

Abstract
Previous evidence demonstrated the ability of quinazoline-based α1-adrenoceptor antagonist doxazosin, to suppress prostate tumor growth via apoptosis. In this study we performed structural optimization of doxazosin’s chemical nucleus and a subsequent structure-function analysis towards the development of a novel class of apoptosis-inducing and angiogenesis-targeting agents. Our lead compound, DZ-50, was effective at reducing endothelial cell viability via a non-apoptotic mechanism. Treatment with DZ-50 effectively prevented in vitro tube formation and in vivo chorioallantoic membrane (CAM) vessel development. Confocal microscopy revealed a significantly reduced ability of tumor cells to attach to extracellular matrix (ECM) and migrate through endothelial cells in the presence of DZ-50. In vivo tumorigenicty studies using two androgen-independent human prostate cancer xenografts, PC-3 and DU-145, demonstrated that DZ-50 treatment leads to significant suppression of tumorigenic growth. Exposure to the drug at the time of tumor cell inoculation led to prevention of prostate cancer initiation. Furthermore, DZ-50 resulted in a reduced formation of prostate-tumor derived metastastic lesions to the lungs in an in vivo spontaneous metastasis assay. Thus, our drug discovery approach led to the development of a class of lead (quinazoline-based) compounds with higher potency than doxazosin in suppressing prostate growth by targeting tissue vascularity. This new class of quinazoline-based compounds provides considerable promise as anti-tumor drugs, not only for the treatment of metastatic disease, but also for the primary prevention of human prostate cancer.
Keywords: quinazoline, prostate cancer, angiogenesis, apoptosis, integrins
INTRODUCTION
Prostate cancer is a major contributor to cancer mortality in American males causing the death of approximately 30,000 men in 2006 (1). Therapeutic modalities such as radical prostatectomy and radiotherapy are considered curative for localized disease, yet no treatments for metastatic prostate cancer are available that significantly increases patient survival (2). Clinical and experimental evidence implicates two components as contributors towards the emergence of the androgen-independent phenotype: activation of survival (apoptosis suppression) pathways and increased tumor neovascularization (3, 4). Consequently, targeting of apoptotic players is of vital therapeutic significance since resistance to apoptosis is not only critical in conferring therapeutic failure to standard treatment strategies, but anoikis (cell death upon detachment from extracellular matrix) also plays an important role in angiogenesis and metastasis of malignant cells (5, 6).
Angiogenesis, the formation of new blood vessels by capillary sprouting from pre-existing vessels, is critical in tumor progression and metastasis, since a functional vascular supply is required for the continued growth of solid tumors, and the spread of cancer cells (7). Small non-growing tumors may remain dormant for years and the angiogenic switch to aggressive metastatic phenotype, involves a change in the local equilibrium between factors inducing blood vessel formation and those inhibiting the process (8, 9). During angiogenesis cells are in a dynamic state, lacking firm attachment to the extracellular matrix, and exceedingly vulnerable to anoikis. Thus, targeting tumor endothelial cell survival by triggering anoikis, may provide a molecular basis for novel therapeutic strategies for metastatic prostate cancer. Two classes of angiogenesis-targeting agents consequently emerge: those preventing the development of neovasculature of tumors, (via inducing apoptosis and/or inhibiting cell proliferation and migration), and those that directly target the existing tumor vasculature (via anoikis of tumor endothelial and epithelial cells) (10, 11).
The quinazoline-based compounds doxazosin and terazosin are known α1-adrenoreceptor antagonists, clinically effective for the relief of benign prostate hyperplasia (BPH) symptoms via their ability to selectively antagonize the α1a-adrenoreceptors, distributed in the bladder neck and prostate gland (12). Recent experimental and clinical evidence however, documented additional antigrowth effects by the quinazoline-based adrenoceptor antagonists, via induction of prostate epithelial and smooth muscle cell apoptosis as one of the molecular mechanisms contributing to their overall long-term clinical efficacy in BPH patients (13, 14). Suppression of prostate tumor growth by these drugs proceeds via an α1-adrenoceptor-independent mechanism, mediated by TGF-β1 apoptotic signaling (15, 16), receptor-mediated apoptosis involving DISC formation and caspase-8 activity (17) and inhibition of Akt activation (17, 18).
The separation of doxazosin’s effect on cancer cell apoptosis from its original pharmacological activity in vascular cells provides an intriguing molecular basis to develop a novel class of apoptosis-inducing agents through lead optimization. Our recent pharmacological exploitation of doxazosin’s quinazoline nucleus led to the development of novel compounds with and without the characteristic “classic” apoptotic activity, but exhibiting potent anti-vascular activity (18). In this study, we report the characterization of targeting, by the new lead quinazoline-based compounds, of prostate tumor epithelial and endothelial cell survival, migration, neovascularization and angiogenesis in vitro and in vivo.
RESULTS
DZ-50 is effective at inducing cell death via a non-apoptotic mechanism
Pharmacological exploitation of doxazosin’s quinazoline nucleus led to the development of several novel agents with varying effects on apoptosis (Fig. 1a). Functional characterization of these compounds revealed two classes of agents: those that are not effective at inducing apoptosis, but elicit their effects by an alternative cell-death mechanism (DZ-50) and those that trigger apoptotic cell death (DZ-3) (Fig. 1a–1c). The ost intriguing compound from the first category was DZ-50 that reduced cell viability in a number of endothelial and epithelial cells lines at both 24 and 48hrs without induction of classic apoptosis (Fig. 1d).
Figure 1. Effect of novel lead quinazoline-derived compound DZ-50 on human prostate cancer cells.


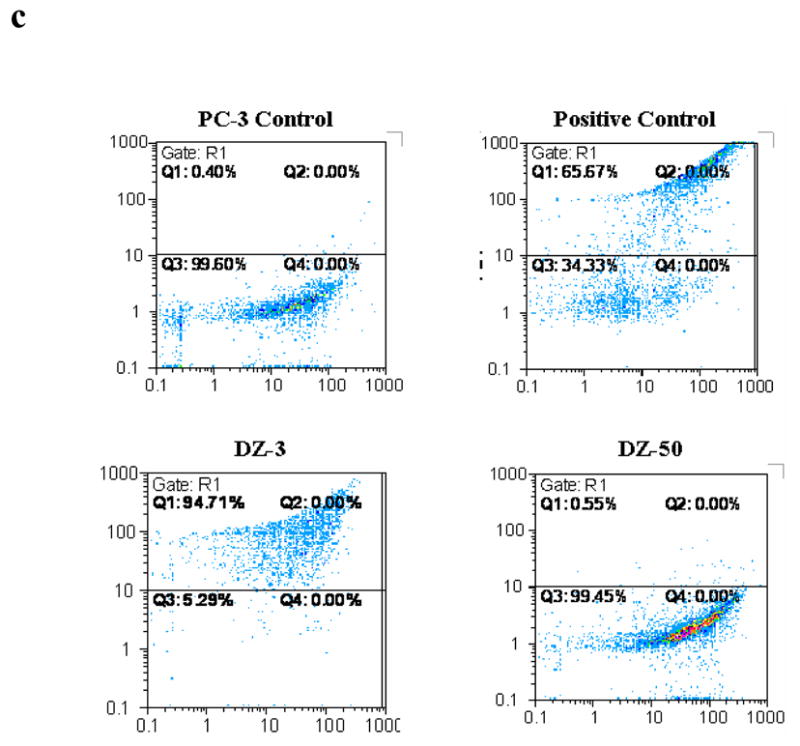

(a) Chemical Structure of DZ-50: The 2,3-difydro-benzo [1,4] dioxane0-carbonyl moitey of doxazisin was replaced with the bi-phenyl aryl sulfonyl substituent; while the methoxy side chains were replaced with isopropyl propoxy functions. (b) Apoptosis induction by novel quinazoline compounds. PC-3 cells were treated (10μM) for 24hrs and apoptosis was measured by Hoechst staining. (c) Apoptosis induction by DZ-3. FACS analysis of PI and BrdU staining was performed on PC-3 cells treated with DZ-3 (10μM) and a negative control, DZ-50 (10μM). (d) Cell death following DZ-50 treatment. Cell death was evaluated in endothelial and epithelial cells lines following a 24hr and 48hr (inset) treatment of DZ-50 (5μM, 10μM, 15μM, 20μM, and 25μM) as described in “Methods”.
Anoikis effect of DZ-50: Inhibition of cell migration and cell adhesion
The ability of DZ-50 to potentially trigger anoikis of tumor epithelial and endothelial cells was subsequently investigated. Treatment with DZ-50 at (non-cytotoxic doses) led to a significant inhibition of endothelial cell and prostate tumor epithelial cell migration (Fig. 2a, S1). Moreover, exposure of PC-3 prostate cancer cells to DZ-50 markedly reduced cellular adhesion to the extracellular matrix proteins fibronectin and collagen at 12hrs (Fig. 2b). While there was no change detected in the attachment of DU-145 prostate epithelial cells to collagen, attachment to fibronectin was significantly inhibited by the drug after 12hrs of treatment (Fig. 2c). Transendothelial migration assays were performed to assess the ability of PC-3 prostate cancer cells to attach and migrate through a monolayer of HMVEC-L following exposure to DZ-50. PC-3 cells were stained with the lipophilic tracer DiI (red) and were subsequently added to a confluent monolayer of HMVEC-L and exposed to DZ-50 for 3 and 9hrs (Fig. 2d). DAPI staining identified the nuclei (blue). As shown on Figure 2d, tumor epithelial cell adhesion to the endothelial cell monolayer was prevented following 9hrs of exposure to the drug (10μM). There was no effect on cell viability/cell death in either cell population (PC-3 nor HMVEC-L cells) in response to the drug DZ-50 (10μM), with the first 24hrs of treatment (Fig. 2e), indicating that the effect on transendothelial tumor cell migration was not due to drug-induced cell death.
Figure 2. DZ-50 prevents cell migration and adhesion to ECM of human prostate tumor epithelial cells and vascular endothelial cells.



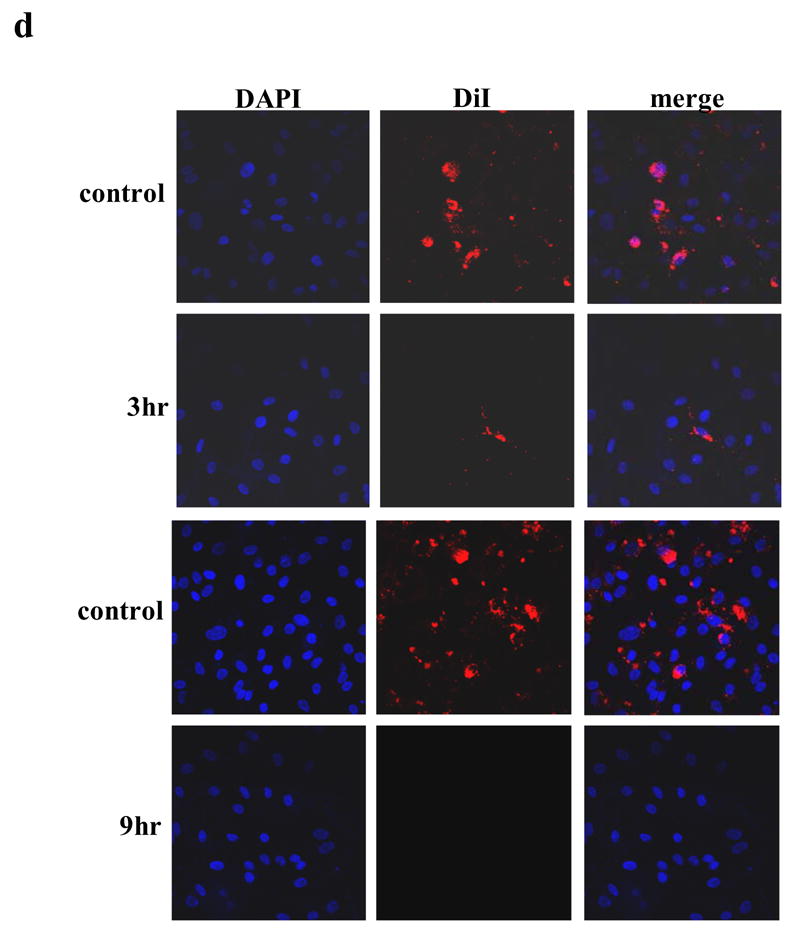

(a) Wounding assays were performed on endothelial and epithelial cells and the number of migratory cells was quantified (as described in the “Methods”). There was a significant reduction in the migratory capacity detected in the vascular endothelial and tumor epithelial cells analyzed; * P < 0.0001, ** P < 0.001, *** P 0.004. (b, c) DZ-50 prevents prostate tumor epithelial cell attachment to ECM components. The ability of prostate cancer cells PC-3 to adhere to ECM protein components was evaluated after exposure to DZ-50 for 6, 9, and 12hrs at concentrations of 5μM and 10μM. Attached prostate cancer cells were counted on fibronectin or collagen coated culture dishes (mean ± SD.). DZ-50 significantly reduced the ability of PC-3 cells to attach to either fibronectin or collagen. (d, e) DZ-50 prevents prostate cancer epithelial cell adhesion to endothelial cells. Transdendothelial migration assays were performed to assess the ability of PC-3 prostate cancer cells to attach and migrate through a monolayer of HMVEC-L following exposure to DZ-50. (d) PC-3 cells were stained with the lipophilic tracer DiI (red) and were subsequently added to a confluent monolayer of HMVEC-L and exposed to DZ-50 for 3 and 9hrs. DAPI staining identified the nuclei (blue). Epithelial cell adhesion to the endothelial cell monolayer was prevented following 9hrs of exposure to the drug (10μM). Cell viability assays were performed on PC-3 and HMVEC-L cells treated with DZ-50 (10μM). No death was detected within the first 24hrs of treatment, indicating that the effect on transendothelial tumor cell migration was not due to drug-induced loss of cell viability (e).
We subsequently investigated the direct effect of our lead drug DZ-50 on angiogenesis in vitro using the tube formation assay. As shown on Figure 3 (panels a, b), following treatment with DZ-50, vascular endothelial cell tube formation was significantly inhibited. Furthermore, exposure to DZ-50 led to a significant suppression of angiogenesis/vascularity in the in vivo CAM blood vessel development assay (Fig. 3c, d). Simultaneous presence of a potent angiogenic factor VEGF and/or bFGF (data not shown) was not able to the rescue the cells from the antiangiogenic effect of DZ-50.
Figure 3. DZ-50 prevents angiogenesis in vitro and in vivo.

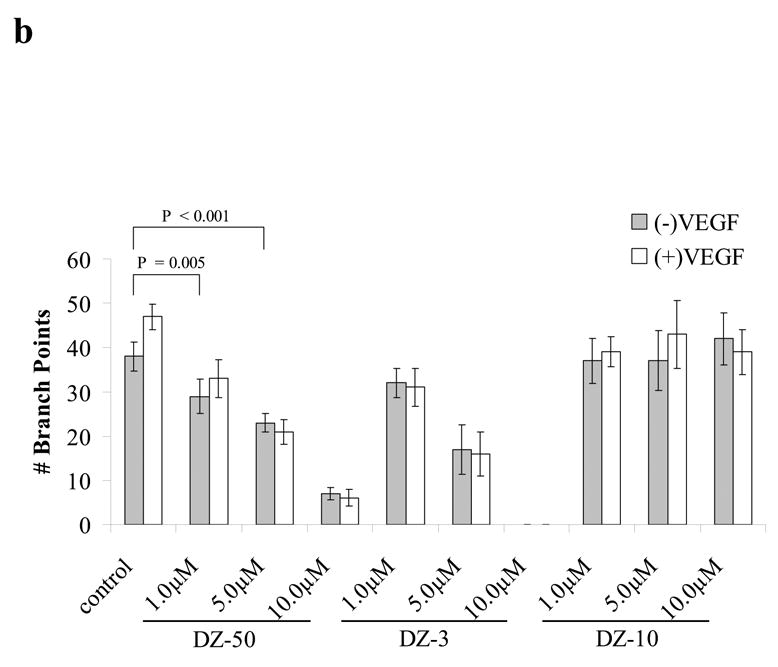

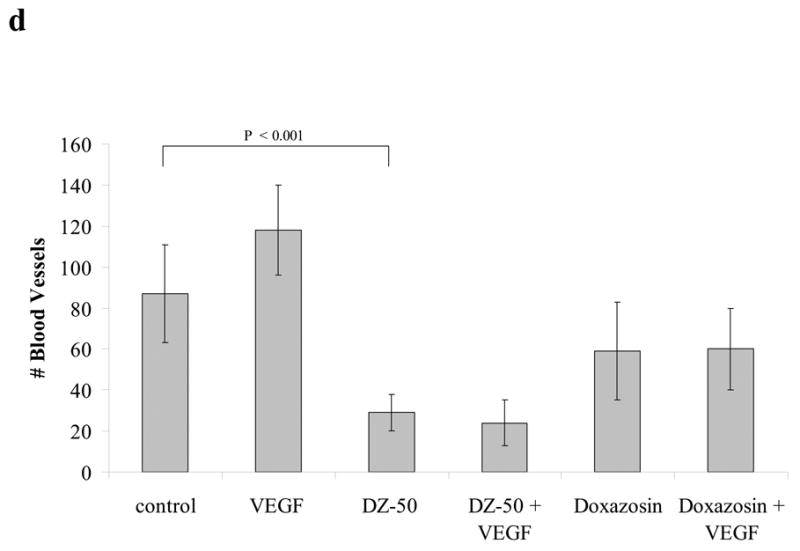
(a,b) In vitro angiogenesis is blocked following exposure to DZ-50. Endothelial cells were seeded in Matrigel in the presence or absence of either DZ-50 or doxazosin at 10μM concentration and tube formation was visualized and quantified in the presence or absence of VEGF, as described in “Methods”. The control (upper panel) shows HUVEC tube formation with decisive branch points while the DZ-50 severely abrogated branch point formation. Panel b, reveals the quantitative analysis of the data; a significant reduction in tube formation is detected in the presence of DZ-50 compared to controls while the novel quinazoline DZ-10 (no effect on cell viability-negative control) does not change the ability of HUVEC cells to form multi-branched tubular networks. VEGF, cannot reverse the antiangiogenic effect of DZ-50, (c,d) In vivo angiogenesis is blocked by DZ-50. CAM assays were performed in the presence or absence of DZ-50 as described in “Methods” and the number of blood vessels were counted.
Reduction of integrin β1 surface expression by DZ-50
To explore the potential mechanism underlying that action of DZ-50 against prostate tumor epithelial cells, analysis of the integrin expression profile was performed. PC-3 untreated control cells were found to express integrin subunits α2, α3, αV, β1, and β3 (Supplementary Data; Fig. S2). Exposure to DZ-50 did not effect the surface expression of integrins α2, α3, αV, and β3 (data not shown). As shown on Figure 4 (panel a), integrin β1 subunit was undetectable in cells treated with DZ-50 for 12–24hrs, compared to vehicle control (Fig. 4a). DU-145 prostate cells exposed to DZ-50, exhibited a significantly smaller shift in integrin β1 expression intensity (Fig. 4b).
Figure 4. DZ-50 targets integrin expression profile in human prostate cancer cells.
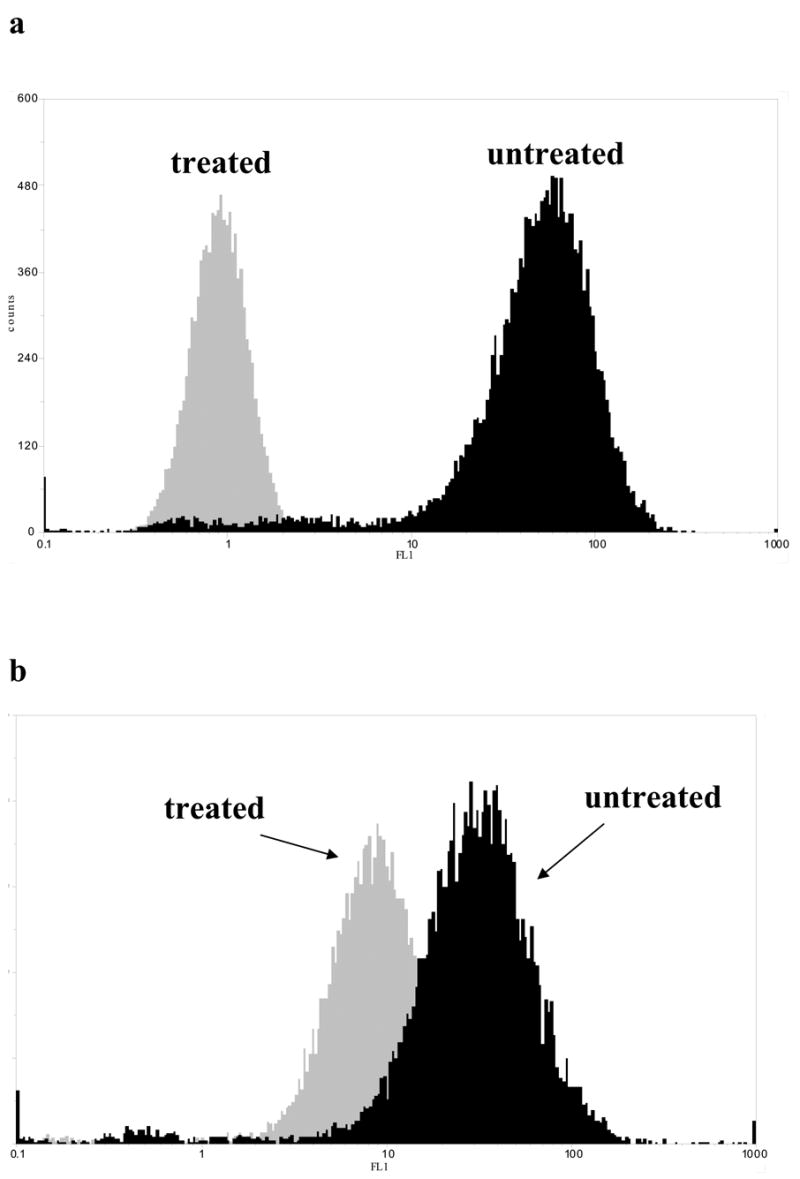
(a) Comparison of integrin β1 expression on PC-3 prostate cells following 12hrs exposure of DZ-50 (10μM) or vehicle control (DMSO). (b) Comparison of integrin β1 expression on DU-145 prostate cells following 12hrs exposure of DZ-50 (10μM) or vehicle control (DMSO).
DZ-50 treatment suppresses prostate tumor growth in vivo
To assess the ability of DZ-50 to suppress prostate cancer growth we subsequently investigated the in vivo anti-tumor efficacy in human prostate cancer xenografts growing in nude mice. Our initial toxicity studies revealed no change in the animal’s behavioral pattern and weight (data not shown). Both gross and histological examination of lung, liver, spleen, and prostate showed no apparent changes compared to control animals (data not shown). The tumorigenicity studies demonstrated a significant reduction in tumor volume in both androgen-independent human prostate cancer PC-3 and DU-145 tumor xenografts following treatment with DZ-50 (200mg/kg) (Fig. 5a, b). The primary prevention efficacy of DZ-50 was examined by inoculation of nude mice with PC-3 prostate cancer cells with simultaneous treatment initiation with DZ-50 (200mg/kg). As shown on Figure 5 (panel c), prostate tumor development was dramatically suppressed with drug exposure (2wks).
Figure 5. Suppression of primary tumor growth and prevention of prostate tumor development in human prostate cancer xenograft model by DZ-50.




(a, b) Tumor volume of prostate xenografts is reduced following DZ-50 treatment. Following subcutaneous inoculation of nude mice (n = 6 per group) with either PC-3 (panel a) or DU-145 (panel b) human prostate cancer cells, DZ-50 (100mg/kg and 200mg/kg) was administered orally (via oral-gavage) to tumor-bearing-hosts for 14 days (subsequent to palpable tumor formation). Tumor volume was measured daily as described in “Methods”; DZ-50 treatment significantly suppressed prostate tumor volume compared to the vehicle control, P < 0.001. (c) Primary prevention of androgen-independent human prostate cancer by DZ-50. To determine the ability of the new lead drug to prevent prostate cancer development, nude mice were subcutaneously inoculated (n = 6 per group) with PC-3 cells with concurrent exposure (orally) to DZ-50 (200mg/kg) for 2-wks. (d) Prostate cancer xenografts were excised from DZ-50-treated and vehicle control tumor-bearing mice, paraffin-embedded, and tissue sections (6μM) were subjected to immunohistochemical analysis for apoptosis, cell proliferation and tumor vascularity, (panels a and b). The three panels represent TUNEL staining for apoptosis, CD31 immunoreactivity for vascularity, and Ki67 expression for cell proliferation, (Magnification 400x). Quantitative analysis of the results is shown on Table 1.
In situ detection of apoptosis in prostate tumor sections revealed no significant change in the apoptotic index of DZ-50 of prostate tumor xenografts from treated tumor-bearing mice compared to control (Fig. 5d, S3) further verifying that this compound does not induce apoptosis. Comparative analysis of the proliferative index of human prostate tumor xenografts from PC-3 and DU-145 cells derived from untreated and DZ-50 treated tumor bearing hosts, revealed no significant changes after treatment with DZ-50. In contrast, treatment with DZ-50 led to a significant suppression of vascularity and angiogenesis, as detected by the reduced CD31 immunoreactivity in both PC-3 and DU-145 derived prostate tumor xenografts compared to the untreated control prostate tissue (from control animals) (Fig. 5d, S2). The results from the immunohistochemical analysis of prostate tumor apoptosis, vascularity and cell proliferation (from untreated and DZ-50 treated tumor-bearing hosts) are summarized on Table 1; these data indicate that the DZ-50-mediated reduction in prostate tumor growth is, at least in part, consequential to targeting and reduction of angiogenesis.
Table 1. Effect of DZ-50 treatment on apoptosis, cell proliferation and vascularity of human prostate cancer xenografts in vivo.
The incidence of apoptosis, endothelial cell/vascularity, and cell proliferation was detected in situ in treated and untreated tumor-bearing hosts. Paraffin-embedded sections (6μm) from human prostate tumor (PC-3 and DU-145) xeongrafts (Fig. 4; panels a and b) were subjected to immunohistochemical analysis as follows: TUNEL staining for apoptosis detection and antibodies against, CD31, and Ki67 antigen for the evaluation of endothelial cell presence and the number of actively proliferating cells respectively. Quantitative analysis of the positive cells was conducted as described in “Methods”. A significant decrease in the number of CD31 positive cells was detected in both PC-3 and DU-145 derived prostate tumors following treatment with DZ-50. Neither the apoptotic nor the proliferative index of the prostate tumor cell populations were affected by DZ-50 treatment. * P ≤ 0.01
| PC-3 | DU-145 | |||||
|---|---|---|---|---|---|---|
| control | 100mg/kg | 200mg/kg | control | 100mg/kg | 200mg/kg | |
| TUNEL (Apoptotic Index) | 1.4 ± 0.3 | 2.0 ± 0.8 | 1.4 ± 0.4 | 3.2 ± 0.8 | 3.5 ± 1.0 | 3.4 ± 1.2 |
| CD31 (+) cells (vascularity) | 14.1 ± 0.8 | 13.5 ± 1.9 | 6.5 ± 0.6* | 18.5 ± 0.9 | 15.1 ± 0.7 | 10.1 ± 0.4* |
| Ki67 (Proliferation Index) | 43.7% | 42.6% | 45.0% | 51.2% | 53.9% | 49.7% |
The ability of DZ-50 to directly affect tumor cell metastasis, was evaluated using the in vivo spontaneous metastasis assay. Following 21 days of DZ-50 treatment, there was a significant reduction in the number of metastatic foci to the lungs compared to the untreated control mice (Fig. 6). These results indicate the ability of DZ-50 to prevent and reduce prostate tumor growth, as well as inhibit invasion and metastatic potential in vivo.
Figure 6. Inhibition of metastasis of human prostate cancer cells by DZ-50.
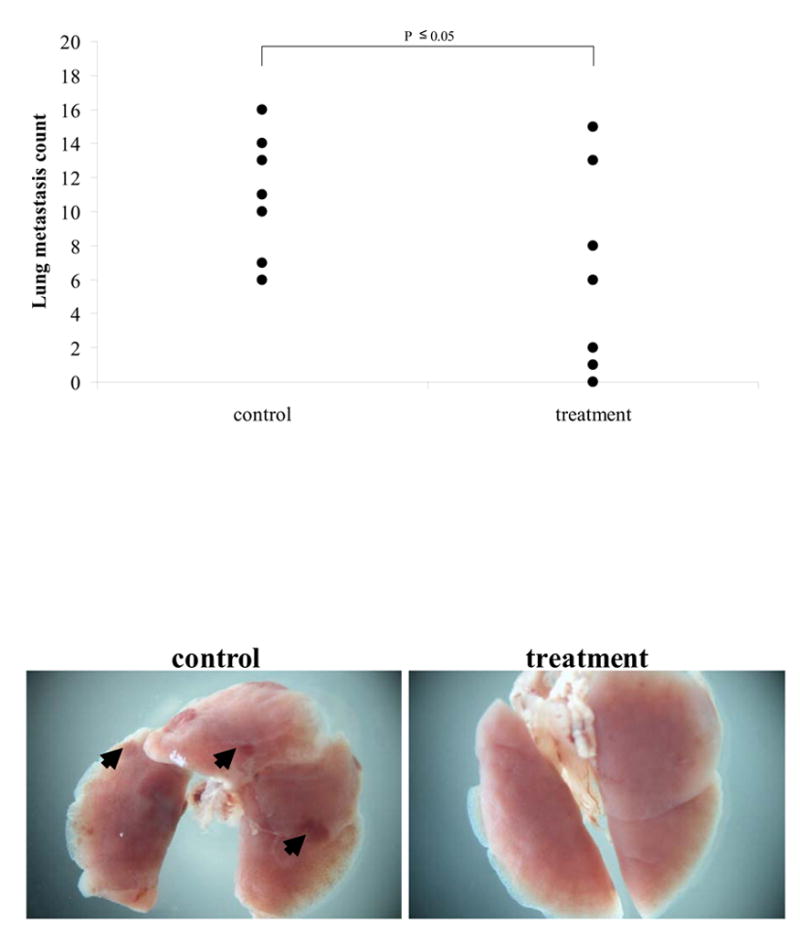
In the experimental metastasis assay, nude mice (n = 7 per group) were injected with prostate cancer cells PC-3 (2x106) through the tail vein DZ-50 treatment (200mg/kg) was initiated at 10 days post-inoculation for 21 days. Evaluation of the lungs (under dissecting microscope) revealed a significant reduction in the number of metastatic lesions to the lungs in the DZ-50 treated group compared to vehicle control mice; P < 0.05. Arrows indicate metastatic foci on the lungs.
DISCUSSION
This study demonstrates that our lead drug, a novel quinazoline-based compound, DZ-50, effectively targets human prostate tumor epithelial cells as well as vascular endothelial cells, without inducing “classic” apoptosis. This unique feature of the anti-tumor action of the new drug, inducing a pattern of cell death that is independent of caspase-activation characteristic of apoptotic signaling, is mechanistically intriguing. The invasion process requires a range of cell-to-cell interactions, primarily through the association of adhesion complexes between tumor cells and the adjacent endothelial cells. The present findings indicate that DZ-50 triggers the anoikis phenomenon, since it can prevent prostate tumor cell migration and attachment to ECM components fibronectin and type I collagen, (most abundant protein in bone). Examination of the ability of tumor cells to extravate by an in vitro model of transendothelial migration revealed that prostate tumor cells upon treatment with DZ-50, lost their ability to attach to the monolayer of endothelial cells; our results indicate that attachment of tumor epithelial cells to an endothelial cell monolayer was significantly inhibited after 6hrs of exposure to DZ-50 and was completely abrogated after 9hrs of treatment at non-cytotoxic doses. These in vitro data point to the ability of the lead compound to effectively minimize the possibility of transendothelial invasion and metastatic behavior of prostate cancer cells.
Collagen I binds the integrin pairs α1β1, α2β1, and α3β1 (19), and although we were unable to detect α1 expression in PC-3 and DU-145 prostate cells, there was strong expression of integrins α2β1 and α3β1. Following exposure to DZ-50, the PC-3 prostate cancer cells (originally isolated from a prostate tumor bone metastasis) exhibited complete loss of integrin β1 surface expression, while the DU-145 prostate cancer cells (derived from a brain metastasis had a minimal loss). Recent evidence suggests that in human prostate cancer cells, characterized by a specific ability for bone metastasis, migrate toward collagen type I in an α2β1-dependent manner and this selection increases in vivo growth within the bone (20). This cell type specific loss of integrin β1 could explain the differences observed in the efficacy of DZ-50 on tumor growth.
Changes in cell-cell and cell-matrix interactions are accompanied by changes in the expression of adhesion receptors like integrins. Down regulation of integrin β1 could provide the molecular basis for the response of prostate cancer cells to DZ-50. The regulation of β1 integrin expression has been shown to be altered by TGF-β1 signaling (21), at the transcriptional level by its attachment to the ECM and post-transcriptional/translational level (22, 23) and during differentiation (24) and cancer progression (25). Moreover, integrin α2β1 has been shown to mediate PC-3 cell adhesion to collagen and fibronectin, both major components of bone microenvironment (19). Thus there is promise in therapeutic modalities targeting the β1 integrin subunit, ultimately leading to changes in adhesion and localization. Indeed, growing evidence suggests that ionizing radiation leads to a significant reduction in β1 integrin levels resulting in decreased cellular adhesion to fibronectin (26). Emodin abrogates human cancer cell adhesion, by inhibiting the lipid raft clustering and subsequent co-localization of integrin β1 (27). The possibility of a novel compound effectively suppressing integrin β1 expression is mechanistically intriguing and warrants dissection.
The present findings indicate that in vivo administration of the novel lead drug DZ-50 (at well-tolerated doses) not only significantly inhibits the growth of established human xenograft prostate tumors, but also prevents the initiation of prostate cancer development in this model. Moreover, exposure to DZ-50 resulted in a considerable suppression of the metastatic capacity of human prostate cancer cells, potentially by targeting the invasion and migration potential of these cells. Initial mechanistic dissection pointed to integrins as primary candidates of drug-targeting. Previous studies have established that integrin β1 knockout mice fail to develop a vasculature (28), so a direct functional link between a reduced tumor growth and a lack of integrin β1 is an attractive possibility. Furthermore, VEGF directly activates integrins α5β1 and α2β1, both implicated in angiogenesis (29). One could easily argue that loss of integrin β1 expression by DZ-50 (as detected in the present study), could interfere with VEGF signaling leading to reduced tumor vascularity, without affecting tumor cell death.
VEGF has been specifically targeted by strategies such as monoclonal antibodies (bevacizumab) and inhibitors of endothelial cell receptor-associated tyrosine kinase activity (9). Other approaches including targeting basement membrane degradation, endothelial cell migration and endothelial cell proliferation have also been clinically evaluated, but success has been variable (30, 31). Increases in patient survival in response to any antiangiogenic therapy have yet to be reported and current antiangiogenic therapy has been clinically ineffective at eliminating or preventing tumor growth. Phase III clinical trial data is lacking for any novel antiangiogenic compound; thus the immediate need for new targeted therapies for metastatic prostate cancer. Ongoing studies focus on dissecting the ability of the lead DZ compounds to target the interactions between integrin β1 with its intracellular signaling partners. Decreased surface expression of integrin β1 might result from down-regulation at the transcriptional or translational level. Alternatively, integrin β1deregulation in response to DZ 50 might be an indirect effect from alterations in the focal adhesion complex [talin, focal adhesion kinase (FAK)], and other key components of the actin microfilaments that determine cell motility and migration. From a therapeutic standpoint either mechanism could prove beneficial, as by reducing the migratory capacity of tumor epithelial cells and/or inducing anoikis of endothelial cells, we could effectively prevent their ability to metastasize.
The observed effect of DZ-50 in preventing prostate tumor development in the xenograft model provides the first evidence that these drugs may also have a prophylactic value. Support for such a concept stems from the recent epidemiological cohort study from our center, indicating that exposure to doxazosin significantly decreases the incidence of prostate cancer among men (32), thus suggesting a chemopreventive role for the quinazoline-based compounds. Finally, a combination of DZ-50 (targeting vascularity) with an apoptosis-inducing regimen for the treatment of metastatic prostate cancer emerges as an attractive therapeutic possibility begging pursuit.
METHODS
Cells Lines and Reagents
The androgen-independent human prostate cancer PC-3 and DU-145 cell lines were obtained from the American Type Tissue Culture Collection (Rockville, MD) and cultured in RPMI-1640 purchased from Invitrogen (Carlsbad, CA) containing 10% fetal bovine serum (Invitrogen) and antibiotics. The human benign prostatic epithelial cell line, BPH-1, [a gift from Dr. Simon W. Hayward (Department of Urological Surgery, Vanderbilt University Medical Center, Nashville TN)] and were cultured in RPMI-1640 (Invitrogen) containing 10% fetal bovine serum and antibiotics. Human vascular endothelial cells (HUVEC) and human lung microvascular endothelial cells (HMVEC-L) were cultured in endothelial basal medium (EGM-2) (Cambrex, East Rutherford, New Jersey) supplemented with endothelial cell growth supplements (EGM-2 and EGM2-MV) (Cambrex). Recombinant human VEGF was purchased from Landing Biotech, (Newton, MA). Doxazosin derivatives (1–23, 38, 40, 42, and 50) were synthesized as described previously (18).
Apoptosis and Cell Viability Evaluation
a) Hoechst Staining
Cells were plated in 6-well culture dishes at 5x104 cells per/well and at subconfluency were treated with increasing concentrations of DZ-1-23, −38, −40, −42, and −50 (0–25μM). After 24 and 48hrs of treatment, cells were fixed with 4% (w/v) paraformaldehyde (Sigma) and stained with 10μg/mL Hoechst 33342 (B2261; Sigma) in the presence of 0.1% Triton X-100 (Sigma) as previously described. Cells were visualized using a Zeiss Axiovert S100 fluorescent microscope (Thornwood, NY) with a UV filter (365nm) and cells with condensed chromatin were designated apoptotic (100x magnification). The apoptotic index was determined by counting three random fields in duplicate wells per group. Each experiment was performed twice.
b) MTT assay
Subconfluent cultures of cells were exposed to increasing concentrations of DZ-1-23, −38, −40, −42, and −50 (0–25μM). After treatment the medium was replaced with 250μl of MTT (Sigma) (1mg/ml) and incubated at 37ºC to form blue crystals. After 2hrs the MTT was removed and replaced with DMSO (250μl) and incubated overnight at 37ºC. The DMSO-crystal solution’s absorbance was read at 540nm in a microplate reader (Bio-Tek Instruments, Winooski, VM). Numerical data represent the average of three independent experiments performed in triplicate.
Cell Migration Assay
(Wounding assay) Confluent monolayers of PC-3, DU-145, HUVEC, or HMVEC-L cells were wounded with a toothpick. After wounding, medium was changed and DZ-3 or DZ-50 (5μM). After incubation for 12 or 24hrs, wounding areas were examined under light microscopy (Axiovert 10, Zeiss). Cells that had migrated to the wounded areas were counted under a microscope for quantification of cell migration. Migration was calculated as the average number of cells observed in five random high power (400x) wounded fields/per well in duplicate wells.
Tube Formation Assay: In vitro Angiogenesis Evaluation
In vitro formation of tubular structures was studied on extracellular matrix using an angiogenesis kit as described by the manufacturer (Chemicon International, Inc., Temecula, CA). HUVEC or HMVEC-L (10x104 cells/well) of 96-well-plates were seeded onto ECMatrigel-coated wells in the presence or absence of DZ-3 or DZ-50 and VEGF. Cells were treated with cytokines as single agents or each in combination (e.g. DZ-50 and VEGF). After 24hrs post-treatment angiogenesis was assessed on the basis of formation of capillary-like structures of HUVEC, according to the manufacturer's protocol. The number of capillary-like tubes was counted (Nikon Eclipse, TE2000-U) in each well and the average was evaluated.
Chicken Chorioallantoic Membrane (CAM) Assay
Fertilized chicken eggs were incubated at 37°C. At E8 a window was created to allow visualization of the egg shell membrane. 6mm blank paper discs (BD) were placed on the egg shell membrane along with VEGF (100ng) or bFGF (100ng) and DZ-50. The windows were sealed with porous adhesive and allowed to incubate 48hrs. At E10 the adhesive was removed along with the egg shell membrane to expose the CAM and 4% paraformaldehyde was added. Following excision the number of vessels per CAM was quantified by counting under a dissecting microscope.
Cell Attachment Assay
Prostate cancer cells PC-3 and DU-145 cells were treated for 3, 6, 9, 12, or 15hrs with DZ-50 (5μM) and harvested. 5x104 cells were added to each well of a 6-well culture dish coated with either collagen or fibronectin and incubated for 30min at 37°C. Following incubation cells were fixed with methanol and the number of cells per well was recorded. Numerical data represent the average of three independent experiments performed in triplicate.
Transendothelial Migration (TEM) Assay
Sterile (12mm diameter) glass coverslips were coated with Matrigel (Becton Dickson, Franklin Lakes, NJ) at a dilution of 1:8 and air dried at room temperature (1hr). Coverslips received approximately 6.25x104 HMVEC-L to form a complete monolayer. The cells were allowed to deposit and spread on the Matrigel for 24hrs prior to the experiment. PC-3 cells were resuspended in EGM-2MV (Cambrex) and added to the HMVEC-L monolayer at a concentration of 8x103 cells/coverslip. Co-cultures were incubated at 37°C at 5% CO2 for 3, 6, 9, 12, and 24hrs. Prior to the addition of prostate epithelial cells, cells were incubated with the lipophilic tracer 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) (Invitrogen, Carlsbad, CA) for 20min at 10μg/ml to stain cell membranes. To label F-actin, PC-3 cells, or co-cultures of PC-3 cells HMVEC-L were fixed for 10min at room temperature in 2% paraformaldehyde in PBS, and were permeabilized for 5min with a buffer containing 15mM Tris, 120mM NaCl, 2mM EDTA, 2mM EGTA, and 0.5% Triton X-100 (pH 7.4). Cells were incubated for 1hr at room temperature with AlexaTM 488-conjugated phalloidin at a dilution of 1:50 in blocking solution, followed by 5min of incubation with 10mM Hoechst 33342 (Sigma) in PBS. Coverslips were mounted with Vectashield (Vector Laboratories, Burlington, Canada) on glass slides and analyzed with confocal microscopy.
Western Blot Analysis
Cultures of PC-3, DU-145, BPH-1, HUVEC, and HMVEC-L cells were treated with DZ-50 (10μM) for various time periods and cell lysates were subsequently generated in RIPA buffer [150mM NaCl, 50mM Tris pH 8.0, 0.5% deoxycholic acid, 1% Nonidet P40 with 1mM phenyl methyl-sulfonyl fluoride (PMSF)]. The total protein concentration in the lysates was quantified by BCA Protein Assay Kit (Pierce, Rockford, IL) and protein samples (30μg) were subjected to sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis, and transferred to Hybond-C membranes (Amersham Pharmacia Biotech., Piscataway, NJ). After blocking with 5% dry milk in TBS-T (Tris-buffered-saline containing 0.05% Tween-20) for 1hr (room temperature), membranes were incubated overnight at 4°C with antibodies against caspase-8, Akt, or phosphorylated Akt (Cell Signaling Technology, Danvers, MA). Following incubation with the respective primary antibody, membranes were exposed to species-specific horseradish peroxidase (HRP)-labeled secondary antibodies. Signal detection was achieved with SuperSignal® West Dura Extended Duration Substrate (Pierce) and visualized using a UVP Bioimaging System (Upland, CA). All bands were normalized to α-actin expression (Oncogene Research Products™, La Jolla, CA).
FACS - Flow Cytometric Analysis
PC-3 cells were treated with DZ-50 (10μM) and harvested with 0.5mM EDTA solution. Prostate cancer epithelial cells were then incubated with hanks balanced salt solution (HBSS) supplemented with 2% BSA and 0.01% sodium azide for 30min at 4°C. Cells were subsequently fixed in 4% (w/v) formaldehyde, washed, and incubated with the designated integrin antibody followed by FITC-conjugated goat anti-mouse secondary. Analysis was performed on a Partec FlowMax (Partec, Munster, Germany).
Tumorigenicity Studies
Human prostate cells (PC-3 and DU-145) suspended in PBS, were inoculated subcutaneously (s.c.) (2.5x106 cells/site) in the flank of male nude mice, 4–6 weeks of age. Tumors were measured every other day using a digital caliber, and tumor volumes were calculated using the formula length x (width)2/2. When tumors reached ≈50mm3 mice were stratified into treatment groups of 6mice/treatment. DZ-50 was administered in the following doses: 0, 50, 100, and 200mg/kg in 0.5% methylcellulose (w/v) + 0.1% Tween-80 (v/v) in sterile water, by oral gavage using a 22-gauge, 1.5-inch gavage needle. Animals were sacrificed after 2wks of treatment unless otherwise indicated. In a separate experiment human prostate cells (PC-3) were inoculated as described above and dosing began (200mg/kg) concurrently for 2wks. Upon termination of the experiment tumors were surgically excised, tissue specimens were fixed in a 10% (v/v) formalin solution (Sigma) and subsequently embedded in Paraplast X-tra paraffin (VWR). Tissue blocks were sectioned at 6μm on a Finesse Microtome (Thermo Shandon, UK) and subsequently subjected to immunohistochemical analysis.
Spontaneous Metastasis Assay
Human prostate cells (PC-3) were injected (2x106 cells in 80μl of PBS) in the tail vein of male nude mice, 4–6wks of age, and mice were maintained in a pathogen-free environment. At 10 days post-inoculation 200 mg/kg of DZ-50 was given daily (via oral gavage as described above) for a period 2wks. Upon termination of treatment, DZ-50 treated and vehicle control mice were sacrificed and lungs, spleen, kidneys, and prostate organs were excised and subjected to examination for metastatic tumor lesions.
Apoptosis Evaluation
Apoptotic cells were detected using the ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA). Briefly, paraffin-embedded sections were treated with Proteinase K (Dako, Carpinteria, CA) and were subsequently incubated with terminal deoxynucleotidyl transferase enzyme at 37°C. Apoptotic cells were counted in five different fields (400x) and the apoptotic index was determined using the number of terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL)-positive cells over the total number of cells.
Vascularity Evaluation
CD31 staining was performed for endothelial cells using enzymatic digestion with Proteinase K (Dako). The primary antibody used was the mouse anti-human CD31 specific for endothelial cells from Dako (overnight incubation at 4°C). CD31-positive endothelial cells were counted in five different fields (400x).
Cell Proliferation
Cell proliferation index was evaluated on the basis of Ki67 nuclear antigen immunoreactivity. Following antigen retrieval slides were incubated with an antibody directed against the Ki67 nuclear antigen (AMAC, Westbrook, MA). Ki67+ cells were counted from five different fields (400x).
Statistical Analysis
One-way analysis of variance (ANOVA) was performed using the StatView statistical program to determine the statistical significance between values. A P value of less than 0.05 was considered statistically significant.
Supplementary Material
Supplementary Figure 1
Image of wounding assay performed on human vascular endothelial cells (HUVEC) following treatment with DZ-50.
Supplementary Figure 2
PC-3 cell analysis for surface expression of integrins α2, α3, αV, and β3.
Supplementary Figure 3
Immunohistochemical analysis of PC-3 human prostate cancer xenografts for evaluation of apoptosis (TUNEL), vascularity (CD31 immunoreactivity), and cell proliferation (Ki67).
Acknowledgments
This work was supported by NIH grants CA107575-01 (N.K.) and CA112250 (C-S. C.) and a Department of Defense Grant (W81XWH-05-1-0089). The authors would like to thank James V. Partin and Samuel Kulp for expert assistance and Dr. Steven Schwarze for critically reading the manuscript.
Abbreviations
- TGF-β1
transforming growth factor-beta 1
- BPH-1
benign prostatic hyperplasia
- HUVEC
human umbilical vein endothelial cell
- HMVEC-L
human microvascular endothelial cell-lung
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- VEGF
vascular endothelial growth factor
- CAM
chorioallantoic membrane
- TUNEL
terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling
References
- 1.Jemal A, et al. Cancer statistics. Cancer J Clin. 2006;56:106–130. doi: 10.3322/canjclin.56.2.106. [DOI] [PubMed] [Google Scholar]
- 2.Hill B, Kyprianou N. Sequencing hormonal ablation and radiotherapy in prostate cancer: A molecular and therapeutic perspective. Oncology Reports. 2002;9:1151–1156. [PubMed] [Google Scholar]
- 3.Garrison JB, Kyprianou N. Novel Targeting of Apoptosis Pathways for Prostate Cancer Therapy: A Review. Current Cancer Drug Targets. 2004;4:85–95. doi: 10.2174/1568009043481623. [DOI] [PubMed] [Google Scholar]
- 4.Weidner N. Intratumoral vascularity as a prognostic factor in cancer of the urogenital tract. Eur J Cancer. 1996;32A:2506–2511. doi: 10.1016/s0959-8049(96)00378-4. [DOI] [PubMed] [Google Scholar]
- 5.Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. Cell Biol. 1994;124:619–626. doi: 10.1083/jcb.124.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rennebeck G, Martelli M, Kyprianou N. Anokis and survival connections in the tumor microenvironment: is there a role in prostate cancer metastasis? Cancer Res. 2005;65:11230–11235. doi: 10.1158/0008-5472.CAN-05-2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 8.Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 9.Ferrara N, Kerbel RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. doi: 10.1038/nature04483. [DOI] [PubMed] [Google Scholar]
- 10.Dameron KM, et al. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 11.Horsman MR, Siemann DW. Pathophysiologic effects of vascular-targeting agents and the implications for combination with conventional therapies. Cancer Res. 2006;66:11520–11539. doi: 10.1158/0008-5472.CAN-06-2848. [DOI] [PubMed] [Google Scholar]
- 12.Kirby RS, Pool JL. Alpha adrenoceptor blockade in the treatment of benign prostatic hyperplasia: past, present and future. Br J Urol. 1997;80:521–532. doi: 10.1046/j.1464-410x.1997.00247.x. [DOI] [PubMed] [Google Scholar]
- 13.Kyprianou N. Doxazosin and terazosin suppress prostate growth by inducing apoptosis: clinical significance. J Urol. 2003;169:1520–1525. doi: 10.1097/01.ju.0000033280.29453.72. [DOI] [PubMed] [Google Scholar]
- 14.Chon JK, et al. Alpha 1-adrenoceptor antagonists terazosin and doxazosin induce prostate apoptosis without affecting cell proliferation in patients with benign prostatic hyperplasia. J Urol. 1999;161:2002–2008. [PubMed] [Google Scholar]
- 15.Partin JV, Anglin IE, Kyprianou N. Quinazoline-based alpha1-adenoreceptor antagonists induce prostate cancer cell apoptosis via transforming growth factor-beta signaling and IkappaB alpha induction. Br J Urol. 2003;88:1615–1621. doi: 10.1038/sj.bjc.6600961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benning CM, Kyprianou N. Quinazoline-derived alpha1 adrenoceptor antagonists induce prostate cancer cell apoptosis via an1-adrenoceptor independent action. Cancer Res. 2002;62:597–602. [PubMed] [Google Scholar]
- 17.Garrison JB, Kyprianou N. Doxazosin induces apoptosis of benign and malignant prostate cancer cells via a death-receptor mediated pathway. Cancer Res. 2006;66:464–472. doi: 10.1158/0008-5472.CAN-05-2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shaw YJ, et al. Pharmacological Exploitation of the α1-Adrenoreceptor Antagonist Doxazosin to Develop a Novel Class of Antitumor Agents That Block Intracellular Akt Activation. J Med Chem. 2004;47:4453–4462. doi: 10.1021/jm049752k. [DOI] [PubMed] [Google Scholar]
- 19.Gullberg D, et al. Analysis of alpha 1 beta 1, alpha 2 beta 1 and alpha 3 beta 1 integrins in cell--collagen interactions: identification of conformation dependent alpha 1 beta 1 binding sites in collagen type I. EMBO J. 1992;11:3865–3873. doi: 10.1002/j.1460-2075.1992.tb05479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall CL, et al. Type I Collagen Receptor ({alpha}2{beta}1) Signaling Promotes the Growth of Human Prostate Cancer Cells within the Bone. Cancer Res. 2006;66:8648–8654. doi: 10.1158/0008-5472.CAN-06-1544. [DOI] [PubMed] [Google Scholar]
- 21.Cervella P, et al. Human beta 1-integrin gene expression is regulated by two promoter regions. J Biol Chem. 1993;268:5148–5155. [PubMed] [Google Scholar]
- 22.Delcommenne M, Streuli CH. Control of integrin expression by extracellular matrix. J Biol Chem. 1995;270:26794–26801. doi: 10.1074/jbc.270.45.26794. [DOI] [PubMed] [Google Scholar]
- 23.Meleady P, Clynes M. Bromodeoxyuridine induces integrin expression at transcriptional (alpha2 subunit) and post-transcriptional (beta1 subunit) levels, and alters the adhesive properties of two human lung tumour cell lines. Cell Commun Adh. 2001;8:45–59. doi: 10.3109/15419060109080706. [DOI] [PubMed] [Google Scholar]
- 24.Hotchin NA, Watt FM. Transcriptional and post-translational regulation of beta 1 integrin expression during keratinocyte terminal differentiation. J Biol Chem. 1992;267:14852–14858. [PubMed] [Google Scholar]
- 25.Paulin Y, et al. Activity of proximal promoter of the human beta(1)-integrin gene was increased in Sezary syndrome. Leuk Res. 2001;25:487–492. doi: 10.1016/s0145-2126(00)00149-1. [DOI] [PubMed] [Google Scholar]
- 26.Simon EL, et al. High dose fractionated ionizing radiation inhibits prostate cancer cell adhesion and beta(1) integrin expression. Prostate. 2005;64:83–91. doi: 10.1002/pros.20227. [DOI] [PubMed] [Google Scholar]
- 27.Huang Q, et al. Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006;66:5807–5815. doi: 10.1158/0008-5472.CAN-06-0077. [DOI] [PubMed] [Google Scholar]
- 28.Fassler &, Meyer M. Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 1995;9:1896–1908. doi: 10.1101/gad.9.15.1896. [DOI] [PubMed] [Google Scholar]
- 29.Byzova TV, et al. Mechanism for Modulation of Cellular Responses to VEGF: Activation of the Integrins. Mol Cell. 2000;6:851–860. [PubMed] [Google Scholar]
- 30.Kerbel R, Folkman J. Clinical translation of angiogenesis inhibitors. Nat Rev Cancer. 2002;2:727–739. doi: 10.1038/nrc905. [DOI] [PubMed] [Google Scholar]
- 31.Eskens FA. Angiogenesis inhibitors in clinical development; where are we now and where are we going? Br J Cancer. 2004;90:1–7. doi: 10.1038/sj.bjc.6601401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harris AM, et al. Effect of α1-adrenoreceptor antagonist exposure on prostate caner incidence: an observational cohort study. 2007. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Image of wounding assay performed on human vascular endothelial cells (HUVEC) following treatment with DZ-50.
Supplementary Figure 2
PC-3 cell analysis for surface expression of integrins α2, α3, αV, and β3.
Supplementary Figure 3
Immunohistochemical analysis of PC-3 human prostate cancer xenografts for evaluation of apoptosis (TUNEL), vascularity (CD31 immunoreactivity), and cell proliferation (Ki67).