

Abstract
Eosinophilic esophagitis (EE) is a newly recognized disease, which has largely been called idiopathic EE, emphasizing the poor understanding of its pathogenesis. EE is a severe disease of the esophagus characterized by an accumulation of eosinophils in the esophageal mucosa. EE is highly associated with atopic disease and emerging evidence suggests a primary role for food antigen sensitization in disease etiology. Nevertheless, the nomenclature “Eosinophilic esophagitis” describes only the surface of the iceberg of a complex disorder. Epithelial cells, fibroblasts, endothelial cells and smooth muscles cells are involved in pathologic features of the disease and numerous leukocyte subtypes are recruited (eosinophils, mast cells, lymphocytes). As such, the pathogenesis of EE involves multiple tissues, cell types, genes and derives from complex genetic and environmental factors. “Pathogenesis” is a fusion of two Greek words pathos (disease) and genesis (development). In this review, we aim to define the fundamental piece of knowledge available today that characterizes the mechanisms by which certain etiological factors cause EE, reviewing human studies, murine models and recent knowledge regarding the involvement of environmental, cellular, molecular and genetic factors in the development of EE.
Eosinophilic esophagitis is characterized by a marked accumulation of eosinophils in the esophageal mucosa suggesting a Th2 disease. In addition to “guilt by association”, experimental models in mice implicate eosinophils in disease pathogenesis. However, eosinophils may neither be the first nor the only critical contributor in EE. Environmental exposure, allergen sensitization, eosinophils and other cells, molecules released and genetic predisposition, all interplay in EE pathogenesis.
Environmental pathogenesis: a Th2 disease
Epidemiologic studies provided the first indirect information about the pathogenesis of EE. The allergic component of EE was apparent from the strong association of EE with allergic diseases (Figure 1). About 70 % of EE patients have current or past allergic diseases or positive skin pricks test especially to a variety of foods (1). Of note, only a minority of EE patients present with food anaphylaxis, indicating distinct mechanisms compared with classical-IgE-mediated mast cell and basophil activation (1, 2). As such, a local esophageal population of allergen specific IgE producing B cells is possible. Indeed, EE patients without allergic disease still respond to an elemental diet (3, 4). Recently, EE has been found in two patients with anticonvulsant hypersensitivity syndrome (5, 6). Notably, withdrawal of carbamazepine, completely restored the endoscopic appearance of the esophagus suggesting that oral agents (in addition to food) could have an etiologic involvement in EE. Of note, like EE, anticonvulsant hypersensitivity syndrome has a male predominance (6).
Figure 1. Etiologic factors involved in EE development.
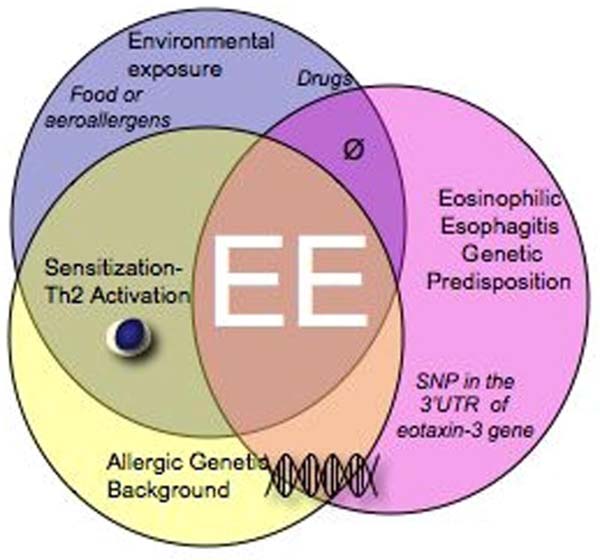
More then 500 gene polymorphisms has been shown to be associated to allergic diseases and the genetic context generated by the presence of these polymorphisms predispose individuals to asthma, atopic dermatitis, food allergy…. Although allergic diseases are common in the population, all allergic patients does not develop EE suggesting that a specific genetic predisposition for EE might be required to develop EE. As such a complex interplay between single nucleotide polymorphisms increase the risk of a Th2 inflammation to occur in the esophagus. To date only one single nucleotide polymorphism (SNP), has been associated with EE. This SNP is located in the 3’UTR of the eotaxin-3 gene (T/G +2496). All together, this complex genetic background and the environmental exposures (food and aeroallergens) contribute to the development of EE.
It is interesting to note that patients with EE, sometimes report seasonal variations in their symptoms; and changes in their esophageal eosinophil levels (7, 8) suggesting a role for aeroallergens. This was indeed recently supported by the study of Yamazaki et al wherein common food and environmental allergens induced cytokine production by peripheral blood mononuclear cells (PBMCs) in adult patients with EE (9). An increased production of IL-5 and IL-13 production by PBMC in EE patients was increased compared to healthy individual after stimulation with aero or food allergens. Thus, the immune responses in EE are characterized by enhanced production of Th2-associated cytokines in response to both food and environmental allergens. Finally, in a recent study examining intracellular staining of PBMC cytokines and cytokine production after stimulation in EE patients, the systemic parameters of EE patients were highly similar to atopic non-EE patients (10).
Treatment such as food avoidance or elemental diet has also reinforced the link between the disease and the allergic etiology (3, 11-13). In several studies, allergen avoidance has been shown to completely restore normal esophageal pathology (3, 4, 11-16). Food allergen and aeroallergen have been shown to be involved in EE pathogenesis, suggesting that direct contact of the allergen with the esophagus may not be required. Additionally, topical or systemic glucocorticoid treatment, used in allergic diseases such as asthma or atopic dermatitis, has been proven to be efficient in the treatment of EE, particularly in non-allergic EE patients (17-19).
Animal models have linked EE and allergic diseases and assess the sensitization pathways that could occur in human EE. Experimental models of EE can be induced in mice by allergen sensitization and exposure, as well as by administration or overexpression of Th2 cytokines (20, 21). A strong link has been established between EE and lung inflammation in mice. Most of published models so far have shown an association between lung and esophageal eosinophilia. Two recent studies have shown that skin sensitization primes for EE (22, 23). EE and atopic dermatitis (AD) share common features, including eosinophil infiltration, eosinophil degranulation, and squamous epithelial cell hyperplasia, suggesting that common pathogenetic mechanisms may be operational. Epicutaneous exposure to the allergens OVA or Aspergillus fumigatus alone induces AD- like skin inflammation but eosinophils do not migrate into the esophagus despite a strong systemic Th2 response, chronic cutaneous antigen exposure and accelerated bone marrow eosinophilopoiesis and circulating eosinophilia. However, when epicutaneously sensitized mice are subsequently exposed only once to intranasal antigen, esophageal eosinophilia (and lung inflammation) is powerfully induced (22, 23). In these studies, mice genetically deficient in signal transducer and activator of transcription 6 (STAT6), IL-13, IL-4 and IL-5 have impaired induction of esophageal eosinophilia in response to allergen (22, 23). It is interesting to note that all murine models of EE had an inflammation of another organ such as the lungs and that the instillation of intratracheal IL-13 in mice induces both airway and esophageal eosinophilia (24, 25) suggesting that the direct contact of the allergen with the esophagus or gastrointestinal tract is not required for development of EE. Collectively, these experimental systems demonstrate an intimate connection between the development of eosinophilic inflammation in the respiratory tract, the skin and esophagus not only in response to external allergic triggers but also to intrinsic Th2 cytokines.
Genetic predisposition
As already described in other Th2 diseases, EE pathogenesis is likely to be associated allergen sensitization in predisposed individuals (figure 1). EE has a strong familial pattern based on the growing clinical literature and our own patient data. Among pediatric patients with EE, ∼8% of them have at least one sibling or parent with EE as well (1). In addition, Patel and Falchuk (26) have recently reported three adult brothers with dysphagia who were found to have EE. Taken together, EE appears to demonstrate a strong familial pattern with a much higher prevalence in siblings.
Gender predisposition and familial clustering emphasize the genetic predisposition of this disease. In both pediatric and adult patients, EE exhibits a remarkable male predilection; about 70% of the patients are male (1). One explanation for such a high prevalence in male may indeed be due, in part, to the presence of a mutation on the sexual chromosome X that would not be corrected by the Y chromosome genes in males, leading to increase susceptibility in males that have only one copy of the affected gene. Of note, two chains for the IL-13 receptor, IL-13 Rα 1 and 2, are on X chromosomes in position Xq13.1-q28. Both population-based case-control comparison and family-based transmission disequilibrium test have shown an association between the allele G of the SNP (rs2302009) locates at the 3’ UTR of the eotaxin-3 gene in EE disease (25). One can hypothesize that the position of this SNP in the 3’UTR might indeed contribute to eotaxin-3 mRNA stability. But at the present time, the mechanism by which this SNP contributes to EE is unclear. Although only one SNP has been associated so far with EE, we predict that multiple other genetic components will be involved (27).
Molecular pathogenesis
Substantial evidence is accumulating that EE is associated with a Th2 type immune response and local or systemic Th2 cytokine overproduction. IL-5 is a cytokine involved in eosinophil production, survival and activation. IL-5 mRNA is increased in the biopsies of EE patients compared to NL patients ((28, 29) and unpublished data). Peripheral CD4+T cells show an increase in intracellular IL-5 in the blood of EE patients compared to non-atopic non-EE patients (10). Determining the role of IL-5 in EE is of importance since two independent studies have shown an improvement in the clinical and/or pathological symptoms of EE after anti-IL-5 treatment (30-32). IL-5 is known to increase proliferation and survival of eosinophils and facilitate their migration from the bone marrow to the blood (20, 33). As IL-5 can be expressed by eosinophils, it is important to elucidate if IL-5, increased in EE biopsies, has a local role in the esophageal eosinophils of EE patients. One can hypothesize that anti-IL-5 therapy may thus have a systemic action on eosinophil trafficking and survival and a local effect in the esophagus.
Using microarray expression profile analysis, an EE transcript signature remarkably conserved across allergic and non-allergic EE patient phenotypes has been identified (25). This demonstrates that the effector phase of the disease is conserved between individuals despite the driving trigger of the inflammation. This EE signature is characterized by 574 dysregulated genes in EE patients compared to normal individuals. Of the entire dysregulated genome, the gene with the greatest overexpression was eotaxin-3, induced ∼50-fold compared with control individuals and highly correlated with eosinophil number in the biopsies (25). Interestingly, while eotaxin-3 was highly upregulated in EE patients, eotaxin-1 and -2 were not significantly upregulated (25). The literature describes the eotaxin family as IL-13-induced molecules in several tissues and cell types. We recently demonstrated that in skin keratinocytes, IL-13 selectively induces eotaxin-3 but not eotaxin-1 and 2 in primary keratinocytes (figure 2). This phenomenon is most likely, mainly transcriptional and dependent upon the transcription factor STAT6 (unpublished data). We also demonstrated that IL-13 mRNA (but not IL-4) is in indeed induced in EE patients compared to NL individuals (unpublished data). It is interesting to note, that murine models have demonstrated that IL-13 and STAT6 are required to develop esophageal eosinophilia in mice (22, 23). Taken together, these results suggest that IL-13 induces eotaxin-3 expression in epithelial cells of the lung through a STAT6 phenomenon in EE.
Figure 2. Molecular pathogenesis of EE.
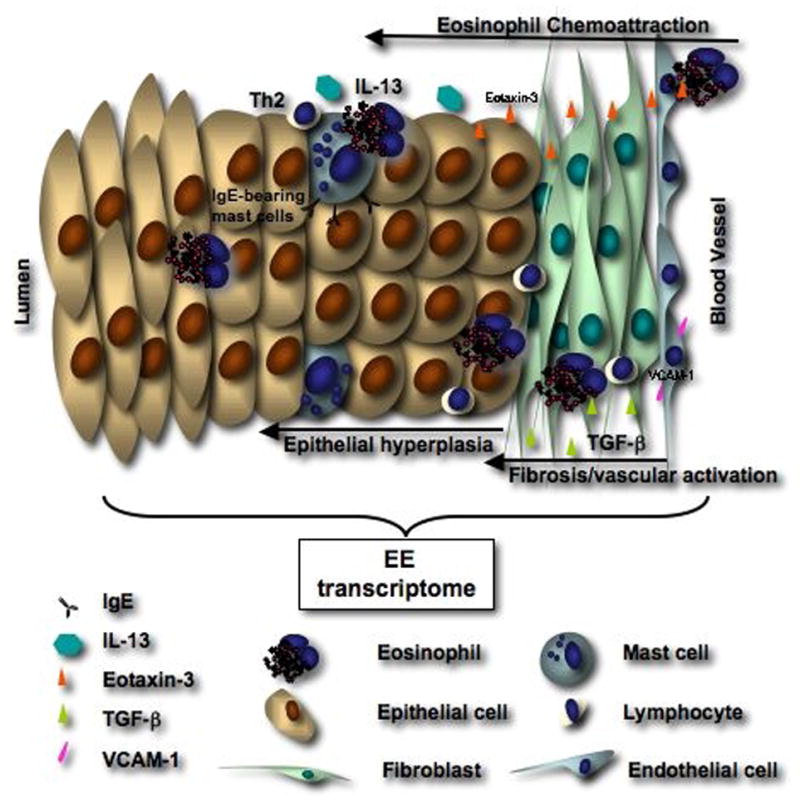
EE involves the complex interaction of genetic factors and environmental exposures leading to a Th2-associated disease localized in the esophagus of predisposed patients. Such processes involve increased expression of Th2 cytokines, chemokines, and chemokine receptors by PBMC (IL-4, IL-13, IL-5) and eosinophils (IL-13, IL-5, CCR3). In the esophagus, increased expression of IL-13 and TGF-beta likely contributes to eotaxin-3 release by epithelial cells, as well as increased collagen production (fibrosis) and vascular activation (VCAM-1 expression). Finally, IgE, systematically or locally synthesized by B cells are detected on the surface of mast cells and likely contributes to mast cell activation by food antigens.
Cellular pathogenesis
While absent in the normal esophagus, eosinophils markedly accumulate in the esophagus of EE patients. A minimum of 15 eosinophils per high power field is now used as clinical diagnostic criteria for EE (34-36). Analysis of the EE transcriptome reveals that, the strong accumulation of esophageal eosinophils is not accompanied by an increase in eosinophil specific transcripts (25). Recently, Locksley’s group demonstrated that eosinophil granule protein mRNAs were detectable in the early development of eosinophils but not once eosinophils infiltrate into the tissues (37). Although not actively transcribed in the esophagus, granule proteins are present in the esophageal eosinophils, and major basic protein (MBP) deposition was detected by immunohistochemistry, in EE patient esophageal biopsies. As such, granule proteins may influence disease via their cytotoxic activity. Eosinophil granules contain a crystalloid core composed of major basic protein (MBP)-1 (and MBP-2), and a matrix composed of eosinophil cationic protein (ECP), eosinophil-derived neurotoxin (EDN), and eosinophil peroxidase (EPO) (38). These cationic proteins share certain pro-inflammatory properties but differ in other ways. For example, MBP, EPO, and ECP have cytotoxic effects on epithelium in concentrations similar to those found in biological fluids from patients with eosinophilia. Additionally, ECP and EDN belong to the Ribonuclease A superfamily and possess antiviral and ribonuclease activity (39-41). ECP can insert voltage insensitive, ion-nonselective toxic pores into the membranes of target cells and these pores may facilitate the entry of other toxic molecules (42). MBP directly increases smooth muscle reactivity by causing dysfunction of vagal muscarinic M2 receptors (43). MBP also triggers degranulation of mast cells and basophils (44). Triggering of eosinophils by engagement of receptors for cytokines, immunoglobulins, and complement can lead to the generation of a wide range of inflammatory cytokines including IL-1, -3, -4, -5, -13, GM-CSF, TGF-β, TNF-α, RANTES, MIP-1α, and eotaxin-1, indicating that they have the potential to modulate multiple aspects of the immune response (45, 46). Additionally, eosinophils can directly activate T cells by antigen presentation, which has been demonstrated in vitro and in vivo (47, 48). Further eosinophil-mediated damage is caused by toxic hydrogen peroxide and halide acids generated by EPO and by superoxide generated by the respiratory burst oxidase enzyme pathway in eosinophils. Eosinophils also generate large amounts of the cysteinyl leukotriene C4 (LTC4) that is metabolized to LTD4 and LTE4 (49). These three lipid mediators increase vascular permeability and mucus secretion, and are potent stimulators of smooth muscle contraction (49). Indeed, leukotriene modifiers appear to improve clinical symptoms in EE patients (50). Notably, dysregulated expression of the mRNA for numerous enzymes involved in arachidonic acid metabolism is present in the esophagus of EE patients (25).
While eosinophil genes are not well represented in the EE transcriptome, mast cell gene expression (such as tryptase or carboxypeptidase 3) is highly increased in EE (25). Several studies have shown that mast cells, present in the normal esophagus, have an increased density in esophageal biopsies of EE patients (25, 28, 51-53). In other tissues, activated mast cells are known to release mediators (such as cytokines, histamine, proteases) able to modify physiological parameter such as smooth muscle contraction phenomenon that may occur in EE (54). Indeed, a recent study demonstrated that esophageal mast cells (as well as eosinophils) are activated in EE patients (Figure 2) compared to GERD patients and were likely to be IgE-bearing cells (52, 53). These findings implicate the presence of a sufficient local Th2 response to activate IgE production by B cells.
Several studies have shown an increase in lymphocytes in pediatric and adult EE patients (19, 25, 28, 52, 53), but only one study so far has described the presence of B cells in EE (53). Using anti-CD20 immunohistochemistry, the authors described the presence of B cells in EE esophageal biopsies. In the same study, immunoreactivity for CD3+, CD8+ and CD4+ cells was demonstrated to be at higher levels in EE biopsies (53). A critical role for T cells in disease induction has been demonstrated in recent murine modeling studies (55).
While infiltrated cell are likely to be involved in EE pathogenesis, the resident cells of the esophageal mucosa are likely to be one of the first effector cells responsible for the chemoattraction of hematopoietic cells. Notably, the esophagus contains CD1a+ dendritic cells that may link innate and adaptive immunity in the esophagus (53). In addition, the esophageal epithelium is likely to contribute to disease induction and propagation, as eotaxin-3 is chiefly produced by esophageal epithelial cells.
The epithelium of the esophagus is a squamous epithelium. In contrast to the skin, the human the esophagus is generally not keratinized and thus keratinocytes are in direct contact with the esophageal content. As such, the permeability, the elasticity, the integrity of the esophagus may contribute to EE disease pathogenesis. Epithelial cells are highly hyperplastic in EE patients (56). Several external factors are known to increase basal cell expansion, such as gastric acid content in GERD disease. But the basal layer expansion observed in EE patients is more extensive in EE patients (56) suggesting that other etiologic factors influence EE disease pathogenesis. We have, for example, demonstrated that the epithelium is the main source of eotaxin-3 chemokines (25), certainly responsible for the chemoattraction of eosinophils. Lymphocytes, eosinophils and mast cells infiltrated into the esophagus may release soluble mediators able to activate the transcription of epithelial cell genes; for example, Th2 cell-derived IL-13 may act directly on epithelial cells to induce the release of eotaxin-3 (figure 2).
In a recent pediatric EE population, remodeling of the esophageal mucosa and especially the lamina propria, has been described (57). In this study, patients with EE had an increased esophageal fibrosis, vascularity, and vascular activation. The authors also described an increased expression of TGF-β1 (figure 2) and its signaling molecule phosphorylated SMAD2/3 (phospho-SMAD2/3). Additionally, esophageal biopsies in patients with EE demonstrate an increased vascular density and an increased expression of the vascular endothelial adhesion molecule, VCAM-1. In addition to its role in fibrosis, TGF-β is known to increase smooth muscle cell hyperplasia and all together these results suggest an implication of TGF-β, VCAM-1, and SAMAD2/3 in the formation of strictures and in the loss of elasticity of the esophageal wall (57).
Conclusion and future direction
EE has a complex pathogenesis, in which multiple etiologic factors interact. The environmental factors and a predisposed genetic background certainly interplay in EE onset and pathogenesis. A gene polymorphism, eotaxin-3, has been shown to be associated with EE (25), and we predict that EE, like most Th2 diseases, is a polygenic disorder with multiple environment factor strongly contributing to disease expression (Figure 1). Although the advances made in the understanding of human pathogenesis, the next challenge will be to understand the molecular mechanisms involved in EE disease development. The overlap with other Th2 diseases tends to complicate the study and interpretation of results on systemic parameters, and the appropriate control, atopic nonEE will have supplanted the normal non-atopic controls. It will thus be of interest to investigate whether the genetics that underlie atopy and EE are partly similar. We hope that from these studies will emerge new biomarkers able to differentiate EE disease from GERD and other atopic diseases.
Although some treatments are effective in EE, the molecular mechanisms involved in the remission have still not been established. The development of in vitro and in vivo models may help to dissect out the molecular mechanisms involved in remission or resistance to therapy; the overall goal being able to molecularly classify patients as a function of their predicted response to treatment. Our current knowledge suggests that targeting the IL-13/eotaxin-3/CCR3 axis may be a promising therapeutic of EE.
Acknowledgments
This work was funded in part by the NIH #AI070235 and #AI45898 (M.E.R.), the Food Allergy and Anaphylaxis Network (FAAN) (M.E.R.), Campaign Urging Research for Eosinophil Disorders (CURED), the Buckeye Foundation (M.E.R.), The Food Allergy Project (M.E.R.), The American Heart Association #0625296B (C.B.) and The Thrasher Research Fund NR-0014 (C.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Noel RJ, Putnam PE, Rothenberg ME. Eosinophilic esophagitis. N Engl J Med. 2004 Sep 26;351(9):940–941. doi: 10.1056/NEJM200408263510924. [DOI] [PubMed] [Google Scholar]
- 2.Garcia-Careaga M, Jr, Kerner JA., Jr Gastrointestinal manifestations of food allergies in pediatric patients. Nutr Clin Pract. 2005 Oct;20(5):526–535. doi: 10.1177/0115426505020005526. [DOI] [PubMed] [Google Scholar]
- 3.Spergel JM. Eosinophilic esophagitis in adults and children: evidence for a food allergy component in many patients. Curr Opin Allergy Clin Immunol. 2007 Jun;7(3):274–278. doi: 10.1097/ACI.0b013e32813aee4a. [DOI] [PubMed] [Google Scholar]
- 4.Markowitz JE, Spergel JM, Ruchelli E, Liacouras CA. Elemental diet is an effective treatment for eosinophilic esophagitis in children and adolescents. Am J Gastroenterol. 2003 Apr;98(4):777–782. doi: 10.1111/j.1572-0241.2003.07390.x. [DOI] [PubMed] [Google Scholar]
- 5.Levy AM, Gleich GJ. Case 26-1996: hypersensitivity to carbamazepine. N Engl J Med. 1997 Jan 30;336(5):376. doi: 10.1056/NEJM199701303360513. author reply 377. [DOI] [PubMed] [Google Scholar]
- 6.Balatsinou C, Milano A, Caldarella MP, et al. Eosinophilic esophagitis is a component of the anticonvulsant hypersensitivity syndrome: Description of two cases. Dig Liver Dis. 2007 Mar 27; doi: 10.1016/j.dld.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 7.Fogg MI, Ruchelli E, Spergel JM. Pollen and eosinophilic esophagitis. J Allergy Clin Immunol. 2003 Oct;112(4):796–797. doi: 10.1016/s0091-6749(03)01715-9. [DOI] [PubMed] [Google Scholar]
- 8.Wang FY, Gupta SK, Fitzgerald JF. Is there a seasonal variation in the incidence or intensity of allergic eosinophilic esophagitis in newly diagnosed children? J Clin Gastroenterol. 2007 May-Jun;41(5):451–453. doi: 10.1097/01.mcg.0000248019.16139.67. [DOI] [PubMed] [Google Scholar]
- 9.Yamazaki K, Murray JA, Arora AS, et al. Allergen-specific in vitro cytokine production in adult patients with eosinophilic esophagitis. Dig Dis Sci. 2006 Nov;51(11):1934–1941. doi: 10.1007/s10620-005-9048-2. [DOI] [PubMed] [Google Scholar]
- 10.Bullock JZ, Villanueva JM, Blanchard C, et al. Interplay of adaptative Th2 immunity with eotaxin-3 C-C chemokine receptor 3 in eosinophilc esophagitis. J Pediatr Gastroenterol Nutr. 2007 doi: 10.1097/MPG.0b013e318043c097. [DOI] [PubMed] [Google Scholar]
- 11.Spergel JM, Andrews T, Brown-Whitehorn TF, Beausoleil JL, Liacouras CA. Treatment of eosinophilic esophagitis with specific food elimination diet directed by a combination of skin prick and patch tests. Ann Allergy Asthma Immunol. 2005 Oct;95(4):336–343. doi: 10.1016/S1081-1206(10)61151-9. [DOI] [PubMed] [Google Scholar]
- 12.Ngo P, Furuta GT. Treatment of eosinophilic esophagitis in children. Curr Treat Options Gastroenterol. 2005 Oct;8(5):397–403. doi: 10.1007/s11938-005-0042-8. [DOI] [PubMed] [Google Scholar]
- 13.Kagalwalla AF, Sentongo TA, Ritz S, et al. Effect of six-food elimination diet on clinical and histologic outcomes in eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2006 Sep;4(9):1097–1102. doi: 10.1016/j.cgh.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 14.Kelly KJ, Lazenby AJ, Rowe PC, Yardley JH, Perman JA, Sampson HA. Eosinophilic esophagitis attributed to gastroesophageal reflux: improvement with an amino acid-based formula. Gastroenterology. 1995 Nov;109(5):1503–1512. doi: 10.1016/0016-5085(95)90637-1. [DOI] [PubMed] [Google Scholar]
- 15.Simon D, Straumann A, Wenk A, Spichtin H, Simon HU, Braathen LR. Eosinophilic esophagitis in adults--no clinical relevance of wheat and rye sensitizations. Allergy. 2006 Dec;61(12):1480–1483. doi: 10.1111/j.1398-9995.2006.01224.x. [DOI] [PubMed] [Google Scholar]
- 16.Pentiuk SP, Miller CK, Kaul A. Eosinophilic esophagitis in infants and toddlers. Dysphagia. 2007 Jan;22(1):44–48. doi: 10.1007/s00455-006-9040-9. [DOI] [PubMed] [Google Scholar]
- 17.Noel RJ, Putnam PE, Collins MH, et al. Clinical and immunopathologic effects of swallowed fluticasone for eosinophilic esophagitis. Clin Gastroenterol Hepatol. 2004 Aug;2(7):568–575. doi: 10.1016/s1542-3565(04)00240-x. [DOI] [PubMed] [Google Scholar]
- 18.Teitelbaum JE, Fox VL, Twarog FJ, et al. Eosinophilic esophagitis in children: immunopathological analysis and response to fluticasone propionate. Gastroenterology. 2002 May;122(5):1216–1225. doi: 10.1053/gast.2002.32998. [DOI] [PubMed] [Google Scholar]
- 19.Konikoff MR, Noel RJ, Blanchard C, et al. A Randomized, Double-Blind, Placebo-Controlled Trial of Fluticasone Propionate for Pediatric Eosinophilic Esophagitis. Gastroenterology. 2006 Nov;131(5):1381–1391. doi: 10.1053/j.gastro.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 20.Mishra A, Hogan SP, Brandt EB, Rothenberg ME. IL-5 promotes eosinophil trafficking to the esophagus. J Immunol. 2002 Apr 1;168(5):2464–2469. doi: 10.4049/jimmunol.168.5.2464. [DOI] [PubMed] [Google Scholar]
- 21.Mishra A, Hogan SP, Brandt EB, Rothenberg ME. An etiological role for aeroallergens and eosinophils in experimental esophagitis. J Clin Invest. 2001 Feb;107(1):83–90. doi: 10.1172/JCI10224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akei HS, Mishra A, Blanchard C, Rothenberg ME. Epicutaneous antigen exposure primes for experimental eosinophilic esophagitis in mice. Gastroenterology. 2005 Sep;129(3):985–994. doi: 10.1053/j.gastro.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Akei HS, Brandt EB, Mishra A, et al. Epicutaneous aeroallergen exposure induces systemic TH2 immunity that predisposes to allergic nasal responses. J Allergy Clin Immunol. 2006 Jul;118(1):62–69. doi: 10.1016/j.jaci.2006.04.046. [DOI] [PubMed] [Google Scholar]
- 24.Mishra A, Rothenberg ME. Intratracheal IL-13 induces eosinophilic esophagitis by an IL-5, eotaxin-1, and STAT6-dependent mechanism. Gastroenterology. 2003 Dec;125(5):1419–1427. doi: 10.1016/j.gastro.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 25.Blanchard C, Wang N, Stringer KF, et al. Eotaxin-3 and a uniquely conserved gene-expression profile in eosinophilic esophagitis. J Clin Invest. 2006 Feb;116(2):536–547. doi: 10.1172/JCI26679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel SM, Falchuk KR. Three brothers with dysphagia caused by eosinophilic esophagitis. Gastrointest Endosc. 2005 Jan;61(1):165–167. doi: 10.1016/s0016-5107(04)02459-9. [DOI] [PubMed] [Google Scholar]
- 27.Blanchard C, Wang N, Rothenberg ME. Eosinophilic esophagitis: pathogenesis, genetics, and therapy. J Allergy Clin Immunol. 2006 Nov;118(5):1054–1059. doi: 10.1016/j.jaci.2006.07.038. [DOI] [PubMed] [Google Scholar]
- 28.Straumann A, Bauer M, Fischer B, Blaser K, Simon HU. Idiopathic eosinophilic esophagitis is associated with a T(H)2-type allergic inflammatory response. J Allergy Clin Immunol ÿ%\⋄□h°. 2001;108(6):954–961. doi: 10.1067/mai.2001.119917. [DOI] [PubMed] [Google Scholar]
- 29.Straumann A, Kristl J, Conus S, et al. Cytokine expression in healthy and inflamed mucosa: probing the role of eosinophils in the digestive tract. Inflamm Bowel Dis. 2005 Aug;11(8):720–726. doi: 10.1097/01.mib.0000172557.39767.53. [DOI] [PubMed] [Google Scholar]
- 30.Garrett JK, Jameson SC, Thomson B, et al. Anti-interleukin-5 (mepolizumab) therapy for hypereosinophilic syndromes. J Allergy Clin Immunol. 2004 Jan;113(1):115–119. doi: 10.1016/j.jaci.2003.10.049. [DOI] [PubMed] [Google Scholar]
- 31.Stein ML, Collins MH, Villanueva JM, et al. Anti-IL-5 (mepolizumab) therapy for eosinophilic esophagitis. J Allergy Clin Immunol. 2006 Dec;118(6):1312–1319. doi: 10.1016/j.jaci.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Simon D, Braathen LR, Simon HU. Anti-interleukin-5 antibody therapy in eosinophilic diseases. Pathobiology. 2005;72(6):287–292. doi: 10.1159/000091326. [DOI] [PubMed] [Google Scholar]
- 33.Rothenberg ME. Eosinophilic gastrointestinal disorders (EGID) J Allergy Clin Immunol. 2004 Feb;113(1):11–28. doi: 10.1016/j.jaci.2003.10.047. quiz 29. [DOI] [PubMed] [Google Scholar]
- 34.Lim JR, Gupta SK, Croffie JM, et al. White specks in the esophageal mucosa: An endoscopic manifestation of non-reflux eosinophilic esophagitis in children. Gastrointest Endosc. 2004 Jun;59(7):835–838. doi: 10.1016/s0016-5107(04)00364-5. [DOI] [PubMed] [Google Scholar]
- 35.Potter JW, Saeian K, Staff D, et al. Eosinophilic esophagitis in adults: an emerging problem with unique esophageal features. Gastrointest Endosc. 2004 Mar;59(3):355–361. doi: 10.1016/s0016-5107(03)02713-5. [DOI] [PubMed] [Google Scholar]
- 36.Gonsalves N, Policarpio-Nicolas M, Zhang Q, Rao MS, Hirano I. Histopathologic variability and endoscopic correlates in adults with eosinophilic esophagitis. Gastrointest Endosc. 2006 Sep;64(3):313–319. doi: 10.1016/j.gie.2006.04.037. [DOI] [PubMed] [Google Scholar]
- 37.Voehringer D, van Rooijen N, Locksley RM. Eosinophils develop in distinct stages and are recruited to peripheral sites by alternatively activated macrophages. J Leukoc Biol. 2007 Mar 5; doi: 10.1189/jlb.1106686. [DOI] [PubMed] [Google Scholar]
- 38.Gleich GJ, Adolphson CR. The eosinophilic leukocyte: structure and function. Adv Immunol. 1986;39:177–253. doi: 10.1016/s0065-2776(08)60351-x. [DOI] [PubMed] [Google Scholar]
- 39.Rosenberg HF, Dyer KD. Eosinophil cationic protein and eosinophil-derived neurotoxin. Evolution of novel function in a primate ribonuclease gene family. J Biol Chem. 1995 Sep 15;270(37):21539–21544. doi: 10.1074/jbc.270.37.21539. [DOI] [PubMed] [Google Scholar]
- 40.Rosenberg HF, Dyer KD, Tiffany HL, Gonzalez M. Rapid evolution of a unique family of primate ribonuclease genes. Nat Genet. 1995 Jun;10(2):219–223. doi: 10.1038/ng0695-219. [DOI] [PubMed] [Google Scholar]
- 41.Slifman NR, Loegering DA, McKean DJ, Gleich GJ. Ribonuclease activity associated with human eosinophil-derived neurotoxin and eosinophil cationic protein. J Immunol. 1986 Nov 1;137(9):2913–2917. [PubMed] [Google Scholar]
- 42.Young JD, Peterson CG, Venge P, Cohn ZA. Mechanism of membrane damage mediated by human eosinophil cationic protein. Nature. 1986 Jun 5-11;321(6070):613–616. doi: 10.1038/321613a0. [DOI] [PubMed] [Google Scholar]
- 43.Jacoby DB, Gleich GJ, Fryer AD. Human eosinophil major basic protein is an endogenous allosteric antagonist at the inhibitory muscarinic M2 receptor. J Clin Invest. 1993 Apr;91(4):1314–1318. doi: 10.1172/JCI116331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Donnell MC, Ackerman SJ, Gleich GJ, Thomas LL. Activation of basophil and mast cell histamine release by eosinophil granule major basic protein. J Exp Med. 1983 Jun 1;157(6):1981–1991. doi: 10.1084/jem.157.6.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kita H. The eosinophil: a cytokine-producing cell? J Allergy Clin Immunol. 1996 Apr;97(4):889–892. doi: 10.1016/s0091-6749(96)80061-3. [DOI] [PubMed] [Google Scholar]
- 46.Shinkai K, Mohrs M, Locksley RM. Helper T cells regulate type-2 innate immunity in vivo. Nature. 2002 Dec 19-26;420(6917):825–829. doi: 10.1038/nature01202. [DOI] [PubMed] [Google Scholar]
- 47.Shi HZ, Humbles A, Gerard C, Jin Z, Weller PF. Lymph node trafficking and antigen presentation by endobronchial eosinophils. J Clin Invest. 2000 Apr;105(7):945–953. doi: 10.1172/JCI8945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mattes J, Yang M, Mahalingam S, et al. Intrinsic defect in T cell production of interleukin (IL)-13 in the absence of both IL-5 and eotaxin precludes the development of eosinophilia and airways hyperreactivity in experimental asthma. J Exp Med. 2002 Jun 3;195(11):1433–1444. doi: 10.1084/jem.20020009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lewis RA, Austen KF, Soberman RJ. Leukotrienes and other products of the 5-lipoxygenase pathway. Biochemistry and relation to pathobiology in human diseases. N Engl J Med. 1990 Sep 6;323(10):645–655. doi: 10.1056/NEJM199009063231006. [DOI] [PubMed] [Google Scholar]
- 50.Attwood SE, Lewis CJ, Bronder CS, Morris CD, Armstrong GR, Whittam J. Eosinophilic oesophagitis: a novel treatment using Montelukast. Gut. 2003 Feb;52(2):181–185. doi: 10.1136/gut.52.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicholson AG, Li D, Pastorino U, Goldstraw P, Jeffery PK. Full thickness eosinophilia in oesophageal leiomyomatosis and idiopathic eosinophilic oesophagitis. A common allergic inflammatory profile? J Pathol. 1997 Oct;183(2):233–236. doi: 10.1002/(SICI)1096-9896(199710)183:2<233::AID-PATH936>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 52.Kirsch R, Bokhary R, Marcon MA, Cutz E. Activated mucosal mast cells differentiate eosinophilic (allergic) esophagitis from gastroesophageal reflux disease. J Pediatr Gastroenterol Nutr. 2007 Jan;44(1):20–26. doi: 10.1097/MPG.0b013e31802c0d06. [DOI] [PubMed] [Google Scholar]
- 53.Lucendo AJ, Navarro M, Comas C, et al. Immunophenotypic characterization and quantification of the epithelial inflammatory infiltrate in eosinophilic esophagitis through stereology: an analysis of the cellular mechanisms of the disease and the immunologic capacity of the esophagus. Am J Surg Pathol. 2007 Apr;31(4):598–606. doi: 10.1097/01.pas.0000213392.49698.8c. [DOI] [PubMed] [Google Scholar]
- 54.Mann NS, Leung JW. Pathogenesis of esophageal rings in eosinophilic esophagitis. Med Hypotheses. 2005;64(3):520–523. doi: 10.1016/j.mehy.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 55.Mishra A, Schlotman J, Wang M, Rothenberg ME. Critical role for adaptive T cell immunity in experimental eosinophilic esophagitis in mice. J Leukoc Biol. 2007 Apr;81(4):916–924. doi: 10.1189/jlb.1106653. [DOI] [PubMed] [Google Scholar]
- 56.Sant’Anna AM, Rolland S, Fournet JC, Yazbeck S, Drouin E. Eosinophilic Esophagitis in Children: Symptoms, Histology and pH Probe Results. J Pediatr Gastroenterol Nutr. 2004 Oct;39(4):373–377. doi: 10.1097/00005176-200410000-00013. [DOI] [PubMed] [Google Scholar]
- 57.Aceves SS, Newbury RO, Dohil R, Bastian JF, Broide DH. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol. 2007 Jan;119(1):206–212. doi: 10.1016/j.jaci.2006.10.016. [DOI] [PubMed] [Google Scholar]