

Abstract
Background & Aims
Krüppel-like factor 5 (KLF5) is a zinc finger-containing transcription factor that regulates cell proliferation. Oncogenic KRAS mutations are commonly found in colorectal cancers. We aimed to determine whether KLF5 mediates KRAS functions during intestinal tumorigenesis.
Methods
The effects of KLF5 on proliferation and transformation were examined in IEC-6 intestinal epithelial cells stably transfected with an inducible KRASV12G. KLF5 expression was examined in intestinal tumors derived from transgenic mice expressing KRASV12G under a villin promoter and in human colorectal cancers with mutated KRAS.
Results
Induction of KRASV12G in IEC-6 cells resulted in increased expression of KLF5, accompanied by an increased rate of proliferation and anchorage-independent growth. Inhibition of KLF5 expression by MEK inhibitors or KLF5-specific small interfering RNA (siRNA) reduced proliferation and anchorage-independent growth despite KRASV12G induction. Human colorectal cancer cell lines with mutated KRAS contained high levels of KLF5 and reduction of KLF5 by MEK inhibitors or KLF5 siRNA also led to reduced proliferation and transformation. In vivo, both intestinal tumors derived from mice transgenic for villin-KRASV12G and human primary colorectal cancers with mutated KRAS contained high levels of KLF5 and increased staining of the proliferative marker, Ki67.
Conclusions
Elevated levels of KLF5 protein are strongly correlated with activating KRAS mutations in intestinal tumors both in vitro and in vivo. Inhibition of KLF5 expression in these tumor cells resulted in significantly reduced rates of proliferation and transforming activities. We conclude that KLF5 is an important mediator of oncogenic KRAS transforming functions during intestinal tumorigenesis.
INTRODUCTION
Proto-oncogenes encode proteins that regulate cell proliferation and are often involved in signal transduction and execution of mitogenic signals. Abnormal activation of a proto-oncogene or its product results in a tumor-inducing agent, an oncogene. RAS genes were among the first to be identified as oncogenes causing human cancers 1, 2. A mutation in one of three codons (12, 13 or 61) of the RAS proto-oncogene was sufficient to cause its activation into an oncogene 2, 3. Three RAS oncogenes (HRAS, KRAS and NRAS) have been well characterized and extensively studied in cancers 1, 2. Several downstream mediators of RAS signaling have also been extensively studied 4-6. It is now well established that the canonical RAS signaling cascade involves the mitogen-activated protein kinase (MAPK) and its downstream effectors in activating cell proliferation 7-9.
Activating mutations in KRAS are present in more than 80% of pancreatic cancers 10, 11 and over 50% of colon cancers 12-14. Previous studies have shown that expression of oncogenic KRAS in intestinal epithelial cells can cause increased proliferation and a transformed state 15. Several distinct mouse models that either over-express KRAS or express KRAS at an endogenous level have been generated 16-19. In the intestine, over-expression of KRASV12G under the control of the villin promoter yielded lesions ranging from aberrant crypt foci to invasive adenocarcinoma in more than 80% of the transgenic animals 19. Expression of KRASV12G caused activation of the MAPK cascade and the tumors were frequently characterized by deregulated cellular proliferation 19. This animal model thus recapitulates the stages of tumor progression as well as represents an important genetic alteration found in human colorectal cancer.
Krüppel-like factors are zinc finger-containing transcription factors with sequence homology to Sp1 and are involved in various cellular functions 20-23. Krüppel-like factor 5 (KLF5), also known as intestinal-enriched Krüppel-like factor (IKLF), is predominantly present in the proliferating crypt epithelial cells of the intestinal epithelium 20, 21, 24. This distribution suggests that KLF5 may function to regulate cell proliferation. Indeed, KLF5 has been shown to promote cell proliferation 25-27 and respond to WNT signaling 28. We previously reported that KLF5 is an important mediator of the oncogenic HRAS signaling pathway in NIH3T3 mouse fibroblasts in promoting cell proliferation and transformation 29, 30. In these studies, oncogenic HRAS activated KLF5 expression through the MAPK signaling cascade, involving extracellular-signal regulated kinase (ERK) and early growth response gene-1 (EGR-1) 30. Furthermore, we demonstrated that the enhanced cell proliferation and transformation was due to activation of downstream target genes that include cyclins D1, B1 and Cdc-2 by KLF5 29, 30. Taken together, these findings support a crucial role for KLF5 in mediating the transforming activities of activated HRAS.
Based on the results from our previous studies on the causal relationship between oncogenic HRAS and KLF5 in NIH3T3 cells, we hypothesize that KLF5 plays an important role in mediating KRAS-induced intestinal tumorigenesis. In the current study, we examined the role of KLF5 on proliferation of IEC-6 intestinal epithelial cells in response to the inducible expression of oncogenic KRAS. We also characterized several human colon cancer cell lines based on their KRAS genotypes and evaluated the pro-proliferative activity of KLF5 in these cell lines. Furthermore, we assessed KLF5 expression in mice over-expressing KRASV12G specifically in the intestinal epithelium under the control of the villin promoter. Lastly, we assembled a panel of matched normal and tumor colonic tissue biopsies from human patients and determined the KRAS genotypes and KLF5 expression. Our results from these studies suggest that KLF5 plays a crucial role in maintaining increased proliferation and transformation resulting from oncogenic KRAS induction of intestinal tumorigenesis.
MATERIALS AND METHODS
Reagents, Cell Lines & Histological Specimens
Cell culture media and fetal bovine serum (FBS) were purchased from Invitrogen (Carlsbad, CA). MEK inhibitors, PD98059 and U0126, were purchased from Promega (Madison, WI). Monoclonal antibody against pan-RAS and β-actin were purchased from Sigma (St. Louis, MO). Polyclonal antibodies against phospho-ERK1/2 and ERK1/2 were acquired from Cell Signaling Technology (Beverly, MA). A rabbit polyclonal antibody targeting KLF5 was generated against amino acids 95−111 of the mouse KLF5 protein by QCB (Hopkinton, MA).
The stable IEC-6 cell line over-expressing KRASV12G (IEC-iKRAS) under the control of an isopropyl-1-thio-β-D-galactopyranoside (IPTG)-inducible promoter was previously established and maintained in the described growth conditions 15. Human colorectal cancer cell lines DLD-1, SW-480, HCT 116, HT-29, Caco-2 and RKO were procured from ATCC (Rockville, MD) and maintained according to supplier's protocol.
Western Blot Analyses and Quantitative Reverse Transcription - Polymerase Chain Reaction (RT-PCR)
Western blot analysis and quantitative RT-PCR were performed as previously described 30. Antibodies and primers used are described in Supplementary Methods.
Small interfering RNA (siRNA) and Transfection
Small interfering RNA (Stealth™ Select) against rat and human KLF5 were obtained from Invitrogen Inc. and reconstituted according to manufacturer's protocol to obtain a stock concentration of 20 μM. The cells were grown to 50% confluence in 6-well plates before transfection with siRNA at a final concentration 20 nM using Lipofectamine RNAiMAX reagent (Invitrogen) for 4 h in Opti-MEM I Reduced Serum Medium (Invitrogen). Following transfection, the cells were supplemented with the appropriate growth media containing 10% FBS and 1% penicillin-streptomycin.
Cell proliferation and anchorage-independent focus formation assays
Cell proliferation was analyzed as previously described 30. For a protocol, please refer to Supplementary Methods.
Immunohistochemical Analysis
Tumor specimens along with matched adjacent normal epithelium were dissected from the intestines of mice over-expressing KRASV12G under the control of the villin promoter. Eight matched normal and cancerous colonic tissue samples were obtained from seven patients without patient identifier at the Emory University Hospital. The protocol has received approval from the Institutional Review Board. Tissues were formalin fixed, paraffin-embedded and cut into 5 μm sections. The sections were then appropriately processed and antigen-retrieval performed using 10 mM citrate buffer (pH 6.0). Tissue sections were blocked using Avidin/Biotin (Vector Labs, Burlingame, CA) and normal goat antiserum. Sections were then stained with KLF5 antibody (1:15,000 dilutions) and developed using Vectastain Elite ABC kit (Vector Labs) with Liquid DAB (Dako, Carpinteria, CA). Sections were then counterstained using Mayer's Hematoxylin (Invitrogen). The tissue histology was verified by a single experienced gastrointestinal pathologist (B.A.B).
RESULTS
KLF5 Mediates the Pro-proliferative and Transforming Activities of Oncogenic KRAS in Intestinal Epithelial Cells
We previously demonstrated that over-expression of oncogenic HRAS in NIH3T3 cells resulted in increased proliferation and anchorage-independent foci formation 29, 30. Since activating KRAS mutations are common in colon cancer, we examined the effect of inducible expression of oncogenic KRAS in rat intestinal epithelial cells, IEC-6, that had been stably transfected with KRASV12G under the control of an IPTG-inducible promoter 15. This cell line, called IEC-iKRAS, was treated with IPTG for up to 3 days. As shown in Figure 1A, the levels of KRAS protein were increased in IPTG-treated cells when compared with control. Correspondingly, we observed a considerable increase in KLF5 protein levels upon IPTG treatment (Figure 1A). Notably, IPTG-treated cells exhibited a higher density (Figure 1B) and rate of proliferation (Figure 1C) when compared to control. In addition, IPTG-treated cells formed a significantly higher number of colonies in soft agar when compared to control (Figure 1D), indicating that KRAS over-expression bestowed the ability for anchorage-independent growth.
Figure 1. Growth characteristics of IEC-6 cells containing inducible KRASV12G.

(A) IEC-iKRAS cells were treated with or without 5 mM IPTG for up to three days. Cells were collected each day and cell lysates subjected to Western blot analyses using KLF5, KRAS and β-actin (loading control) antibodies. (B) Representative images of IEC-iKRAS cells with and without IPTG induction for 3 days. (C) Cell proliferation assay performed on cells treated or not with IPTG. N = 6; * p < 0.05 by two-tailed t test. (D) Anchorage-independent assay was conducted in soft agar in cells treated or not with IPTG for 21 days. N = 6; * p < 0.01.
In oncogenic HRAS-transformed NIH3T3 cells, KLF5 level is high and can be reduced by treatment with MEK inhibitors, indicating that KLF5 is subjected to MAPK/ERK regulation 30. Here we examined MAPK signaling in IEC-iKRAS cells. As seen in Figure 2A, the levels of p-ERK were significantly elevated in cells treated with IPTG when compared to control (lanes 2 and 1, respectively). This increase in p-ERK was reduced to the baseline level in IPTG-induced cells treated with the MEK inhibitor, PD98059, but not the vehicle DMSO (lanes 4 and 3, respectively). The levels of KLF5 under different treatment conditions directly followed those of p-ERK (Figure 2A). The rate of proliferation in IPTG-induced cells treated with PD98059 was also significantly reduced when compared to DMSO-treated cells (Figure 2B; compare dark circles and dark triangles). Of note, the IPTG-induced and PD98059-treated cells (Figure 2B; dark circles) had a greater than expected reduction in proliferation on day 3 than the control untreated cells (Figure 2B; open diamonds). This is likely due to the slight increase in KRAS levels over time in the absence of IPTG (Figure 1A). Our results therefore indicate that MAPK/ERK signaling is important in mediating the pro-proliferative effect of KRAS.
Figure 2. MAPK/ERK signaling mediates the pro-proliferative effect of oncogenic KRAS.
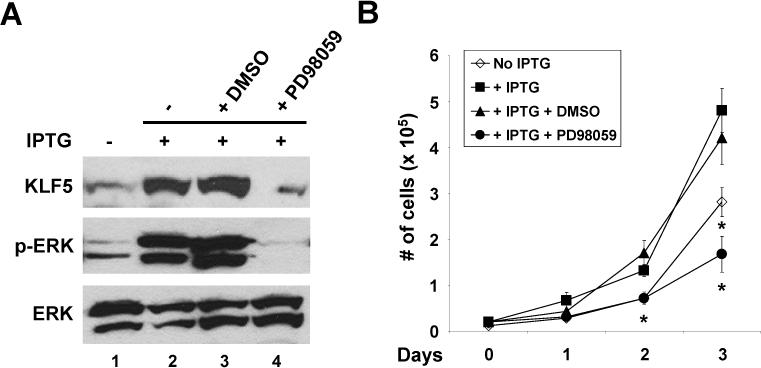
(A) IEC-iKRAS cells were treated with or without 5 mM IPTG for 48 h. Fifty μM PD98059 or DMSO (vehicle control) were added 24 h following the addition of IPTG. Western blot analyses were performed using KLF5, phospho-ERK (p-ERK) and ERK antibodies. (B) Cell proliferation was measured under the treatment conditions depicted in the panel. N = 6; * p < 0.05 when compared to DMSO-treated cells.
We next determined the effect of KLF5 inhibition, using small interfering RNA (siRNA), on proliferation and anchorage-dependent growth in IEC-iKRAS cells inducible by IPTG. Upon transfection of three different KLF5 siRNAs, we detected significant decreases in KLF5 mRNA levels as detected by quantitative RT-PCR when compared to cells transfected with non-specific (NS) siRNA control (Figure 3A). This decrease in mRNA levels was mirrored by a significant reduction in KLF5 protein levels (Figure 3B). Treatment of IPTG-induced iKRAS cells with KLF5-specific siRNA significantly reduced the rate of cell proliferation when compared with cells treated with non-specific siRNA (Figure 3C). Moreover, the number of colonies formed in soft agar was significantly reduced in IPTG-induced cells treated with KLF5-specific siRNA when compared to non-specific siRNA-treated cells (Figure 3D). In comparison, in the absence of IPTG, IEC-iKRas cells transfected with KLF5-specific siRNA showed only a modest decrease in proliferation when compared to untransfected or NS siRNA-transfected cells (Supplementary Figure 1). These results suggest that KLF5 is crucially involved in controlling the rate of cell proliferation and anchorage-independent growth in IEC-6 cells upon KRAS induction.
Figure 3. KLF5 mediates the pro-proliferative effect of oncogenic KRAS in IEC-6 cells.

IEC-iKRAS cells were induced with IPTG for 24 h and then transfected or not with KLF5 siRNA or non-specific (NS) siRNA. RNA and protein were extracted from cells 24 h after siRNA transfection. (A) Real-time PCR was performed on cDNA generated by KLF5-specific primers. The relative KLF5 mRNA levels were determined after adjusting for the levels of control β-actin, with the level in IPTG-treated iKRAS cells set at 1.0. Shown are the means of three independent experiments. (B) Proteins were analyzed by Western blotting using KLF5, KRAS and β-actin antibodies. The treatment conditions were identical to (A). (C) Cell proliferation was measured by cell counting. N = 6 and * p < 0.05, comparing between KLF5 siRNA3- and NS siRNA-treated cells. (D) Anchorage-independent colony formation in soft agar 21 days post-transfection. N = 6; * p < 0.01.
We next established several independent clones of KRAS-transformed cells by selecting colonies formed on soft agar upon induction of KRAS in IEC-iKRAS cells. IPTG treatment of these clones increased KRAS protein to levels higher than those seen in the parental cell line, and correspondingly, KLF5 protein levels were also increased (Figure 4A). Time course analysis of cell proliferation revealed that a representative clone (clone 1) proliferated faster compared to the parental IEC-iKRAS cells (Figure 4B). The rate of proliferation was further increased upon KRAS induction in clone 1. This enhanced proliferation was reduced to baseline when the induced clone 1 was treated with KLF5-specific siRNA (Figure 4C). Enhanced colony formation on soft agar after IPTG induction of clone 1 was also significantly reduced by KLF5-specific siRNA treatment (Figure 4D). Combining the results of the studies thus far, we showed that oncogenic KRAS induction led to increased KLF5 protein levels and enhanced proliferation and transformation. These two phenotypes were suppressed upon treatment with either MEK inhibitors or KLF5-specific siRNA. In addition, stable clones selected from KRAS-induced cells had enhanced basal levels of KLF5 protein and increased proliferation compared to parental cells and this could be further augmented with IPTG induction.
Figure 4. Stable clones of KRAS-transformed cells exhibit higher rates of proliferation that can be reduced by inhibiting KLF5.

Colonies of IEC-iKRAS cells formed on soft agar after induction with IPTG were selected and expanded. A total of 7 clones were examined and they exhibited similar behavior by the following assays. Shown are results from representative clones. (A) Two selected clones (1 and 2) were grown along with parental iKRAS cells with or without IPTG induction for 24 h, after which cell lysates were analyzed for KLF5, KRAS and β-actin proteins. (B) Cell proliferation was measured in parental iKRAS cells and a representative clone (Clone 1) in the presence and absence of IPTG treatment. N = 6; * p < 0.05, comparing IPTG-treated and untreated cells in Clone 1. (C) Cells from Clone 1 were grown with or without IPTG for 24 h and then transfected with KLF5-specific siRNA or non-specific (NS) siRNA for the stated time in the presence of the inducer. N = 6 and * p < 0.05, comparing KLF5- and NS-siRNA-transfected cells. (D) Results of soft agar assays 21 days post-transfection by the stated siRNA in Clone 1 in the presence of IPTG. N = 6; * p <0.01.
KLF5 Levels are Elevated in Human Colorectal Cancer Cell Lines with Mutated KRAS
Since KRAS is mutated in over 50% of human colorectal cancer 12-14, we examined six different human colorectal cancer cell lines for mutated KRAS. By DNA sequencing, we found that DLD-1, SW-480 and HCT116 contained activating mutations, while HT-29, Caco-2 and RKO possessed wild type KRAS (Figure 5A). The levels of KLF5 in the six cell lines correlated with the KRAS genotypes, with those containing mutated KRAS having higher levels (Figure 5A and Supplementary Figure 2). It is of interest to note that HT-29, which contains a mutated BRAF 31, had a modest amount of KLF5 (Figure 5A). DLD-1 cells, with an activated KRASG13D, contained a relatively high baseline level of p-ERK, which was reduced after treatment with MEK inhibitors, PD98059 or U0126 (Figure 5B). Importantly, KLF5 levels in DLD-1 cells were also reduced with inhibitor treatment (Figure 5B). We were able to reduce the level of KLF5 in DLD-1 cells by treatment with siRNA against KLF5 but not non-specific siRNA (Figure 5C). However, p-ERK levels were not reduced after treatment with either siRNA, suggesting that KLF5 does not control p-ERK activity. Treatment with either MEK inhibitors (Figure 5D) or KLF5-specific siRNA (Figure 5E) significantly decreased proliferation rates when compared to their respective controls. Similar results were observed for both SW-480 and HCT116 with either MEK inhibitors or KLF5 siRNA treatment (Supplementary Figures. 3 and 4, respectively). In contrast, RKO cells, which had wild type KRAS, contained negligible levels of KLF5 (Figure 5A) and p-ERK proteins (result not shown). Consequently, treatment of RKO cells with KLF5-specific siRNA had no significant change on cell proliferation (Supplementary Figure 5). Lastly, while the three cell lines with mutated KRAS (DLD-1, SW-480 and HCT116), had significant decreases in the number of colonies in soft agar upon when treated with KLF5-specific siRNA as compared to control, there was no change observed in RKO cells (Figure 5F). These results suggest that KLF5 plays an important role in controlling proliferation and transformation in colorectal cancer cell lines with oncogenic KRAS mutation. Moreover, upon over-expression of KLF5 in two of the cell lines (DLD-1 and HCT116), there was an increased in the number of colonies when compared to control (Supplementary Figure 6), further supporting a pro-proliferative effect of KLF5 on colorectal cancer cells.
Figure 5. Human colorectal cancer cell lines containing oncogenic KRAS have high KLF5 levels.

(A) Six human colorectal cancer cell lines were cultured and cell lysates extracted when sub-confluent. Proteins were analyzed by Western blotting using KLF5 and β-actin antibodies. KRAS codons 12, 13 and 61 were sequenced and oncogenic mutations, if present, were noted. WT is wild type. (B) DLD-1 cells were treated with 50 μM MEK inhibitors, PD98059 and U0126, or DMSO for 24 h or left untreated. Proteins were extracted and analyzed by Western blotting for KLF5, p-ERK, ERK and β-actin. (C) DLD-1 cells were either untreated or transfected with KLF5-specific siRNA or non-specific (NS) siRNA and cell lysates prepared 24 h after transfection. Western blot analysis was performed for KLF5, p-ERK, ERK and β-actin. (D) Cell proliferation was measured in DLD-1 cells treated with MEK inhibitors, DMSO or left untreated for up to three days. N = 6; * p < 0.05, comparing MEK inhibitor-treated and DMSO-treated cells. (E) Cell proliferation was determined in DLD-1 cells transfected with KLF5-specific or NS siRNA for up to three days post transfection. N = 6; * p < 0.05, comparing KLF5 siRNA and NS siRNA-transfected cells. (F) Anchorage-independent growth was examined in soft agar for DLD-1, SW-480, HCT 116 and RKO cells. Untransfected, KLF5 siRNA or NS siRNA-transfected cells were evaluated 21 days post-treatment for colony number. N = 6; * p <0.01.
Increased KLF5 Expression in Intestinal Neoplasm from Villin-KRASV12G Transgenic Mice
Transgenic mice that over-express KRASV12G in the intestine under control of the villin promoter were previously shown to develop intestinal neoplasm 19. We therefore examined KLF5 levels by immunohistochemistry in intestinal tumors developed in the villin-KRASV12G transgenic mice. In normal small intestinal epithelium, KLF5 is present in the nuclei of epithelial cells lining the crypts with a diminishing gradient toward the villus (Figure 6A). In contrast, KLF5 is uniformly present throughout intestinal adenoma and carcinoma (Figures 6B, 6C and 6D). This staining pattern is representative of multiple adenomas and four different carcinomas examined. Quantification of staining intensities showed that KLF5 levels were approximately 20% higher in neoplastic tissues than normal tissues (Supplementary Figure 8A). This suggests that oncogenic KRAS expression in the intestinal epithelium caused tumor formation with associated elevation in KLF5 levels.
Figure 6. KLF5 expression in intestinal tumors from villin-KRASV12G transgenic mice.

Tissues were dissected from mice and formalin fixed, paraffin-embedded and sectioned in 5 μm thickness. Histochemical sections were analyzed for KLF5 protein (brown color) and nuclei counterstained using Mayer's Hematoxylin (blue color). (A) Representative normal intestinal tissue from mouse jejunum analyzed (10× magnification) showing localized KLF5 staining in the crypt epithelium. (B) Adenomatous tissue (10× magnification) from jejunum showing intense KLF5 staining throughout the tumor section. (C) Histological section showing invasive carcinoma in the muscularis mucosa (4× magnifications), adjacent to adenomatous tissues, showing intense KLF5 staining. (D) 10× magnification of invasive carcinoma from panel (C).
Analysis of KLF5 in Human Colorectal Cancer Specimens
We obtained 8 matched normal and cancer specimens from patients who underwent surgical resection and genotyped them for KRAS. Seven of 8 tumor specimens contained activating mutations in codon 12 or 13 of KRAS (Supplementary Table). KLF5 staining in the normal human colonic epithelium was primarily in the crypt epithelial cells in a manner that is similar to the mouse small intestine (Figures 7A, 7B and C). In contrast, KLF5 staining was much more extensive within the tumors with mutated KRAS (Figures. 7D, 7E and 7F). Upon quantification, KLF5 staining intensities were approximately 20% higher in tumor tissues compared to normal tissues (Supplementary Figure 8B). Since KLF5 is involved in proliferation, we also stained tissues for Ki67, a proliferation marker. We observed that the Ki67 staining pattern closely correlated to KLF5 in both normal (results not shown) and tumor tissues (Supplementary Figure 7). In the lone tumor specimen with wild-type KRAS from patient 3, KLF5 staining was present in the cytoplasm of tumor cells (Figure 8E) as opposed to the typical nuclear staining in the crypt cells of matched normal colonic epithelium from the same patient (Figure 8B). Immunohistochemical staining of tissues from patient 3 in the presence of KLF5 blocking peptide resulted in the disappearance of KLF5 staining from nuclei of the normal tissue (Figure 8C) and cytoplasm of the tumor tissue (Figure 8F), suggesting that the cytoplasmic localization of KLF5 in this particular tumor specimen is specific.
Figure 7. Immunohistochemical analysis of KLF5 in human colorectal cancer specimens with mutated KRAS.

Matched normal and tumor tissues resected from patients with colorectal carcinoma were stained with an antibody specific to KLF5 and the nuclei counterstained with Mayer's Hematoxylin. (A, B and C) Normal colonic sections from patients 1, 5 and 7. (D, E and F) Tumor tissues from patients 1, 5 and 7.
Figure 8. Immunohistochemical analysis of KLF5 in human colorectal cancer specimens with wild type KRAS.

Matched normal and tumor tissues resected from patients were stained with KLF5 antibodies and the nuclei counterstained with Mayer's Hematoxylin. (A and B) Normal colon tissues resected from patients 1 and 3. (D and E) Tumor specimens from patient 1 (carrying KRASG13D mutation) and patient 3 (containing wild type KRAS) stained for KLF5. (C and F) Normal and tumor tissues from patient 3 stained with KLF5 antibodies in the presence of an excessive amount of KLF5 peptide used to generate the antibody.
DISCUSSION
Oncogenic RAS mutations occur in about 30% of all human cancers 2, 3, 13 and 50% of colorectal cancers 12-14. Since KRAS has been shown to be essential for normal development 32, 33, strategies targeting downstream effectors of the RAS signaling pathway may be important in the treatment of cancer with KRAS mutations. Results of our previous studies showed that oncogenic HRAS over-expression in NIH3T3 cells resulted in enhanced levels of KLF5, which promoted cell proliferation and transformation 29, 30. Results of our current study extended these findings to intestinal tumors containing oncogenic KRAS mutations. These studies suggest that KLF5 may potentially be a therapeutic target in managing colorectal cancer with KRAS mutations.
The correlation between KRAS mutation and KLF5 expression is demonstrated both in vitro and in vivo in the current study. In vitro, both IEC-6 intestinal epithelial cells following induction of KRASV12G and established colorectal cancer cell lines with endogenous KRAS mutations contained high levels of KLF5. The increased KLF5 levels correlated with elevated MAPK activity as reflected by the levels of p-ERK. Treatment of cells containing oncogenic KRAS with MEK inhibitors resulted in diminished proliferation and anchorage-independent growth. Significantly, the inhibitory effect on cell proliferation and transformation was replicated in cells treated with siRNA that inhibited KLF5 expression. These findings are highly indicative of a causal relationship between oncogenic KRAS mutation and KLF5 activation. Moreover, KLF5 manifests itself as an essential downstream mediator for the oncogenic activity of KRAS. A potential mechanism by which KLF5 exerts this important effect is to activate expression of genes involved in cell cycle progression such as cyclin D1, cyclin B1 and Cdc2, as previously shown in oncogenic HRAS-transformed NIH3T3 cells 29, 30. The potential for KLF5 to serve as a therapeutic target in the treatment of colorectal cancer has also been previously illustrated by the ability of all-trans retinoic acid (ATRA) to inhibit KLF5 expression and proliferation of colorectal cancer cell lines with high endogenous levels of KLF5 34.
The current study also demonstrated an in vivo correlation between oncogenic KRAS mutations and KLF5 expression. Thus, both intestinal tumors derived from villin-KRASV12G transgenic mice and human colorectal cancer specimens with endogenous KRAS mutations displayed high levels of nuclear KLF5 staining. Moreover, the relative intensity of nuclear KLF5 was higher in the tumors than in the matched normal tissues in both mice and humans. Thus, enhanced and perhaps even dysregulated KLF5 expression may be a characteristic biological marker for tumors with KRAS mutation. To this end, it is of interest to note a previous report stating that expression of KLF5 is directly correlated with cell proliferation in vivo and is a prognostic factor for patients with breast cancer 35. Specifically, patients with higher KLF5 expression have shorter disease-free and overall survival than patients with lower KLF5 expression 35. In the case of colorectal cancer, several studies have demonstrated that KRAS mutation is associated with a poorer survival 36-39. It would be interesting to determine whether KLF5 levels may also influence prognosis of colorectal cancer.
Similar to the findings of our current study, a previously published paper also established a pro-proliferative role for KLF5 in non-transformed intestinal epithelial cells including IEC-18 and IMCE 40. However, their results indicated that over-expression of KLF5 by transfection led to decreased cell proliferation and transformation in colon cancer cell lines including Ward and DLD-1, and that siRNA-silencing of KLF5 resulted in enhanced rate of proliferation in HCT116 40. They also reported that IMCE cells stably transfected with oncogenic H-Ras when exogenously induced to express KLF5 showed reduced proliferation and transformation. Finally, their results showed that adenomas from ApcMin/+ mice and a patient with familial adenomatous polyposis showed decreased levels of KLF5 mRNA when compared with the corresponding normal crypt epithelium. Our study did not address the final point of the Bateman paper as intestinal adenomas derived from APC mutations evolve by a different genetic and biochemical mechanism from those with KRAS mutations. Another major difference between our present work and the Bateman paper is that we focused on the effect of activation of the endogenous KLF5 gene by an overactive KRAS, while the Bateman study primarily involved over-expression of an exogenous KLF5. We did attempt to replicate the results of the Bateman paper by over-expressing KLF5 in several different colorectal cancer cell lines including DLD-1 and HCT116 but obtained the opposite outcome (Supplementary Figure 6). The reason behind this discrepancy is not clear at this time but could be due to clonal variations or differences in the level of expression of the transfected KLF5 gene.
The villin-KRASV12G transgenic mice used in this study probably offer an excellent opportunity to further explore the relationship between KRAS activation and KLF5 induction in vivo and the potential for therapeutic interventions of KRAS-induced intestinal tumorigenesis by manipulating KLF5 levels. However, there are potential limitations to the model. One is the relatively long latency (often > 9 months) by which tumors develop. The second is that over-expression from a villin promoter may not recapitulate the physiologic nature of intestinal tumorigenesis 41. To this end, other mouse models of KRAS-mediated carcinogenesis that rely on tissue-specific conditional mutational activation of the endogenous KRAS locus 17, 18, 42, 43 may provide additional opportunities to examine the biological relevance of KLF5 in mediating the effect of oncogenic KRAS in intestinal tumorigenesis. As results of our in vitro studies showed a strong correlation between reduction in KLF5 expression and decreased proliferation and transformation, identification of small molecules with which to inhibit KLF5 expression or activity and subsequent testing in animals could lead to the development of novel therapeutic agents for colorectal cancer.
Acknowledgments
Grant Support: This work was in part supported by NIH grants DK52230, DK64399 and CA84197.
Abbreviations
- APC
adenomatous polyposis coli
- ERK
extracellular signal-regulated kinase
- IKLF
intestinal-enriched Krüppel-like factor
- IPTG
isopropyl-1-thio-β-D-galactopyranoside
- KLF5
Krüppel-like factor 5
- MAPK
mitogen-activated protein kinase
- MEK
MAPK/ERK kinase
- RT-PCR
reverse transcription-polymerase chain reaction
- siRNA
small interfering RNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
No conflicts of interest exit.
Supplementary Material
REFERENCES
- 1.Barbacid M. ras genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 3.Bos JL. The ras gene family and human carcinogenesis. Mutat Res. 1988;195:255–271. doi: 10.1016/0165-1110(88)90004-8. [DOI] [PubMed] [Google Scholar]
- 4.Feig LA, Schaffhausen B. Signal transduction. The hunt for Ras targets. Nature. 1994;370:508–509. doi: 10.1038/370508a0. [DOI] [PubMed] [Google Scholar]
- 5.Kodaki T, Woscholski R, Hallberg B, et al. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4:798–806. doi: 10.1016/s0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 6.Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- 7.Marais R, Marshall CJ. Control of the ERK MAP kinase cascade by Ras and Raf. Cancer Surv. 1996;27:101–125. [PubMed] [Google Scholar]
- 8.Marshall CJ. Ras effectors. Curr Opin Cell Biol. 1996;8:197–204. doi: 10.1016/s0955-0674(96)80066-4. [DOI] [PubMed] [Google Scholar]
- 9.Marshall MS. Ras target proteins in eukaryotic cells. Faseb J. 1995;9:1311–1318. doi: 10.1096/fasebj.9.13.7557021. [DOI] [PubMed] [Google Scholar]
- 10.Almoguera C, Shibata D, Forrester K, et al. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 11.Smit VT, Boot AJ, Smits AM, et al. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res. 1988;16:7773–7782. doi: 10.1093/nar/16.16.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arber N, Shapira I, Ratan J, et al. Activation of c-K-ras mutations in human gastrointestinal tumors. Gastroenterology. 2000;118:1045–1050. doi: 10.1016/s0016-5085(00)70357-x. [DOI] [PubMed] [Google Scholar]
- 13.Bos JL, Fearon ER, Hamilton SR, et al. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- 14.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 15.Sheng H, Shao J, Dubois RN. K-Ras-mediated increase in cyclooxygenase 2 mRNA stability involves activation of the protein kinase B1. Cancer Res. 2001;61:2670–2675. [PubMed] [Google Scholar]
- 16.Tuveson DA, Shaw AT, Willis NA, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 17.Johnson L, Mercer K, Greenbaum D, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 18.Guerra C, Mijimolle N, Dhawahir A, et al. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 19.Janssen KP, el-Marjou F, Pinto D, et al. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]
- 20.McConnell BB, Ghaleb AM, Nandan MO, et al. The diverse functions of Krüppel-like factors 4 and 5 in epithelial biology and pathobiology. BioEssays. 2007;29:549–557. doi: 10.1002/bies.20581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghaleb AM, Nandan MO, Chanchevalap S, et al. Krüppel-like factors 4 and 5: the yin and yang regulators of cellular proliferation. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dang DT, Pevsner J, Yang VW. The biology of the mammalian Krüppel-like family of transcription factors. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaczynski J, Cook T, Urrutia R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003;4:206.1–206.8. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conkright MD, Wani MA, Anderson KP, et al. A gene encoding an intestinal-enriched member of the Krüppel-like factor family expressed in intestinal epithelial cells. Nucleic Acids Res. 1999;27:1263–1270. doi: 10.1093/nar/27.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun R, Chen X, Yang VW. Intestinal-enriched Krüppel-like factor (Krüppel-like factor 5) is a positive regulator of cellular proliferation. J Biol Chem. 2001;276:6897–6900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chanchevalap S, Nandan MO, McConnell BB, et al. Krüppel-like factor 5 is an important mediator for lipopolysaccharide-induced proinflammatory response in intestinal epithelial cells. Nucleic Acids Res. 2006;34:1216–1223. doi: 10.1093/nar/gkl014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen C, Benjamin MS, Sun X, et al. KLF5 promotes cell proliferation and tumorigenesis through gene regulation and the TSU-Pr1 human bladder cancer cell line. Int J Cancer. 2006;118:1346–1355. doi: 10.1002/ijc.21533. [DOI] [PubMed] [Google Scholar]
- 28.Ziemer LT, Pennica D, Levine AJ. Identification of a mouse homolog of the human BTEB2 transcription factor as a beta-catenin-independent Wnt-1-responsive gene. Mol Cell Biol. 2001;21:562–574. doi: 10.1128/MCB.21.2.562-574.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nandan MO, Chanchevalap S, Dalton WB, et al. Krüppel-like factor 5 promotes mitosis by activating the cyclin B1/Cdc2 complex during oncogenic Ras-mediated transformation. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nandan MO, Yoon HS, Zhao W, et al. Krüppel-like factor 5 mediates the transforming activity of oncogenic H-Ras. Oncogene. 2004;23:3404–3413. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 32.Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koera K, Nakamura K, Nakao K, et al. K-ras is essential for the development of the mouse embryo. Oncogene. 1997;15:1151–1159. doi: 10.1038/sj.onc.1201284. [DOI] [PubMed] [Google Scholar]
- 34.Chanchevalap S, Nandan MO, Merlin D, et al. All-trans retinoic acid inhibits proliferation of intestinal epithelial cells by inhibiting expression of the gene encoding Krüppel-like factor 5. FEBS Lett. 2004;578:99–105. doi: 10.1016/j.febslet.2004.10.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong D, Czerwenka K, Heinze G, et al. Expression of KLF5 is a prognostic factor for disease-free survival and overall survival in patients with breast cancer. Clin Cancer Res. 2006;12:2442–2448. doi: 10.1158/1078-0432.CCR-05-0964. [DOI] [PubMed] [Google Scholar]
- 36.Lee JC, Wang ST, Lai MD, et al. K-ras gene mutation is a useful predictor of the survival of early stage colorectal cancers. Anticancer Res. 1996;16:3839–3844. [PubMed] [Google Scholar]
- 37.Samowitz WS, Curtin K, Schaffer D, et al. Relationship of Ki-ras mutations in colon cancers to tumor location, stage, and survival: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000;9:1193–1197. [PubMed] [Google Scholar]
- 38.Nemunaitis J, Cox J, Hays S, et al. Prognostic role of K-ras in patients with progressive colon cancer who received treatment with Marimastat (BB2516). Cancer Invest. 2000;18:185–190. doi: 10.3109/07357900009031822. [DOI] [PubMed] [Google Scholar]
- 39.Belly RT, Rosenblatt JD, Steinmann M, et al. Detection of mutated K12-ras in histologically negative lymph nodes as an indicator of poor prognosis in stage II colorectal cancer. Clin Colorectal Cancer. 2001;1:110–116. doi: 10.3816/CCC.2001.n.011. [DOI] [PubMed] [Google Scholar]
- 40.Bateman NW, Tan D, Pestell RG, et al. Intestinal tumor progression is associated with altered function of KLF5. J Biol Chem. 2004;279:12093–12101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 41.Sansom OJ, Meniel V, Wilkins JA, et al. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc Natl Acad Sci U S A. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jackson EL, Willis N, Mercer K, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez-Mancera PA, Tuveson DA. Physiological analysis of oncogenic K-ras. Methods Enzymol. 2005;407:676–690. doi: 10.1016/S0076-6879(05)07053-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.