10.1093/genetics/159.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharples GJ, Leach DR. Structural and functional similarities between the SbcCD proteins of Escherichia coli and the RAD50 and MRE11 (RAD32) recombination and repair proteins of yeast. Mol Microbiol. 1995;17:1215–1217. doi: 10.1111/j.1365-2958.1995.mmi_17061215_1.x. [DOI] [PubMed] [Google Scholar]
- Symington LS. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol Mol Biol Rev. 2002;66:630–670. doi: 10.1128/MMBR.66.4.630-670.2002. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KM, Roh DH, Chen L, Van Komen S, Tomkinson A, Sung P. Yeast xrs2 binds DNA and helps target rad50 and mre11 to DNA ends. J Biol Chem. 2003;278:48957–48964. doi: 10.1074/jbc.M309877200. [DOI] [PubMed] [Google Scholar]
- Trujillo KM, Sung P. DNA structure-specific nuclease activities in the Saccharomyces cerevisiae Rad50/Mre11 complex. J Biol Chem. 2001;13:13. doi: 10.1074/jbc.M105482200. [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Ogawa H. A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol Cell Biol. 1998;18:260–268. doi: 10.1128/mcb.18.1.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- Yu J, Marshall K, Yamaguchi M, Haber JE, Weil CF. Microhomology-dependent end joining and repair of transposon-induced DNA hairpins by host factors in Saccharomyces cerevisiae. Mol Cell Biol. 2004;24:1351–1364. doi: 10.1128/MCB.24.3.1351-1364.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.