

Abstract
Background & aims
Fractalkine (FKN/CX3CL1) is a unique chemokine combining adhesive and chemotactic properties. We investigated FKN production by the mucosal microvasculature in inflammatory bowel disease (IBD), its capacity for leukocyte recruitment into the gut, and the number of CX3CR1+ cells in the circulation and mucosa of IBD patients.
Methods
Expression of FKN by human intestinal microvascular endothelial cells (HIMEC) and CX3CR1 by circulating cells was evaluated by flow cytometry, and mucosal CX3CR1+ cells were enumerated by immunohistochemistry. The capacity of FKN to mediate leukocyte binding to HIMEC was assessed by immunoblockade, and to induce HIMEC transmigration by a Transwell system.
Results
The spontaneously low HIMEC FKN expression was markedly enhanced by TNF-α plus IFN-γ stimulation, or direct leukocyte contact. This effect was significantly stronger in IBD than control HIMEC. Upregulation of HIMEC FKN expression was dependent on p38 and ERK phosphorylation, as was abrogated by selective MAP kinase inhibitors. Circulating T-cells contained significantly higher numbers of CX3CR1+ cells in active IBD than inactive IBD or healthy subjects, and IBD mucosa contained significantly more CX3CR1+ cells than control mucosa. Antibody blocking experiments showed that FKN was a major contributor to T- and monocytic-cell adhesion to HIMEC. Finally, FKN enhanced expression of active β1 integrin on leukocytes and mediated leukocyte HIMEC transmigration.
Conclusions
In view of FKN capacity to mediate leukocyte adhesion, chemoattraction and transmigration, its increased production by mucosal microvascular cells and increased numbers of circulating and mucosal CX3CR1+ cells in IBD point to a significant role of FKN in disease pathogenesis.
Keywords: FKN/CX3CL1, CX3CR1, VCAM-1, HIMEC, inflammatory bowel disease, ulcerative colitis, Crohn’s disease
INTRODUCTION
Migration of leukocytes from the vascular compartment into sites of inflammation is a dynamic process mediated by a complex series of interactions between leukocytes and the endothelium. Margination and rolling of leukocytes along the endothelial surface are followed by leukocyte activation, firm adhesion and, finally, transmigration into the interstitium 1. These cell-cell interactions are finely tuned by several cell adhesion molecules (CAMs) expressed on the surface of both endothelial cells and leukocytes 2, 3. Leukocyte migration also depends on the existence of a chemoattractant gradient across the blood vessel wall created by a large family of molecules known as chemokines 4. Under physiological conditions these substances selectively target various types of leukocytes, a process that ensures an appropriate distribution of discrete leukocyte subsets in all tissues and organs 5. Under inflammatory conditions, including IBD, altered levels and types of chemokines lead to improper leukocyte distribution and accumulation accounting for the presence of inflammatory infiltrates in the target tissues 6.
Unlike most chemokines, that are small and exclusively secreted, fractalkine (FKN/CX3CL1) is a large, transmembrane molecule, consisting of a chemokine domain bound to the cell surface by a long mucin-like stalk 7. Transfection studies have revealed that the key function of the mucin portion of FKN is to mediate cell adhesion 8, 9. This portion extends the chemokine domain away from the endothelial cell surface and enables its presentation to leukocytes, thus directly supporting their adhesion to the endothelium 8, 9. In addition to this membrane-bound form, a soluble form of FKN is secreted through cleavage at a membrane-proximal site 10, 11, and this soluble form exhibits efficient chemotactic activity for multiple cell types 12. Therefore, due to its unique properties, FKN combines a dual function as an adhesion and chemotactic molecule 13. FKN is produced by a large number of cell types, including neurons 14, glomeruli 15, and epithelial cells 16, but is particularly prominent in endothelial cells 17, 18.
In addition to its distinctive structure and function, FKN also differs from other chemokines because has a single receptor, CX3CR1, which is expressed primarily on the surface of monocytes, NK cells and CD8+ T-cells, and mediates both adhesive and chemoattractive functions 19, 20. Like other chemokine receptors, CX3CR1 signals through pertussis toxin-sensitive G proteins to induce migration but not adhesion 21.
Several reports indicate that FKN is involved in the pathogenesis of numerous inflammatory conditions, including atherosclerosis 22, glomerulonephritis 23, cardiac allograft rejection 24, psoriasis 18, 25, rheumatoid arthritis 26, systemic sclerosis 27, and primary biliary cirrhosis 28. FKN may also play a role in inflammatory bowel disease (IBD). Immunohistochemistry reveals increased FKN expression in Crohn’s disease (CD) mucosa 16, and studies with transformed epithelial cells lines show that CX3CR1 mediates epithelial cell activation and neutrophil migration 29. In addition, a recent study demonstrates that lamina propria dendritic cells requires CX3CR1 to form transepithelial dendrites necessary to sample luminal antigens 30, an event potentially relevant to intestinal inflammation. These studies have investigated the pathophysiological role of FKN primarily in regard to epithelial cell function, but whether endothelial cell-derived FKN also contributes to gut inflammation has yet to be explored. Thus, the aim of this study was to investigate whether FKN contributes to IBD pathogenesis by mediating leukocyte adhesion to or transmigration through mucosal microvascular cells, or both.
MATERIALS AND METHODS
Reagents, antibodies, and cell lines
The following antibodies (Ab) were purchased from R&D Systems (Minneapolis, MN): blocking anti-FKN, biotinylated anti-FKN, anti-intercellular adhesion molecule 1 (ICAM-1) (CD54a), and anti-vascular cell adhesion molecule 1 (VCAM-1) (CD106). Antibodies against CX3CR1 were purchased from Torrey Pines Biolabs (Houston, TX) and from Abcam (Cambridge, MA). Phycoerythrin (PE)-conjugated anti-goat Ab was purchased from Caltag (Burlingame, CA) and FITC-conjugated anti-rabbit Ab from BD Biosciences (San Jose, CA). Goat, sheep, and rabbit immunoglobulin (Ig) isotype controls were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Recombinant human FKN was purchased from R&D Systems. PE-conjugated anti-CD3, PerCp-conjugated anti-CD4, and PE-conjugated anti-CD8 were purchased from Dako (Carpinteria, CA), and PE-conjugated anti-CD14 was purchased from BD Biosciences.
CD40L-positive D1.1 T-cells 31 and CD40L-negative Jurkat T-cells were obtained from the American Type Culture Collection (ATCC, Rockville, MD), cultured in RPMI 1640 with 10% fetal bovine serum (FBS) and fed twice weekly. The monocytic cell line THP1 was also obtained from ATCC and cultured in medium containing antibiotics, 1% L-glutamine, 1% non-essential amino acids, 1% sodium piruvate and 5×10−5 M mercaptoethanol.
Isolation and culture of human intestinal microvascular endothelial cells (HIMEC) and peripheral blood mononuclear cells (PBMC)
Isolation of HIMEC was performed as previously reported 32. Briefly, HIMEC were obtained from surgical specimens of patients with CD, UC and, as control, from normal areas of the intestine of patients admitted for bowel resection because of colon cancer, polyps, or diverticulosis. HIMEC were isolated by enzymatic digestion of intestinal mucosal strips followed by gentle compression to extrude endothelial cell clumps which adhered to fibronectin-coated plates, and were subsequently cultured in MCDB131 medium (Sigma, St. Louis, MO) supplemented with 20% FBS, antibiotics, heparin, and endothelial cell growth factor. Cultures of HIMEC were maintained at 37°C in 5% CO2, fed twice a week and split at confluence. HIMEC were used between passages 3 and 12.
Blood samples were collected from patients with active and inactive UC and CD, and healthy subjects. All diagnoses were confirmed by clinical, radiologic, endoscopic, and histologic criteria, and clinical disease activity was assessed by the Harvey-Bradshaw Activity Index 33 and the Colitis Activity Index 34. PBMC were isolated from heparinized venous blood, using a Ficoll-Hypaque density gradient, as previously reported 35. This project was approved by the Institutional Review Board of University Hospitals of Cleveland.
Stimulation of HIMEC with cytokines, leukocytes and CD40L
To study FKN expression on HIMEC surface, cells were plated on fibronectin-coated 24-well cluster plates at a density of 105 HIMEC/well. The effect of cytokines on FKN expression was assessed by stimulation of confluent HIMEC monolayers with 100 U/mL of IL-1β, 100 U/mL of TNF-α, 500 U/mL of IFN-γ, 20 U/mL IL-4, 20 U/mL IL-10, or a combination of the previous cytokines, for periods of time ranging from 4 to 72 hours. To evaluate whether the MAP kinases p38 and ERK-1 and -2 were involved in FKN upregulation, additional experiments were performed in which HIMEC monolayers were pretreated for 1 hour with the p38 specific inhibitor SB203580 (Calbiochem, San Diego, CA) or the ERK-1 and -2 specific inhibitor PD98059 (Calbiochem), both at 10 μg/mL, prior to combined TNF-α and IFN-γ stimulation, for 24 hours.
To assess the effect of leukocyte contact on HIMEC surface FKN expression, T-cells (Jurkat) or monocytic cells (THP1) were added to the HIMEC monolayers at a concentration of 1 × 106 cells/mL and incubated for 2 or 24 hours. In some experiments HIMEC had been preincubated with 500 U/mL of IFN-γ for 48 hours. In an additional set of experiments 0.2 μm porous membranes were placed between leukocytes and endothelial cells to ascertain whether leukocyte-induced FKN upregulation requires physical contact between both cell types or is mediated by soluble molecules released by leukocytes.
To study the contribution of the costimulatory pathway CD40/CD40L to FKN expression, the CD40L-positive D1.1 T-cells and CD40L-negative Jurkat T-cells were added to HIMEC monolayers at a concentration of 1 × 106 cells/mL. In addition, soluble CD40L was added to HIMEC in some wells at 1 μg/mL.
Flow cytometric characterization of HIMEC and PBMC
After exposure to cytokines, leukocytes or soluble CD40L, HIMEC monolayers were washed twice and suspended by a quick trypsin treatment. In some experiments, single HIMEC suspensions were obtained using a gentle detachment buffer (PBS, 20 mmol/L HEPES, pH 7.4, 10 mmol/L ethylenediaminetetraacetic acid, and 0.5% bovine serum albumin) instead of trypsin, as previously described 36. HIMEC were then incubated with anti-FKN antibody for 30 minutes in ice, washed, and incubated with a PE-conjugated anti-goat Ig Ab. After 30 minutes, cells were washed twice, fixed in 1% paraformaldehyde and FKN expression on HIMEC surface was analyzed by flow cytometry.
Expression of the FKN receptor CX3CR1 was studied in PBMC obtained from CD and UC patients and healthy subjects. PBMC were incubated with the anti-CX3CR1 Ab for 30 minutes in ice, washed, and incubated with a FITC-conjugated anti-rabbit Ig Ab at 4°C. After 30 minutes, cells were washed and stained with either PE-conjugated anti-CD3, PerCp-conjugated anti-CD4, PE-conjugated anti-CD8, or PE-conjugated anti-CD14. Cells were fixed in 1% paraformaldehyde, analyzed, and the percentage of positive cells for CX3CR1 in the CD3+, CD4+, CD8, and CD14+ subpopulations was calculated by concomitantly gating their forward and side double stain distribution for CX3CR1+CD3, CX3CR1+CD4, CX3CR1+CD8, and CX3CR1+CD14. The isotype staining was negligible for all cytometric analyses.
All samples were analyzed by quantitative flow cytometry using a Coulter Epics XL Flow Cytometer (Beckman Coulter Inc., Fullerton, CA). Each analysis was performed on at least 10,000 events. Quantification of FKN and CX3CR1 was performed using the Winlist software program (Verity Software House, Topsham, ME).
Quantification of CX3CR1-positive cells in the intestinal mucosa
The number of cells expressing CX3CR1 in control and IBD mucosa was assessed by routine immunohistochemistry using two different Ab under conditions pre-tested to yield optimal staining: the first was a rabbit polyclonal antibody from Torrey Pines Biolabs (# TP502) used on frozen sections at 1:500 dilution; the second was a rabbit polyclonal antibody from Abcam (# 8021) used on paraffin-fixed sections at 1:300 dilution without antigen retrieval. As negative control, the same tissue sections were processed omitting the primary antibody. Multiple sections from 14 control, 9 CD and 11 UC specimens were evaluated by a single investigator (C.F.) blinded to the final diagnosis. All positively stained and unstained lamina propria mononuclear cells were counted in 2–4 randomly selected high power fields, and CX3CR1+ cells calculated as percent of the total number of counted cells. The relative counts using the two Abs were essentially identical in control and IBD tissues, and were combined in the final analysis.
FKN, p38 and ERK immunoblotting
Cytokine-induced expression of FKN on HIMEC surface was also evaluated by Western blotting. Using the experimental conditions described above, HIMEC were stimulated with medium alone, 100 U/mL of TNF-α, 500 U/mL of IFN-γ, or a combination of TNF-α and IFN-γ for 24 hours, lysed and immunoblotting performed using anti-FKN antibody (R & D; # AF365). Recombinant human FKN (R & D) was used as a positive control. To study activation (phosphorylation) of the MAP kinases p38 and ERK-1 and -2 in HIMEC, confluent HIMEC monolayers were exposed to 100 U/mL of TNF-α, 500 U/mL of IFN-γ, alone and combined, or medium alone for 15 minutes. This time point was chosen taking into account the results of previous studies assessing optimal phosphorylation of p38 and ERK 36, 37. HIMEC monolayers were washed twice and lysed using a buffer containing 50 mmol/L HEPES, pH 7.5, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 10% glycerol, 1% Triton X-100, and 50 mmol/L protease- an 50 mmol/L phosphatase-inhibitor cocktail. Total protein was extracted and its concentration in each lysate measured using the Bio-Rad protein assay (Bio-Rad Laboratories, Hercules, CA). Equivalent amounts of proteins (20 μg) were fractioned on a 10% Tris-glycine gel and electrotransferred to a nitrocellulose membrane (Novex, San Diego, CA). Nonspecific binding was blocked by incubation with 5% milk in 0.1% Tween 20/Tris-buffered saline, incubated with the appropriate horseradish peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology) and incubated with the chemiluminescent substrate (Super Signal; Pierce, Rockford, IL) for 5 minutes before exposure to film (Amersham, Arlington Heights, IL).
Effect of FKN on active β1 integrin expression
Chemokines can induce changes in integrin conformation on leukocyte surface, leading to a higher integrin activation state and enhanced leukocyte adhesion. To ascertain whether FKN is able to modify the conformation of β1 integrin, we used a monoclonal antibody, 12G10 (Serotec, Oxford, UK), which specifically recognizes the activated form of β1 integrin 38, 39. PBMC from healthy donors, Jurkat T-cells, and THP1 monocytic cells were cultured with 1, 10, and 50 nM of human recombinant FKN, for different periods of time (1, 5, 10, 30 and 60 minutes). Cells were collected, washed, and expression of active β1 integrin on the cell surface was assessed by flow cytometry. To verify the participation of the G-protein coupled intracellular signaling cascade on β1 integrin conformational changes, PBMC, Jurkat, and THP1 were pretreated with the G-protein inhibitor pertussis toxin (Sigma, St Louis, MI), at 0.4 nM for 45 minutes prior to incubation with FKN. Similarly, in additional experiments cells were pretreated with PP2 (Calbiochem), an Ab directed against the Src kinase p56lck, prior to incubation with FKN to assess whether this kinase plays a role in the regulation of β1 integrin affinity 39.
Cell adhesion assay
Endothelial-leukocyte adhesion assays were performed as previously described 40, 41. Confluent HIMEC monolayers cultured in 24-well cluster plates were left alone or stimulated with a combination of 100 U/mL of TNF-α and 500 U/mL of IFN-γ for 24 hours. To assess the contribution of FKN, CX3CR1, VCAM-1, and ICAM-1 to leukocyte adhesion, 10 μg/mL of either anti-FKN Ab, anti-CX3CR1 Ab, anti-ICAM-1 Ab, anti-VCAM-1 Ab, or a combination of the previous Abs were added to the wells, at 37°C for 1 hour. Extra wells received 10 μg/mL of rabbit, goat or sheep IgG as control Abs. Jurkat or THP1 cells, kept under exponential growth conditions, were labeled with calcein (Molecular Probes, Eugene, OR), added to HIMEC monolayers at 1 × 106 leukocytes/well, and allowed to adhere to HIMEC at 37°C in a 5% CO2 incubator. After 1 hour of co-culture, wells were gently rinsed 4 times with calcium- and magnesium-containing phosphate buffered saline to remove all non adherent leukocytes. Fluorescent adherent leukocytes were quantified by an imaging system (Image Pro Plus; Media Cybernetics, Silver Spring, MD) connected to an Optronics Color digital camera (Olympus) on an inverted fluorescence microscope. Ten random fields were analyzed for each well and results were expressed as number of adherent cells/mm2.
Quantification of soluble FKN by ELISA
HIMEC supernatants were harvested by centrifugation at 800xg for 10 minutes at 4°C, aliquoted and stored at −70°C. Supernatants were thawed once and analyzed for soluble FKN content in duplicate using a non-commercially available ELISA. Briefly, plates (Costar, Corning, NY) were coated with anti-FKN Ab by adding to each well 100 μl of anti-FKN Ab, at a concentration of 2 μg/ml in PBS, overnight. Plates were washed and the non-specific binding was blocked with Super Block blocking buffer (Pierce, Rockford, IL) for 2 hours. After subsequent washing, two 100 μl aliquots of each sample were added to their corresponding wells and the plate was incubated for 2 hours. Plates were washed again and 100 μl of biotinylated anti-FKN Ab, at a concentration of 250 ng/ml, were added to each well for 2 hours. After additional washing, 100 μl of 1:100 streptavidin HRP (R&D Systems) dissolved in tris buffer containing 1% bovine serum albumin and 0.05% tween 20 was added to each well for 20 minutes. After this period of time, 100 μl of substrate (R&D Systems) were added and plates were incubated for other 20 minutes in the dark. All procedures were carried out at room temperature.
Chemotaxis and transmigration assays
A modification of the leukocyte transmigration system described by Roth et al. was employed 42. The method is based on a cluster transwell plate containing polycarbonate 5 μm porous filter inserts (Costar) separating the upper and lower chambers. A HIMEC monolayer was established on the upper chamber filter by seeding 25 × 103 HIMEC in MCDB131 containing 20% FBS. Monolayers were grown for 5–7 days until complete confluence was reached and verified by microscopic evaluation of the histochemically stained monolayer (Diff Quick Stain Set, Dade Diagnostics, Aguada, PR). On the day of the assay, the HIMEC monolayer was thoroughly rinsed with MCDB131 medium to remove all serum. THP1 cells were suspended at 2 × 106/mL in PBS with 5% FBS, and labeled at 37°C with 4 μM calcein (Molecular Probes). After 20 minutes monocytes were rinsed twice, resuspended in transmigration medium consisting of 50% RPMI 1640 and 50% MCDB131 medium with 0.5% BSA, and added to the upper chamber at 2,5 × 105/0.1 mL/insert. The insert was placed in a well of the 24-well cluster plate containing 0.5 mL of transmigration medium with or without recombinant FKN or MCP-1, the latter being used as a positive control for monocyte migration 43, 44. Plates were incubated at 37°C, in 5% CO2 for 4 hours, after which the inserts were removed, and the bottom surface of the filter was gently scraped to recover cells that had not completely migrated into the lower chamber. The cell suspension in the lower chamber was allowed to settle, and fluorescent cells were quantified using the same imaging system described for the adhesion assay. THP1 were chosen as migrating leukocytes to assess the maximal chemotactic effect of FKN, since essentially 100% of them express CX3CR1. Optimal doses of FKN and MCP1 were determined in preliminary chemotaxis experiments, using the previously described transwell plate, without a HIMEC monolayer between the upper and the lower chamber (data not shown).
Statistical analysis
Data were analyzed by Stat View software (SAS Institute Inc., Cary, NC) using analysis of variance followed by the appropriate post-hoc test or the Mann-Whitney test. Repeated measures for the same subject were analyzed by using Student’s paired t test. Values are expressed as mean ± SEM. Statistical significance was set at p< 0.05.
RESULTS
Enhanced cytokine-induced FKN surface expression by IBD HIMEC
Production of chemokines by endothelial cells can be spontaneous or regulated by inflammatory mediators, and it is usually increased in inflammatory conditions like IBD 44, 45. We initially investigated the capacity of control and IBD HIMEC to express FKN under resting and cytokine-stimulated conditions. Resting HIMEC from control, UC and CD patients displayed low FKN surface expression, but this increased progressively in a time-dependent fashion upon exposure to IL-1β, IFN-γ, or TNF-α (Fig. 1). Interestingly, the combination of the Th1 cytokines IFN-γ and TNF-α induced a marked and selective synergistic effect, resulting in a significantly higher FKN expression in both CD and UC than control HIMEC (Fig. 1 and 2). In addition to the differences in the percentage of FKN+ cells, upregulation of FKN on HIMEC surface was also documented by the correspondent increase in the mean fluorescence intensity (MFI) of HIMEC stimulated with IFN-γ and TNF-α. Synergism was also observed in HIMEC stimulated by IFN-γ and IL-1β combined (data not shown). When the detachment buffer was used to suspend control, UC and CD HIMEC prior to cytokine stimulation, no differences in FKN expression were observed compared to trypsin-treated cells (data not shown).
Figure 1.

Enhanced upregulation of FKN expression on HIMEC surface by proinflammatory cytokines. Control, UC, and CD HIMEC monolayers were left untreated or stimulated with IL1-β, IFN-γ, TNF-α, or TNF-α plus IFN-γ for different time periods, after which the percentage of FKN-expressing HIMEC was assessed by flow cytometric analysis. Each bar represents 4–5 separate HIMEC lines. * p<0.05 and # p<0.01 for UC and CD compared to control HIMEC.
In contrast to the results seen with Th1 cytokines, stimulation with the Th2 cytokines IL-4 or IL-10 did not increase FKN expression in control or IBD HIMEC (data not shown). Because IL-4 or IL-10 display a potent immunoregulatory activity, we ascertained whether they could modulate FKN upregulation induced by the combination of IFN-γ and TNF-α. However, pre-treatment with IL-4 or IL-10 or concomitant addition of these cytokines failed to inhibit the IFN-γ plus TNF-α-induced upregulation of HIMEC FKN expression (data not shown).
Enhanced leukocyte-induced FKN surface expression by IBD HIMEC
In addition to cytokines, we investigated whether other stimuli could also modulate HIMEC FKN expression. Compared to unstimulated HIMEC, contact with T (Jurkat) or monocytic (THP1) cells for a 2-hour period was sufficient to induce a significant (p<0.05) increase in FKN expression on UC and CD, but not control HIMEC. When contact time was extended to 24 hours, a significant enhancement of FKN expression was seen in all HIMEC, but IBD HIMEC further increased their expression to a significantly greater level than control HIMEC (Fig. 3A). Because IFN-γ appeared to enhance the FKN enhancing capacity of TNF-α (Fig. 1), leukocytes were put in contact with IFN-γ-pre-treated HIMEC, but this did not result in FKN expression greater than that seen with untreated HIMEC (Fig. 3A). We have previously shown that leukocyte contact mediated by the CD40/CD40L system causes HIMEC to produce other chemokines such as IL-8 and RANTES 36, 41. Therefore, we investigated whether contact with CD40-positive D1.1 cells could also induce FKN expression. FKN was induced, but to a similar degree similar than that of CD40-negative Jurkat T-cells (Fig. 3A). As an additional control, soluble CD40L failed to significantly upregulate FKN expression in both control and IBD HIMEC. When the THP1 monocytic cell line was used the results were comparable to those obtained with Jurkat and D1.1 cells (Fig. 3A). The same results were obtained when MFI was measured instead of % positive cells (Fig. 3B). In all cell contact experiments, the magnitude of FKN upregulation was significantly higher in IBD than control HIMEC. Finally, to prove that the above results were strictly dependent on cell-to-cell contact, and not on soluble factors potentially released during the co-culture, parallel experiments were performed using a Transwell system to physically separate leukocytes from HIMEC. As shown in Fig. 4, no increase in HIMEC FKN expression was noted with any of the leukocytes used.
Figure 3.

Enhanced upregulation of FKN expression on HIMEC surface by leukocyte contact. Control, UC, and CD HIMEC monolayers were left untreated or cultured with CD40L-negative Jurkat T-cells, CD40L-positive D1.1 T-cells, THP1 monocytic cells, or sCD40L for 24 hours, after which leukocytes were removed and the percentage of FKN-expressing HIMEC (3A) and MFI (3B) were assessed by flow cytometric analysis. Where indicated, HIMEC were pretreated with IFN-γ for 48 hours, prior to co-culture. Each bar represents 4–5 separate HIMEC lines. * p<0.05–0.01 for stimulated compared to unstimulated (none) HIMEC; ** p<0.05 for CD and UC compared to control HIMEC.
Figure 4.
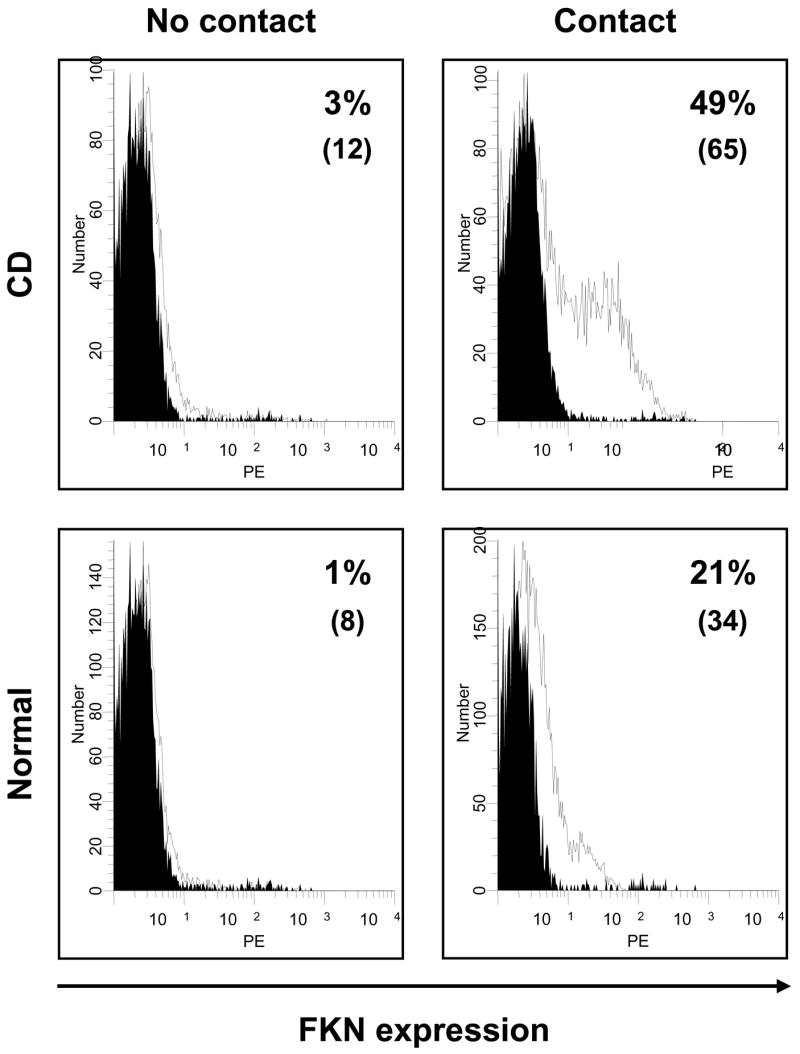
Leukocyte contact-induced upregulation of FKN surface expression by HIMEC. HIMEC monolayers were cultured with Jurkat T-cells for 24 hours, after which the percentage of FKN-expressing HIMEC was assessed by flow cytometric analysis. In some experiments (No contact), a 0.2 μm porous filter was placed between leukocytes and endothelial cells to prevent physical contact. Numbers in parentheses indicate the corresponding MFI. The black curve represents the background signal from the isotype control. This figure is representative of 12 separate experiments (4 normal, 4 UC and 4 CD HIMEC).
Secretion of soluble FKN by HIMEC
The results so far demonstrated that FKN is expressed on the surface of HIMEC, where it can function as an adhesion molecule for leukocytes 46. Because FKN can also function as a leukocyte chemoattractant 43, we used the Th1 cytokine-dependent experimental system described above to determine whether HIMEC could actually secrete the soluble form of this chemokine. FKN was not detected in unstimulated HIMEC cultures. After activation of HIMEC with the combination of IFN-γ and TNF-α, FKN was detected in the culture supernatants and its level was significantly higher in UC and CD than control cultures (Fig. 5). This difference was not observed when HIMEC were stimulated individually with TNF-α or IFN-γ. The secretion pattern of soluble FKN by HIMEC essentially paralleled that seen with FKN surface expression in the same cultures (data not shown).
Figure 5.

Cytokine-induced secretion of soluble FKN by HIMEC. Control, UC and CD HIMEC monolayers were left untreated or stimulated with TNF-α, IFN-γ, or TNF-α plus IFN-γ. Supernatants were harvested at 48 hours and FKN content analyzed by ELISA. Each bar represents 4 separate HIMEC lines. * p<0.05 for UC and CD compared to control HIMEC.
Involvement of p38 and ERK-1/2 in HIMEC FKN production
To explore some of the signaling pathways involved in FKN production, HIMEC were stimulated with either IFN-γ alone, TNF-α alone, or IFN-γ plus TNF-α. TNF-α, alone or in combination with IFN-γ, increased phosphorylation of MAP kinases p38, ERK-1 and -2 in both control and IBD HIMEC, while IFN-γ alone had no affect the baseline phosphorylation status (Fig. 6A). To determine whether activation of these MAP kinases was directly involved in FKN upregulation, the selective inhibitors SB203580 and PD98059 were added to the HIMEC cultures to block p38 and ERK-1/2, respectively. Flow cytometric analysis demonstrated that both inhibitors caused a significant reduction in the number of FKN-expressing HIMEC, in control, UC and CD cultures (Fig. 6B). Notably, even though the degree of FKN expression was significantly higher in cytokine-treated IBD than control HIMEC, treatment with the inhibitors reduced FKN expression by IBD to the level of control HIMEC.
Figure 6.

A) Cytokine-induced phosphorylation of p38 and ERK in HIMEC. HIMEC monolayers were left untreated or stimulated with IFN-γ, TNF-α, or IFN-γ plus TNF-α for 15 minutes, after which cells were washed, lysed, and total protein extracted for hybridization with a phopho-p38- or phospho-ERK specific antibody. The figure is representative of 3 separate experiments.
B) Decrease of FKN-expressing HIMEC by p38 and ERK inhibitors. Control, UC and CD HIMEC monolayers were left untreated or pretreated for 1 hour with the p38 inhibitor SB203580 or the ERK-1 and -2 inhibitor PD98059 prior to stimulation with IFN-γ plus TNF-α. After 24 hours, the percentage of FKN-expressing HIMEC was assessed by flow cytometric analysis. Each bar represents 3–4 separate cell lines. * p<0.05 compared to inhibitor untreated cells.
Increased FKN receptor-positive cells in the circulation and mucosa of IBD patients
The increased production of FKN by HIMEC in IBD prompted the question of whether the expression of CX3CR1 was also increased on leukocytes of IBD patients. Flow cytometric analysis showed that CX3CR1 expression was markedly and significantly higher in CD3+, CD4+, and CD8+ circulating T-cells of patients with active disease than T-cells from healthy control subjects (Fig. 7). Notably, when IBD patients in remission were evaluated the number of circulating CX3CR1+ T-cells was similar to that of healthy controls (Fig. 7). As expected, essentially all circulating monocytes were CX3CR1+, with no significant differences among active and inactive IBD patients and healthy subjects (data not shown). Because CX3CR1 can mediate adhesion of leukocytes 7, 9, we also measured its expression level on the cell types used in the leukocyte-HIMEC adhesion assays (see below). Expression of FKN receptor was found on a substantial number of Jurkat (19.4±4.8%) and practically all THP1 cells (97.2±0.1%).
Figure 7.


A) Differential CX3CR1 expression by T-cells from healthy and IBD subjects. PBMC were isolated from healthy subjects (normal, n=10) and patients with active CD (n=8), inactive CD (n=8), active UC (n=9), and inactive UC (n=7). Cells were co-stained for CX3CR1 and CD3, CD4 and CD8, and the percentage of positive cells for each subpopulation calculated. * p<0.05 for active UC and CD compared to normal and inactive UC and CD.
B) Flow cytometry analysis showing differential surface expression of the FKN receptor CX3CR1 on peripheral bloodT-cells from healthy subjects (normal) and active CD patients. Values represent the percentage of CX3CR1 positive cells in each scatter plot. This figure is representative of 18 experiments (10 healthy subjects and 8 patients with active CD).
When the number of CX3CR1+ cells was assessed in the intestine, a markedly greater proportion of positive cells was detected in IBD mucosa (8.78±1.0 %) compared to histologically normal control mucosa (1.77±0.3 %) (p<0.0001). When assessed separately, both CD (8.58±1.1 %) and UC (8.94±1.7 %) mucosa contained a significantly (p<0.0001 for both) greater percentage of CX3CR1+ cells than control mucosa, with no significant difference between CD and UC (Fig. 8). On note, numerous CX3CR1+ cells were easily detectable in the lumen of or adherent to the wall of the local microvasculature in actively inflamed IBD mucosa (insert in Fig. 8).
Figure 8.

Detection of CX3CR1+ cells in normal and IBD mucosa. The panels show dark brown immunohistochemical staining for CX3CR1 in mononuclear cells of the colonic lamina propria of histologically normal control, active CD and active UC tissue sections. The insert in the UC panel shows CX3CR1+ cells in the lumen of or adherent to the wall of the mucosal microvasculature in actively inflamed areas of the same UC specimen. The panels are representative of 14 control, 9 CD, and 11 UC samples.
Contribution of FKN to leukocyte-HIMEC adhesion
Having shown that activated HIMEC upregulate FKN surface expression, we investigated its leukocyte adhesive capacity compared to potent adhesion molecules like VCAM-1 and ICAM-1. Using a previously reported assay 32, unstimulated HIMEC retained a low number of Jurkat cells, but this number dramatically increased after HIMEC stimulation with IFN-γ plus TNF-α (Fig. 9). As anticipated, both UC and CD HIMEC adhered significantly (p<0.05) more T-cells than control HIMEC 32 (Fig. 9). As multiple molecules participate in leukocyte-endothelial adhesion, extensive antibody blocking experiments were performed to assess the relative contribution of FKN to this process. Blockade of either FKN or CX3CR1 resulted in a significant decrease in the ability of control, UC and CD HIMEC to retain Jurkat cells, ranging from 18 to 27% (p<0.05 vs. no blockade, Fig. 9). Blocking VCAM-1 reduced the adhesion of Jurkat cells to a greater degree than blocking FKN or CX3CR1, whereas ICAM-1 blockade had not effect (Fig. 9). Simultaneous blockade of FKN and VCAM-1 resulted in further inhibition of T-cell adhesion equivalent to an additive effect. Blocking FKN and ICAM-1 showed a degree of inhibition equal to that of blocking FKN alone, and blockade of all three molecules resulted in an inhibition comparable to that of FKN and VCAM-1 blockade (Fig. 8). When THP1 cells were used, overall comparable results were observed except that the contribution of FKN to monocyte adhesion was greater, ranging from 32 to 35% (data not shown). As a final control, anti-CD3/CD28-activated normal PBMC were also used in the same assay. About 30% of these cells expressed CX3CR1, and 20–30% of their adhesion to HIMEC was FKN-dependent (data not shown). Control goat, rabbit, or sheep immunoglobulin had no effect on adhesion (data not shown), demonstrating the specificity of FKN and VCAM-1 blockade on leukocyte adhesion.
Figure 9.

Relative contribution of FKN to HIMEC-T-cell adhesion. Confluent HIMEC monolayers were left unstimulated (white bars) or stimulated (back bars) with TNF-α and IFN-γ. After 24 hours, calcein-labeled Jurkat T-cells were co-cultured with the HIMEC monolayers for 1 hour after which the monolayers were rinsed and retained leukocytes quantified using a computerized imaging system on an inverted fluorescence microscope. To assess the relative contribution of FKN, CX3CR1, VCAM-1, and ICAM-1 to adhesion, antibodies specific for each molecule were added alone (gray bars) or in combination to HIMEC or Jurkat cells (shaded bars). Each bar represents 4 separate HIMEC lines. * p<0.05 for antibody-treated compared to -untreated HIMEC.
Induction of active β1 integrin expression by FKN
As part of the global process of leukocyte attraction and retention, chemokines also increase leukocyte integrin affinity 47. In particular, induction of the active form of β1 integrin on leukocytes increases affinity for VCAM-1 and several extracellular matrix proteins, contributing to a more efficient adhesion to these substrates 48. To investigate whether FKN could modulate expression of active β1 integrin, we exposed normal PBMC, Jurkat and THP1 cells to several concentrations of FKN, for different periods of time, and measured the expression level of active β1 with a monoclonal antibody that exclusively recognizes the high affinity form of this integrin 49. Maximal expression of active β1 was obtained using either 10 or 50 nM FKN for 30 minutes. Spontaneous expression of active β1 was low in resting PBMC, and essentially doubled upon exposure to FKN (Fig. 10). Expression in Jurkat and THP1 cells was substantially higher, reflecting the activated state of these tumor cells, but still significantly increased after exposure to FKN (Fig. 10). The induction of active β1 on the surface of all three types of leukocytes was almost entirely prevented by the Src kinase p56lck inhibitor PP2, and totally abrogated by the G protein inhibitor pertussis toxin (data not shown).
Figure 10.

FKN-induced enhancement of active β1 integrin expression on leukocytes. PBMC isolated from healthy subjects, Jurkat cells and THP1 cells were cultured with 50 nM of human recombinant FKN. After 30 minutes cells were washed, stained with 12G10, a monoclonal antibody that specifically recognizes the activated form of β1 integrin, and assessed by flow cytometry. Each bar represents 4 separate experiments. * p<0.05 compared to FKN-untreated cells.
Effect of FKN on HIMEC transmigration
After demonstrating the active contribution of FKN to leukocyte adhesion, we finally investigated the capacity of this chemokine to attract and transmigrate leukocytes through resting HIMEC monolayers. In preliminary experiments, where MCP-1 was used as positive control, we demonstrated that FKN was able to induce chemotaxis of TPH1 cells in a dose-dependent fashion (data not shown). Subsequently, using an optimal dose of FKN in a Transwell system, we tested its ability to induce transmigration of THP1 cells through control, UC and CD HIMEC. With MCP-1 as positive control, FKN triggered THP1 cell transmigration through HIMEC to a magnitude of 30–40% compared to that of MCP-1 (Fig. 11), with no significant differences among control, UC and CD HIMEC (data not shown).
Figure 11.

FKN-induced transmigration of THP-1 cells through HIMEC monolayers. HIMEC monolayers were grown to confluence on filter inserts separating the upper and lower chambers of a Transwell system. Calcein-labeled THP1 cells were placed in the upper chamber overlaying the HIMEC monolayer, while medium alone, MCP-1 or FKN were added to the lower chamber. After a 4 hour incubation period, migrated cells were quantified using a computerized imaging system on an inverted fluorescence microscope. Each bar represents 3 separate HIMEC lines. * p<0.05 for FKN compared to unstimulated, and MCP-1 compared to FKN.
DISCUSSION
This study demonstrates that microvascular cells from IBD mucosa produce greater amounts of FKN than those from normal mucosa, and greater numbers of T-cells express CX3CR1 in the circulation of IBD patients than healthy subjects. Thus, the study indicates that FKN is a major mediator of leukocyte-endothelial interaction in gut inflammation.
Both the surface and secreted form of FKN are markedly upregulated by HIMEC upon stimulation with Th1 cytokines, particularly the combination of TNF-α with IFN-γ. Of note, this upregulation was significantly stronger in CD and UC compared to control HIMEC. Although some controversy exists on the expression of FKN by the IBD microvasculature in vivo 50, our results are in keeping with previous immunohistochemical evidence of increased endothelial cell FKN expression and mRNA in CD tissue 16. Paralleling the surface expression results, HIMEC stimulation with combined TNF-α and IFN-γ also resulted in a marked increase in the secreted form of FKN, IBD HIMEC once again producing significantly greater amounts of this chemokine than control HIMEC. Considering that levels of most pro-inflammatory cytokines are high in IBD-involved tissue 51, this could explain the increased production of FKN by the mucosal microvasculature in CD and UC.
Immunoregulatory cytokines can downregulate excessive chemokine production by endothelial cells, as documented by the ability of IL-4 and IL-13 to inhibit FKN production by human umbilical vein endothelial cells 18. Surprisingly, this did not occur when IBD HIMEC were exposed to IL-4 or IL-10. One explanation could be that this unique cell type is less susceptible to immunoregulatory signals. Alternatively, chronic exposure to the inflammatory milieu of the IBD mucosa may select in vivo for a hyper-producer endothelial cell phenotype which is conserved and quantifiable in culture, similarly to what has been previously shown for the hyper-adhesive phenotype of IBD HIMEC 32. If these two characteristics of the IBD microvasculature synergize in vivo, this could result in heightened and persistent chemokine production and leukocyte recruitment, and thus contribute to chronicity of inflammation. Apart from cytokines, HIMEC activation mediated by direct contact with T-cells and monocytes was also a potent inducer of FKN upregulation on the endothelial cell surface, similar to what has been observed with antigen-specific CD4+ T-cells 52. Once again, leukocyte contact with CD and UC HIMEC produced higher FKN levels than control HIMEC, reinforcing the notion that the heightened degree of responsiveness of IBD microvascular cells is a phenomenon independent of the type of stimulatory signal.
The mechanism of FKN production by HIMEC may differ from that of other chemokines. In fact, we previously reported that both IL-8 and RANTES were comparably induced in HIMEC by cell-bound as well as soluble CD40L 41. In contrast, CD40L-positive D1.1 cells were able to trigger FKN production but not the soluble form of CD40L. The reason for this difference is not apparent, but HIMEC FKN production may be dependent on complex physical cell surface interactions but independent of the CD40/CD40L pathway.
When some of the signaling pathways governing FKN upregulation in HIMEC were investigated, enhanced phosphorylation of the MAP kinases p38, ERK-1 and -2 was detected early after HIMEC stimulation by TNF-α alone or TNF-α combined with IFN-γ, but not IFN-γ alone. This observation, combined with the ineffectiveness of CD40 signaling may suggest that broad pro-inflammatory rather than selective stimuli are needed for efficient signaling leading to FKN production. FKN upregulation on HIMEC surface was markedly reduced when these cells were pretreated with a specific inhibitor of p38 (SB203580) or ERK (PD98059), demonstrating that both MAP kinases have an important regulatory role on FKN expression. These results agree with those of a previous study, in which we showed that inhibiting p38 significantly reduces CD40-mediated IL-8 production by HIMEC 36. Interestingly, the degree of inhibition of FKN production by both inhibitors was clearly greater in both UC and CD than control HIMEC, suggesting a modulatory potential of these substances for inhibition of FKN and other chemokines in gut inflammation 53. This notion is supported by two recent studies showing that SB203580 is able to attenuate DSS-54 as well as TNBS-induced murine colitis 55.
If the increased HIMEC FKN production in IBD mucosa is involved in disease pathogenesis, this cytokine should attract to the gut and engage a large population of CX3CR1+ leukocytes to mediate inflammation. This scenario is supported by two complementary observations. First, both CD and UC patients with active disease have a significantly increased number of CX3CR1+ T-cells in the peripheral circulation, CD8+ T-cells predictably predominating over CD4+ T-cells 56. Notably, increased CX3CR1 expressing T-cells were detected exclusively in patients with clinically active disease, whereas in patients in remission the number of FKN receptor-bearing cells was comparable to that of healthy subjects. Second, a fivefold increase in the percentage of CX3CR1+ was detected in the lamina propria of both CD and UC mucosa compared to normal control mucosa, a finding compatible with an enhanced recruitment of CX3CR1+ cells in response to an increased production of FKN by the local microvasculature. Large numbers of CX3CR1+ cells are seen in patients with other inflammatory conditions such as rheumatoid arthritis 26, systemic sclerosis 27, and allergic asthma and rhinitis 57, all of which also display high endothelial cell FKN expression in the affected tissues. These reports, combined with the results we found in IBD, makes likely that a high CX3CR1 expression by circulating leukocytes reflects an elevated FKN production by the inflamed microcirculation of the target organ.
Given the increased number of FKN+ endothelial cells and of CX3CR1+ T-cells, a physical interaction between them must occur in IBD mucosa. Considering the biological properties of FKN, leukocyte attraction and adhesion could occur, and both were specifically investigated in our study. Using an established adhesion assay and multiple blocking antibodies we initially defined the relative contribution of FKN to leukocyte-endothelial adhesion. By neutralizing the activity of FKN or blocking its receptor, it became evident that a sizeable proportion of leukocyte adhesion to HIMEC was FKN-dependent. For T-cells, this proportion was not as high as that dependent on VCAM-1, while for monocytes FKN-dependent adhesion was as high as that of VCAM-1. In contrast, the contribution of FKN to T-cell and monocyte adhesion was clearly more important that that of ICAM-1. In view of these results, it seems justified to conclude that FKN acts a major adhesion molecule involved in leukocyte recruitment into IBD mucosa. Our findings are in keeping with other studies demonstrating the capacity of FKN to capture and adhere various CX3CR1-expressing leukocyte subsets in other experimental systems, like immobilized FKN or FKN fusion proteins 21, 43, 46, 58–60, FKN-transfected endothelial cells 46, 58, 60, or cytokine-stimulated HUVEC 46, 58, 60.
In addition to its intrinsic adhesive function, there is indirect evidence that FKN also cooperates with other adhesion molecules, presumably by augmenting integrin affinity 43, 58. VCAM-1 is essential for leukocyte adhesion to endothelium, but so far there is no direct evidence that FKN upregulates the active form of β1 integrin 38, 49, a component of the VCAM-1 counter-receptor α4β1 integrin. We measured the expression of the active form of β1 integrin on different leukocytes exposed to FKN, and observed a significant increase in its expression, a specific effect abrogated by Src kinase p56lck and G protein blockade. Thus, FKN produced in the microcirculation likely contributes to recruitment of leukocytes in the mucosa not simply by mediating the CX3CR1-FKN physical interaction on the endothelial surface, but also enhancing their integrin affinity which strengthens leukocyte interaction with other adhesion molecules like VCAM-1.
Finally, we assessed the capacity of FKN to act as a pure chemotactic molecule by inducing transendothelial migration of THP1 cells. A significant degree of THP1 cell migration through HIMEC was observed, though to a lesser degree than that induced by MCP-1, the prototypical monocyte chemoattractant 43, 44. These results indicate that FKN, although displaying features different from those of other classes of chemokines 43, 44, 61, 62, can also play a traditional chemotactic role in IBD, particularly for CD16+ monocytes 60, 63.
In summary, translating our in vitro results to the in vivo situation, the milieu of the IBD mucosa, with its high content of pro-inflammatory cytokines and high density of leukocytes, seems to be an ideal environment to stimulate FKN production by the local microvasculature. In turn, endothelial cell-derived FKN exerts a localized proinflammatory response through multiple complementary functions that include leukocyte retention, integrin affinity upregulation, chemoattraction and transmigration. These crucial activities of FKN have not been previously reported in the context of IBD. Interference with the chemokine system is currently considered an effective approach to inflammatory diseases 64, and disrupting FKN/CX3CR1 interactions could represents a promising therapeutic approach for IBD. Preliminary evidence supporting this concept is emerging from animal models of inflammation, where blockade of FKN with specific antibodies results in improvement of experimental collagen-induced arthritis and autoimmune myositis 65, 66.
Figure 2.

A) Flow cytometric analysis of FKN surface expression by HIMEC after cytokine stimulation. HIMEC monolayers were left untreated or stimulated with TNF-α, IFN-γ, or TNF-α plus IFN-γ for 48 hours, after which the percentage (%) of FKN-expressing HIMEC was assessed by flow cytometric analysis. Numbers in parentheses indicate the corresponding MFI. The black curve represents the background signal from the isotype control. This figure is representative of 14 experiments (5 control, 5 UC, and 4 CD HIMEC).
B) Enhanced FKN production by IBD HIMEC. Control, UC and CD HIMEC monolayers were left untreated or stimulated with IFN-γ plus TNF-α for 48 hours, after which cells were lysed, and total proteins extracted for FKN assessment by immunoblotting. This figure is representative of 6 separate experiments. The molecular weight of recombinant human FKN (rhFKN) is slightly smaller than that of the native molecule due to differences in glycosylation.
Acknowledgments
This work was supported by grants from the Fulbright/Generalitat de Catalunya, Ministerio de Educación y Ciencia (Programa Ramón y Cajal, SAF2005-00280 and C03/02) and Fundación Ramón Areces to M.S., and the National Institutes of Health (DK30399 and DK 50984) to C.F. The authors thank Dr. S. Kessler and Mr. R.M. Sramkoski for technical assistance. The contribution of the Institute of Pathology of University Hospitals of Cleveland and the Department of Colorectal Surgery of the Cleveland Clinic Foundation is acknowledged. Tissue samples were provided by the Human Tissue Procurement Facility of University Hospitals of Cleveland.
Abbreviations
- Ab
antibody
- CAM
cell adhesion molecule
- CD
Crohn’s disease
- FKN
fractalkine
- HIMEC
human intestinal microvascular endothelial cells
- IBD
inflammatory bowel disease
- ICAM-1
intercellular adhesion molecule 1
- PBMC
peripheral blood mononuclear cells
- PE
Phycoerythrin
- UC
ulcerative colitis
- VCAM-1
vascular cell adhesion molecule 1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cook-Mills JM, Deem TL. Active participation of endothelial cells in inflammation. J Leukoc Biol. 2005;77:487–495. doi: 10.1189/jlb.0904554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327–334. doi: 10.1016/s1471-4906(03)00117-0. [DOI] [PubMed] [Google Scholar]
- 3.Panés J, Granger DN. Leukocyte-endothelial cell interactions: molecular mechanisms and implications in gastrointestinal disease. Gastroenterology. 1998;114:1066–1090. doi: 10.1016/s0016-5085(98)70328-2. [DOI] [PubMed] [Google Scholar]
- 4.Proudfoot AE, Power CA, Rommel C, Wells TN. Strategies for chemokine antagonists as therapeutics. Semin Immunol. 2003;15:57–65. doi: 10.1016/s1044-5323(02)00128-8. [DOI] [PubMed] [Google Scholar]
- 5.van Buul JD, Hordijk PL. Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol. 2004;24:824–833. doi: 10.1161/01.ATV.0000122854.76267.5c. [DOI] [PubMed] [Google Scholar]
- 6.Papadakis KA. Chemokines in inflammatory bowel disease. Curr Allergy Asthma Rep. 2004;4:83–89. doi: 10.1007/s11882-004-0048-7. [DOI] [PubMed] [Google Scholar]
- 7.Umehara H, Bloom E, Okazaki T, Domae N, Imai T. Fractalkine and vascular injury. Trends Immunol. 2001;22:602–607. doi: 10.1016/s1471-4906(01)02051-8. [DOI] [PubMed] [Google Scholar]
- 8.Haskell CA, Cleary MD, Charo IF. Unique role of the chemokine domain of fractalkine in cell capture. Kinetics of receptor dissociation correlate with cell adhesion. J Biol Chem. 2000;275:34183–34189. doi: 10.1074/jbc.M005731200. [DOI] [PubMed] [Google Scholar]
- 9.Fong AM, Erickson HP, Zachariah JP, Poon S, Schamberg NJ, Imai T, Patel DD. Ultrastructure and function of the fractalkine mucin domain in CX(3)C chemokine domain presentation. J Biol Chem. 2000;275:3781–3786. doi: 10.1074/jbc.275.6.3781. [DOI] [PubMed] [Google Scholar]
- 10.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 11.Tsou CL, Haskell CA, Charo IF. Tumor necrosis factor-alpha-converting enzyme mediates the inducible cleavage of fractalkine. J Biol Chem. 2001;276:44622–44626. doi: 10.1074/jbc.M107327200. [DOI] [PubMed] [Google Scholar]
- 12.Bazan JF, Bacon KB, Hardiman G, Wang W, Soo K, Rossi D, Greaves DR, Zlotnik A, Schall TJ. A new class of membrane-bound chemokine with a CX3C motif. Nature. 1997;385:640–644. doi: 10.1038/385640a0. [DOI] [PubMed] [Google Scholar]
- 13.Umehara H, Bloom ET, Okazaki T, Nagano Y, Yoshie O, Imai T. Fractalkine in vascular biology: from basic research to clinical disease. Arterioscler Thromb Vasc Biol. 2004;24:34–40. doi: 10.1161/01.ATV.0000095360.62479.1F. [DOI] [PubMed] [Google Scholar]
- 14.Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 15.Cockwell P, Chakravorty SJ, Girdlestone J, Savage CO. Fractalkine expression in human renal inflammation. J Pathol. 2002;196:85–90. doi: 10.1002/path.1010. [DOI] [PubMed] [Google Scholar]
- 16.Muehlhoefer A, Saubermann LJ, Gu X, Luedtke-Heckenkamp K, Xavier R, Blumberg RS, Podolsky DK, MacDermott RP, Reinecker HC. Fractalkine is an epithelial and endothelial cell-derived chemoattractant for intraepithelial lymphocytes in the small intestinal mucosa. J Immunol. 2000;164:3368–3376. doi: 10.4049/jimmunol.164.6.3368. [DOI] [PubMed] [Google Scholar]
- 17.Imaizumi T, Yoshida H, Satoh K. Regulation of CX3CL1/fractalkine expression in endothelial cells. J Atheroscler Thromb. 2004;11:15–21. doi: 10.5551/jat.11.15. [DOI] [PubMed] [Google Scholar]
- 18.Fraticelli P, Sironi M, Bianchi G, D’Ambrosio D, Albanesi C, Stoppacciaro A, Chieppa M, Allavena P, Ruco L, Girolomoni G, Sinigaglia F, Vecchi A, Mantovani A. Fractalkine (CX3CL1) as an amplification circuit of polarized Th1 responses. J Clin Invest. 2001;107:1173–1181. doi: 10.1172/JCI11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imai T, Hieshima K, Haskell C, Baba M, Nagira M, Nishimura M, Kakizaki M, Takagi S, Nomiyama H, Schall TJ, Yoshie O. Identification and molecular characterization of fractalkine receptor CX3CR1, which mediates both leukocyte migration and adhesion. Cell. 1997;91:521–530. doi: 10.1016/s0092-8674(00)80438-9. [DOI] [PubMed] [Google Scholar]
- 20.Combadiere C, Salzwedel K, Smith ED, Tiffany HL, Berger EA, Murphy PM. Identification of CX3CR1. A chemotactic receptor for the human CX3C chemokine fractalkine and a fusion coreceptor for HIV-1. J Biol Chem. 1998;273:23799–23804. doi: 10.1074/jbc.273.37.23799. [DOI] [PubMed] [Google Scholar]
- 21.Haskell CA, Cleary MD, Charo IF. Molecular uncoupling of fractalkine-mediated cell adhesion and signal transduction. Rapid flow arrest of CX3CR1-expressing cells is independent of G-protein activation. J Biol Chem. 1999;274:10053–10058. doi: 10.1074/jbc.274.15.10053. [DOI] [PubMed] [Google Scholar]
- 22.Greaves DR, Hakkinen T, Lucas AD, Liddiard K, Jones E, Quinn CM, Senaratne J, Green FR, Tyson K, Boyle J, Shanahan C, Weissberg PL, Gordon S, Yla-Hertualla S. Linked chromosome 16q13 chemokines, macrophage-derived chemokine, fractalkine, and thymus- and activation-regulated chemokine, are expressed in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2001;21:923–929. doi: 10.1161/01.atv.21.6.923. [DOI] [PubMed] [Google Scholar]
- 23.Furuichi K, Wada T, Iwata Y, Sakai N, Yoshimoto K, Shimizu M, Kobayashi K, Takasawa K, Kida H, Takeda S, Matsushima K, Yokoyama H. Upregulation of fractalkine in human crescentic glomerulonephritis. Nephron. 2001;87:314–320. doi: 10.1159/000045936. [DOI] [PubMed] [Google Scholar]
- 24.Robinson LA, Nataraj C, Thomas DW, Howell DN, Griffiths R, Bautch V, Patel DD, Feng L, Coffman TM. A role for fractalkine and its receptor (CX3CR1) in cardiac allograft rejection. J Immunol. 2000;165:6067–6072. doi: 10.4049/jimmunol.165.11.6067. [DOI] [PubMed] [Google Scholar]
- 25.Raychaudhuri SP, Jiang WY, Farber EM. Cellular localization of fractalkine at sites of inflammation: antigen-presenting cells in psoriasis express high levels of fractalkine. Br J Dermatol. 2001;144:1105–1113. doi: 10.1046/j.1365-2133.2001.04219.x. [DOI] [PubMed] [Google Scholar]
- 26.Nanki T, Imai T, Nagasaka K, Urasaki Y, Nonomura Y, Taniguchi K, Hayashida K, Hasegawa J, Yoshie O, Miyasaka N. Migration of CX3CR1-positive T cells producing type 1 cytokines and cytotoxic molecules into the synovium of patients with rheumatoid arthritis. Arthritis Rheum. 2002;46:2878–2883. doi: 10.1002/art.10622. [DOI] [PubMed] [Google Scholar]
- 27.Hasegawa M, Sato S, Echigo T, Hamaguchi Y, Yasui M, Takehara K. Up regulated expression of fractalkine/CX3CL1 and CX3CR1 in patients with systemic sclerosis. Ann Rheum Dis. 2005;64:21–28. doi: 10.1136/ard.2003.018705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, Nakanuma Y. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–516. doi: 10.1002/hep.20582. [DOI] [PubMed] [Google Scholar]
- 29.Brand S, Sakaguchi T, Gu X, Colgan SP, Reinecker HC. Fractalkine-mediated signals regulate cell-survival and immune-modulatory responses in intestinal epithelial cells. Gastroenterology. 2002;122:166–177. doi: 10.1053/gast.2002.30329. [DOI] [PubMed] [Google Scholar]
- 30.Niess JH, Brand S, Gu X, Landsman L, Jung S, McCormick BA, Vyas JM, Boes M, Ploegh HL, Fox JG, Littman DR, Reinecker HC. CX3CR1-mediated dendritic cell access to the intestinal lumen and bacterial clearance. Science. 2005;307:254–258. doi: 10.1126/science.1102901. [DOI] [PubMed] [Google Scholar]
- 31.Lederman S, Yellin MJ, Krichevsky A, Belko J, Lee JJ, Chess L. Identification of a novel surface protein on activated CD4+ T cells that induces contact-dependent B cell differentiation (help) J Exp Med. 1992;175:1091–1101. doi: 10.1084/jem.175.4.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binion DG, West GA, Ina K, Ziats NP, Emancipator SN, Fiocchi C. Enhanced leukocyte binding by intestinal microvascular endothelial cells in inflammatory bowel disease. Gastroenterology. 1997;112:1895–907. doi: 10.1053/gast.1997.v112.pm9178682. [DOI] [PubMed] [Google Scholar]
- 33.Harvey RF, Bradshaw JM. A simple index of Crohn’s-disease activity. Lancet. 1980;1:514. doi: 10.1016/s0140-6736(80)92767-1. [DOI] [PubMed] [Google Scholar]
- 34.Sutherland LR, Martin F, Greer S, Robinson M, Greenberger N, Saibil F, Martin T, Sparr J, Prokipchuk E, Borgen L. 5-Aminosalicylic acid enema in the treatment of distal ulcerative colitis, proctosigmoiditis, and proctitis. Gastroenterology. 1987;92:1894–1898. doi: 10.1016/0016-5085(87)90621-4. [DOI] [PubMed] [Google Scholar]
- 35.Sturm A, Itoh J, Jacobberger JW, Fiocchi C. p53 negatively regulates intestinal immunity by delaying mucosal T cell cycling. J Clin Invest. 2002;109:1481–1492. doi: 10.1172/JCI14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Danese S, de la Motte C, Sturm A, Vogel JD, West GA, Strong SA, Katz JA, Fiocchi C. Platelets trigger a CD40-dependent inflammatory response in the microvasculature of inflammatory bowel disease patients. Gastroenterology. 2003;124:1249–1264. doi: 10.1016/s0016-5085(03)00289-0. [DOI] [PubMed] [Google Scholar]
- 37.Ropert C, Almeida IC, Closel M, Travassos LR, Ferguson MA, Cohen P, Gazzinelli RT. Requirement of mitogen-activated protein kinases and I kappa B phosphorylation for induction of proinflammatory cytokines synthesis by macrophages indicates functional similarity of receptors triggered by glycosylphosphatidylinositol anchors from parasitic protozoa and bacterial lipopolysaccharide. J Immunol. 2001;166:3423–3431. doi: 10.4049/jimmunol.166.5.3423. [DOI] [PubMed] [Google Scholar]
- 38.Thodeti CK, Albrechtsen R, Grauslund M, Asmar M, Larsson C, Takada Y, Mercurio AM, Couchman JR, Wewer UM. ADAM12/syndecan-4 signaling promotes beta 1 integrin-dependent cell spreading through protein kinase Calpha and RhoA. J Biol Chem. 2003;278:9576–9584. doi: 10.1074/jbc.M208937200. [DOI] [PubMed] [Google Scholar]
- 39.Feigelson SW, Grabovsky V, Winter E, Chen LL, Pepinsky RB, Yednock T, Yablonski D, Lobb R, Alon R. The Src kinase p56(lck) up-regulates VLA-4 integrin affinity. Implications for rapid spontaneous and chemokine-triggered T cell adhesion to VCAM-1 and fibronectin. J Biol Chem. 2001;276:13891–13901. doi: 10.1074/jbc.M004939200. [DOI] [PubMed] [Google Scholar]
- 40.Musso A, Condon TP, West GA, De La MC, Strong SA, Levine AD, Bennett CF, Fiocchi C. Regulation of ICAM-1-mediated fibroblast-T cell reciprocal interaction: implications for modulation of gut inflammation. Gastroenterology. 1999;117:546–556. doi: 10.1016/s0016-5085(99)70447-6. [DOI] [PubMed] [Google Scholar]
- 41.Vogel JD, West GA, Danese S, De La MC, Phillips MH, Strong SA, Willis J, Fiocchi C. CD40-mediated immune-nonimmune cell interactions induce mucosal fibroblast chemokines leading to T-cell transmigration. Gastroenterology. 2004;126:63–80. doi: 10.1053/j.gastro.2003.10.046. [DOI] [PubMed] [Google Scholar]
- 42.Roth SJ, Carr MW, Rose SS, Springer TA. Characterization of transendothelial chemotaxis of T lymphocytes. J Immunol Methods. 1995;188:97–116. doi: 10.1016/0022-1759(95)00208-1. [DOI] [PubMed] [Google Scholar]
- 43.Umehara H, Goda S, Imai T, Nagano Y, Minami Y, Tanaka Y, Okazaki T, Bloom ET, Domae N. Fractalkine, a CX3C-chemokine, functions predominantly as an adhesion molecule in monocytic cell line THP-1. Immunol Cell Biol. 2001;79:298–302. doi: 10.1046/j.1440-1711.2001.01004.x. [DOI] [PubMed] [Google Scholar]
- 44.Chapman GA, Moores KE, Gohil J, Berkhout TA, Patel L, Green P, Macphee CH, Stewart BR. The role of fractalkine in the recruitment of monocytes to the endothelium. Eur J Pharmacol. 2000;392:189–195. doi: 10.1016/s0014-2999(00)00117-5. [DOI] [PubMed] [Google Scholar]
- 45.Papadakis KA, Targan SR. The role of chemokines and chemokine receptors in mucosal inflammation. Inflamm Bowel Dis. 2000;6:303–313. doi: 10.1002/ibd.3780060408. [DOI] [PubMed] [Google Scholar]
- 46.Fong AM, Robinson LA, Steeber DA, Tedder TF, Yoshie O, Imai T, Patel DD. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J Exp Med. 1998;188:1413–1419. doi: 10.1084/jem.188.8.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Constantin G, Majeed M, Giagulli C, Piccio L, Kim JY, Butcher EC, Laudanna C. Chemokines trigger immediate beta2 integrin affinity and mobility changes: differential regulation and roles in lymphocyte arrest under flow. Immunity. 2000;13:759–769. doi: 10.1016/s1074-7613(00)00074-1. [DOI] [PubMed] [Google Scholar]
- 48.Chan JR, Hyduk SJ, Cybulsky MI. Detecting rapid and transient upregulation of leukocyte integrin affinity induced by chemokines and chemoattractants. J Immunol Methods. 2003;273:43–52. doi: 10.1016/s0022-1759(02)00417-9. [DOI] [PubMed] [Google Scholar]
- 49.Mould AP, Askari JA, Barton S, Kline AD, McEwan PA, Craig SE, Humphries MJ. Integrin activation involves a conformational change in the alpha 1 helix of the beta subunit A-domain. J Biol Chem. 2002;277:19800–19805. doi: 10.1074/jbc.M201571200. [DOI] [PubMed] [Google Scholar]
- 50.Lucas AD, Chadwick N, Warren BF, Jewell DP, Gordon S, Powrie F, Greaves DR. The transmembrane form of the CX3CL1 chemokine fractalkine is expressed predominantly by epithelial cells in vivo. Am J Pathol. 2001;158:855–866. doi: 10.1016/S0002-9440(10)64034-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Podolsky DK, Fiocchi C. Cytokines, chemokines, growth factors, eicosanoids and other bioactive molecules in IBD. In: Kirsner JB, editor. Inflammatory bowel disease. Philadelphia: W.B. Saunders; 1999. pp. 191–207. [Google Scholar]
- 52.Bolovan-Fritts CA, Trout RN, Spector SA. Human cytomegalovirus-specific CD4+-T-cell cytokine response induces fractalkine in endothelial cells. J Virol. 2004;78:13173–13181. doi: 10.1128/JVI.78.23.13173-13181.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hommes DW, Peppelenbosch MP, van Deventer SJ. Mitogen activated protein (MAP) kinase signal transduction pathways and novel anti-inflammatory targets. Gut. 2003;52:144–151. doi: 10.1136/gut.52.1.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hollenbach E, Neumann M, Vieth M, Roessner A, Malfertheiner P, Naumann M. Inhibition of p38 MAP kinase- and RICK/NF-kappaB-signaling suppresses inflammatory bowel disease. FASEB J. 2004;18:1550–1552. doi: 10.1096/fj.04-1642fje. [DOI] [PubMed] [Google Scholar]
- 55.Hollenbach E, Vieth M, Roessner A, Neumann M, Malfertheiner P, Naumann M. Inhibition of RICK/nuclear factor-kappaB and p38 signaling attenuates the inflammatory response in a murine model of Crohn disease. J Biol Chem. 2005;280:14981–14988. doi: 10.1074/jbc.M500966200. [DOI] [PubMed] [Google Scholar]
- 56.Foussat A, Coulomb-L’Hermine A, Gosling J, Krzysiek R, Durand-Gasselin I, Schall T, Balian A, Richard Y, Galanaud P, Emilie D. Fractalkine receptor expression by T lymphocyte subpopulations and in vivo production of fractalkine in human. Eur J Immunol. 2000;30:87–97. doi: 10.1002/1521-4141(200001)30:1<87::AID-IMMU87>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 57.Rimaniol AC, Till SJ, Garcia G, Capel F, Godot V, Balabanian K, Durand-Gasselin I, Varga EM, Simonneau G, Emilie D, Durham SR, Humbert M. The CX3C chemokine fractalkine in allergic asthma and rhinitis. J Allergy Clin Immunol. 2003;112:1139–1146. doi: 10.1016/j.jaci.2003.09.041. [DOI] [PubMed] [Google Scholar]
- 58.Goda S, Imai T, Yoshie O, Yoneda O, Inoue H, Nagano Y, Okazaki T, Imai H, Bloom ET, Domae N, Umehara H. CX3C-chemokine, fractalkine-enhanced adhesion of THP-1 cells to endothelial cells through integrin-dependent and -independent mechanisms. J Immunol. 2000;164:4313–4320. doi: 10.4049/jimmunol.164.8.4313. [DOI] [PubMed] [Google Scholar]
- 59.Kerfoot SM, Lord SE, Bell RB, Gill V, Robbins SM, Kubes P. Human fractalkine mediates leukocyte adhesion but not capture under physiological shear conditions; a mechanism for selective monocyte recruitment. Eur J Immunol. 2003;33:729–739. doi: 10.1002/eji.200323502. [DOI] [PubMed] [Google Scholar]
- 60.Ancuta P, Rao R, Moses A, Mehle A, Shaw SK, Luscinskas FW, Gabuzda D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197:1701–1707. doi: 10.1084/jem.20022156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dichmann S, Herouy Y, Purlis D, Rheinen H, Gebicke-Harter P, Norgauer J. Fractalkine induces chemotaxis and actin polymerization in human dendritic cells. Inflamm Res. 2001;50:529–533. doi: 10.1007/PL00000230. [DOI] [PubMed] [Google Scholar]
- 62.Papadopoulos EJ, Fitzhugh DJ, Tkaczyk C, Gilfillan AM, Sassetti C, Metcalfe DD, Hwang ST. Mast cells migrate, but do not degranulate, in response to fractalkine, a membrane-bound chemokine expressed constitutively in diverse cells of the skin. Eur J Immunol. 2000;30:2355–2361. doi: 10.1002/1521-4141(2000)30:8<2355::AID-IMMU2355>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 63.Ancuta P, Moses A, Gabuzda D. Transendothelial migration of CD16+ monocytes in response to fractalkine under constitutive and inflammatory conditions. Immunobiology. 2004;209:11–20. doi: 10.1016/j.imbio.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 64.Johnson Z, Schwarz M, Power CA, Wells TN, Proudfoot AE. Multi-faceted strategies to combat disease by interference with the chemokine system. Trends Immunol. 2005;26:268–274. doi: 10.1016/j.it.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 65.Nanki T, Urasaki Y, Imai T, Nishimura M, Muramoto K, Kubota T, Miyasaka N. Inhibition of fractalkine ameliorates murine collagen-induced arthritis. J Immunol. 2004;173:7010–7016. doi: 10.4049/jimmunol.173.11.7010. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki F, Nanki T, Imai T, Kikuchi H, Hirohata S, Kohsaka H, Miyakasa N. Inhibition of CX3CL1 (fractalkine) improves experimental autoimmune myositis in SJL/J mice. J Immunol. 2005;175:6987–6996. doi: 10.4049/jimmunol.175.10.6987. [DOI] [PubMed] [Google Scholar]