

Abstract
The Tn10 transpososome has symmetrical components on either side: there are two transposon ends each of which has binding sites for a monomer of transposase and an IHF heterodimer. The DNA bending activity of IHF stimulates assembly of an intermediate with tightly folded transposon ends in which transposase has additional ‘subterminal’ DNA contacts, located distal to the IHF site. These subterminal contacts are required to activate later steps in the reaction. Quantitative hydroxyl radical footprinting and gel retardation unfolding experiments show that the transpososome is fundamentally asymmetric, despite having identical components on either side. Major differences between the transposon ends define α and β sides of the complex. IHF can dissociate from the transposon arm on the β side of the complex in the absence of metal ion. However, IHF is locked onto the α side of the complex, probably by the subterminal transposase contacts, until released by a metal ion-dependent conformational change. Later in the reaction, IHF inhibits target interactions. Using a very short transposon arm, target interactions are demonstrated at a saturating IHF concentration. This suggests that inhibition of target interactions is due to steric hindrance of the target binding site by a single IHF-folded transposon arm.
INTRODUCTION
Transposons are probably ubiquitous and have been found in all organisms examined with notable exceptions such as the specialized intracellular parasite Encephalitozoon cuniculi (1). Transposons are important in the spread of antibiotic resistance and as natural genomic engineers: most pathogenic bacteria, for example, have many transposons which function in phase variation and mobilization of pathogenicity islands [e.g. (2)]. During the course of evolution, transposons have diversified to provide functions ranging from HIV integrase to the beneficial V(D)J recombination machinery (3,4).
Tn10 is a composite transposon in which the flanking IS10 elements cooperate to mobilize the tetracycline resistance genes in between (5,6). The ends of IS10 are not identical and are defined as outside ends and inside ends with respect to their orientation in Tn10. Each end of IS10 encodes a terminal inverted repeat which are the recognition sites for transposase. However, adjacent to the outside end is an additional binding site for the host protein IHF (integration host factor). Since the IS10 elements are configured as inverted repeats, Tn10 is flanked by two identical outside ends both of which have IHF binding sites.
Tn10 transposition in vitro depends on the presence of negative supercoiling in the DNA. This requirement is relieved by the presence of IHF and the specific IHF binding site in the outside end of the transposon. IHF is a metabolically stable histone-like protein, ubiquitous in the eubacteria. Upon binding to the specific recognition site it bends the DNA by almost 180° and acts as an architectural component in the assembly of several higher order protein–DNA complexes (7). IHF mutations are highly pleiotropic because it participates in a large number of fundamental biological processes such as DNA replication, transposition and phase variation by DNA inversion (8,9). IHF binding sites are also found in a large number of promoters and a gene expression profile in Escherichia coli identified 150 genes differentially expressed in IHF+ and IHF–-strains (10). In eukaryotes, IHF is present in chloroplasts, African swine virus and shares sequence and structural homology with the ubiquitous TATA box binding protein (9,11,12).
DNA recombination generally takes place within large, highly ordered nucleoprotein complexes which often contain a multitude of different protein–DNA interactions. A good example is bacteriophage λ recombination in which there are 10 or more accessory sites in addition to the two core sites where recombination takes place (13). Indeed, IHF was discovered and named because it is required during bacteriophage λ integrative recombination (14). The purpose of many of these accessory sites and proteins is presumably to provide a high degree of specificity and to regulate the recombination reaction. This is supported by the fact that other members of the tyrosine recombinase family such as Cre and Flp function in the complete absence of accessory factors (13). This variation in the degree of dependency on accessory factors is similar to the relationship between Tn10 and Tn5. Whereas Tn10 depends on IHF or negative supercoiling, Tn5 is completely independent of these factors and functions efficiently even when the transposon ends are encoded on 20 bp linear DNA fragments (15).
Amongst the well characterized IS elements, IS10 is unique in having an IHF binding site adjacent to the inverted repeat. However, there are IHF binding sites in many bacterial MITES (miniature inverted repeat transposable elements) and there is an important IHF binding site in the phage Mu enhancer (2,16). In MITES, the purpose of the IHF binding site is unknown. However, it is probably required to help synapse the ends of the elements which can be as little as 100 bp apart. In phage Mu, the function of the IHF site is similar to Tn10 in that it rescues the transposition reaction in the absence of negative supercoiling (17).
Although all of the chemical steps of Tn10 transposition are known and many mutants have been documented, very little is know about assembly of the protein–DNA complex or changes in the relationship between structure and function as the reaction progresses. For Tn10, the higher order complex, or transpososome, is remarkably versatile and accommodates several different configurations of the DNA during synapsis of the ends, cleavage, flanking DNA release, target capture and strand transfer. However, one of the most interesting and difficult to understand aspects of Tn10 transposition is how these functional steps are correlated with the different configurations of the protein and DNA as the reaction progresses.
The protein–DNA contacts of several intermediates in the Tn10 transposition reaction were previously determined by hydroxyl radical footprinting. To display the protection pattern, a molecular model of the IHF-folded arm of Tn10 was developed by combining the structure of the Tn5 transpososome with that for the IHF co-crystal (Fig. 1A) (18). The 180° bend in the DNA imposed by IHF provides a set of ‘subterminal’ transposase contacts, located distal to the IHF binding site. The contacts in the model fit well with the hydroxyl radical protection pattern and together with several other lines of evidence support the model as an accurate representation of the general shape of the Tn10 transpososome.
Figure 1.

Molecular model of the IHF-folded Tn10 transpososome. (A) The Tn10 transpososome was modeled by superimposing the DNA from the IHF co-crystal structure on the structure of the Tn5 transpososome (7,43). Superimposition of the IHF-folded DNA was achieved by minimizing the RMS difference in the position of the equivalent atoms in the Tn5 DNA (18). One transposon end, IHF and flanking DNA have been omitted for clarity. Regions of transposase and IHF mediated hydroxyl radical protection are shown in red and green, respectively (18). Every tenth nucleotide on the transferred strand is shown in white. The transposon end is seen embedded in the turquoise monomer of transposase. The subterminal transposase contacts are located on the top of the structure illustrated on the left. Note that the IHF-folded arm of the transposon contacts both transposase monomers. (B) A slight adjustment of the geometry between transposase and the transposon end is sufficient to make the IHF-folded transposon arm pass across only one of the transposase monomers. (C) A space filling representation of the structure of the Tn5 transpososome (43). The putative target binding groove is illustrated running vertically in the plane of the page. The 3′-OH groups at the end of the transposon are colored yellow and can be seen at the bottom of the groove. (D) A section of B DNA (gold) was docked in the putative target binding groove of the model in (A).
The molecular model for Tn10 clearly illustrates the spatial relationship between the terminal and subterminal transposase contacts. It also highlights a fascinating congruence between the structural role of IHF in Tn10 transposition and phage λ excision (18). The 180° bend imposed by IHF allows a single molecule of phage λ integrase to bind simultaneously to the C′ and P′1 sites in attL (7). In Tn10 and phage λ, recombination is initiated by a nick at the transposon end and in the C′ site, respectively. In both systems, this position is exactly 30 bases 3′ to the start of the IHF consensus. Furthermore, the Tn10 subterminal contacts and the λ integrase P′1 contacts are exactly the same distance from the recombination sites. The precise congruence of the three subsites in Tn10 and phage λ is not due to descent from a common ancestor. Rather it suggests that the involvement of IHF imposes powerful structural and functional constraints on recombination reactions. Thus, insights into the function of IHF in one system may be directly applicable to many others.
From the molecular model in Figure 1A, it is easy to visualize why Tn10 transposase would require a contribution from the energy of IHF binding or negative supercoiling to bend the DNA and establish the subterminal contacts. These contacts are required to activate the chemical steps during the cleavage reaction and must be disengaged to allow target interactions later in the reaction (18,19). In this paper, we investigate the symmetry of the IHF-folded arms of the transposon during transpososome assembly, and the properties of the conformational change leading to disengagement of the subterminal contacts and ejection of IHF from the complex. We find that the transpososome is fundamentally asymmetric and can accommodate only one fully folded transposon arm. Furthermore, the IHF on the fully folded arm is locked into the complex, probably by the subterminal contacts, until ejected by a conformational change as metal ion enters into the reaction. This is important because it shows that unfolding of a single transposon arm activates the cleavage step of the reaction at each transposon end and reflects mechanistic coupling between both sides of the complex. We also test and confirm the prediction of the molecular model that negative regulation of transposition, by the IHF-dependent inhibition of target interactions, is the result of simple steric hindrance of the target binding groove by a single IHF-folded arm of the transposon.
MATERIALS AND METHODS
Materials and reagents were generally of the best quality available. Most chemicals were from Sigma. Enzymes were from New England Biolabs or Boehringer–Roche. Molecular models were created on a Crystal Graphics workstation using InsightII software. The structures for the Tn5 transpososome and the IHF co-crystal were from PDB database files 1MUH and 1IHF, respectively.
DNA, proteins and assembly of complexes
Transposase was purified according to (20). Transposon end fragments were purified by electrophoresis in polyacrylamide gels (PAG) and recovered by the crush and soak method as described in (21). Transpososomes were assembled and purified using gel shift assays as described in (18,21). IHF was purified as described in (22).
The transposon end fragments were prepared by digesting pRC98, pRC100, pRC351 and pPKC4. pRC98 is the BglII–SalI fragment from pNK1935, containing an 87 bp IS10 outside end and 50 bp of flanking DNA, cloned into BamHI–SalI digested pBluescript. pRC100 contains the even-end fragment and was described previously (18). pRC351 is identical to pRC98 but with a XhoI site introduced by PCR at bp+51 of the transposon end. pPKC4 contains the 70 bp PvuII–BamHI fragment from pNK3287 cloned in pUC19 HincII–BamHI. Details of the restriction digests used to generate particular fragments are given in the figure legends. Where indicated, calcium and heparin were added to 4 mM and 250 ng/ml, respectively. Competitor DNA was 1 µg of pBluescript per 20 µl reaction.
Hydroxyl radical footprinting
DNA footprints were generated by hydroxyl radical treatment of protein–DNA complexes which were subsequently purified in the mobility shift assay (18). The resultant ladders were compared to Maxam-Gilbert G+A sequence ladders and plotted using NIH Image software.
RESULTS
Molecular models of the Tn10 transpososome
In the model for the Tn10 transpososome in Figure 1A, the path of the transposon arm across the dimer interface means that only one transposon end can engage the subterminal contacts. The second arm will still bind IHF but steric hindrance would prevent it engaging the same set of subterminal contacts. If this is true, it would mean that the Tn10 transpososome is fundamentally asymmetric despite having identical components on either side of the complex. However, as shown in Figure 1B, a small change in the geometry between transposase and the transposon end would accommodate two fully folded transposon arms, one on either side of the dimer interface.
The crystal structure of the Tn5 transpososome has a deep groove which is the correct size to accommodate the target DNA (Fig. 1C). Figure 1D shows a molecular model of Tn10 in which target DNA has been docked in this groove. The model immediately suggests a mechanism to explain the inhibition of target interactions by IHF. The IHF-folded transposon arm simply occludes the target binding groove as it passes over the top at an angle of almost 90°. An alternative mechanism would be that direct interaction between IHF and transposase produces a conformational change in transposase that precludes target interactions. Limited support for a direct protein–protein interaction is provided by the observation that IHF has a higher affinity for the IHF binding site in the Tn10 transpososome than for the free transposon end (19).
Quantitative footprinting of the subterminal transposase contacts
To address the problem of whether one or both transposon arms are able to engage the subterminal transposase contacts, the hydroxyl radical footprints for a Tn10 paired ends complex (PEC) and an IS10 PEC were compared (Fig. 2). Tn10 PECs are the standard experimental system and are assembled with two outside ends, both of which have an IHF binding site (21). IS10 PECs have one outside end and an inside end which lacks an IHF binding site. One technical problem with this experiment is that the inside end of IS10 is naturally distorted and has a low affinity for IHF which confuses interpretation of the results. To circumvent this problem a new transposon end was constructed in which the DNA between bp+19 and +47 was replaced by a tandem repeat of the bases 5′-CTGA. This is referred to as an ‘even-end’ because of the evenness of the hydroxyl radical footprint in the presence of a high concentration of IHF (18). The even-end fails to form PEC on its own because it lacks an IHF binding site. However, it is efficiently recruited into a mixed PEC when the assembly reaction is supplemented with an outside end fragment. In addition to the mixed PEC, such a reaction will of course produce a standard outside end PEC. However, provided that the outside end and even end are of different sizes, the different PECs can be resolved in the gel mobility shift assay.
Figure 2.

Quantitative hydroxyl radical footprinting of IS10 and Tn10 PECs. Hydroxyl radical footprints of the transferred strand of the indicated complexes were performed as described in Materials and Methods. The footprints were resolved on a DNA sequencing gel, recorded on a phosphoimager and the traces were generated using NIH Image software. The long arrow indicates the position of the transposon end with the position in bp above. Light and dark rectangles indicate IHF and transposase contacts, respectively. The transposon ends used to form the complexes are illustrated. The asterisk indicates the location of the radioactive label. The arrowhead is the end of the transposon and the length of the transposon arm and the flanking DNA are indicated. The OE and EE fragments were generated from pRC98 and pRC100, respectively. OE, outside end; EE, even-end; T’ase, transposase.
Tn10 and IS10 PECs were assembled and the hydroxyl radical protection pattern was determined (Fig. 2). To ensure a valid comparison, the footprints were performed in a single experiment and displayed on adjacent lanes of a sequencing gel. Furthermore, the strength of the signals was carefully normalized to the amplitude of the transposase footprint between bp+10 and –2 which should not be affected by IHF binding to the transposon arm. The footprint for the Tn10 PEC has sets of IHF and transposase contacts, arrayed approximately in phase with the 10 bp helical repeat of DNA (Fig. 2, top panel). The footprint of the IS10 PEC is very similar, but the subterminal transposase contacts between bp+48 and +75 are twice as strong (middle panel). This is the result expected if only one transposon arm is engaged with the subterminal contacts. The signal from the Tn10 PEC is diluted by the second outside end which does not engage the subterminal contacts. Finally, as expected, the footprint of the even-end in the IS10 PEC had no signal whatsoever from IHF or the subterminal transposase contacts. Taken together, these results support the model in Figure 1A which suggests that although both transposon arms may be able to bind IHF, only one can engage the subterminal contacts.
Release of IHF from the PEC requires divalent metal ion
Tn10 transposition depends on the presence of the catalytic metal ion Mg2+. Ca2+ appears immediately below Mg2+ in the periodic table and can replace the catalytic metal ion in some reactions such as the strand transfer steps in phage Mu and V(D)J transposition (23,24). However, it will not support any of the catalytic steps in Tn10 transposition. Nevertheless, Ca2+ does function as an analog of the catalytic metal ion in some aspects of Tn10 transposition: (i) it is required to stabilize target interactions in the absence of Mg2+ (25); (ii) in common with phage Mu and V(D)J recombination, it stabilizes the transpososome per se (23,25,26); (iii) Ca2+ produces a conformational change in the Tn10 PEC that renders two nucleotides on the transferred strand of the transposon hyperreactive to hydroxyl radicals (18). The position of the hyperreactive nucleotides, on either side of the scissile phosphate, suggests that there has been a conformational change that distorts the DNA at the transposon end in preparation for cleavage.
In the presence of Ca2+ ions, transposase assembles a bottom-PEC (bPEC) which contains two IHF-bound transposon ends and a dimer of transposase (6). Treatment with competitor DNA or heparin removes IHF and produces the top-PEC (tPEC) which is retarded in the gel mobility assay because the unfolded transposon arms have an extended structure (18,19). Together with cleavage of the flanking DNA, IHF release is a prerequisite for interaction with target later in the reaction (19,25).
When bPEC is prepared in the absence of divalent metal ion, IHF binding is resistant to competitor DNA or heparin (Fig. 3A). Treatment with heparin or competitor DNA strips IHF from the IHF-shifted DNA fragment, but the bPEC maintains its position in the gel indicating that IHF remains associated (lanes 3 and 4). Dissociation of IHF in the presence of competitor requires the addition of 4 mM Ca2+, an analog of Mg2+, the catalytic metal ion (lane 5). It therefore appears that during assembly of the bPEC, IHF is somehow locked onto its binding site and that the trigger for release or expulsion from the complex is the entry of the divalent metal ion into the reaction.
Figure 3.

Asymmetric divalent metal ion-dependent unfolding of the PEC. PEC was assembled and visualized in a mobility shift assay using a 5% PAG as previously described (18,21). Autoradiograms are shown. The transposon ends are shown below each panel. These were generated by the indicated restriction digestion of pRC98 in part (A) and pPKC4 in part (B). The length of the transposon and flanking DNA sequences are shown to the left and right, respectively, of the transposon end (arrowhead). The location of the 32P label is indicated by an asterisk. The concentration of IHF is indicated above the autoradiogram. The concentrations of other additions are given in Materials and Methods. tPEC, top-PEC; ffbPEC, fully-folded bottom-PEC; sfbPEC, semi-folded bottom-PEC; IHF, IHF-bound outside end; free DNA, unbound outside end; ‡, uncharacterized species believed to contain two tPECs that are weakly associated (44).
Two forms of the bPEC can be distinguished at low IHF concentrations
When the PEC assembly reaction is titrated with IHF in the presence of Ca2+, bPEC and tPEC are both produced (19). However, if the bPEC has two IHF-folded arms, and the tPEC is completely unfolded, then an intermediate form of the complex with one IHF-folded transposon arm is also expected. This species has not been detected in previous experiments.
In the PEC, transposase holds the two transposon ends in a pseudo four-way junction from which the transposon ends and the flanking DNA emerge at unknown angles. Since the mobility of protein–DNA complexes is dominated by the shape of the DNA, we reasoned that the intermediate complex with one IHF-folded arm might be resolved from bPEC or tPEC by changing the length of the flanking DNA segments. Indeed, when the flanking DNA was reduced to 42 bp or less, the intermediate form of the complex with one arm folded is detected migrating slightly faster than the fully folded bPEC. Further evidence for the identity of these complexes is described below, but henceforth they will be referred to as the ‘fully-folded bottom PEC’ (ffbPEC) when both arms are folded and the ‘semi-folded bottom PEC’ (sfbPEC) when only one arm is folded.
An IHF titration of PEC assembly was performed using a transposon end with 22 bp of flanking DNA (Fig. 3B). At low IHF concentration, when most of the DNA remains unshifted by IHF, a small amount of sfbPEC is produced (lane 6). At intermediate IHF concentrations, both forms of the bPEC are present (lanes 7 and 8), whilst at the highest IHF concentration, only ffbPEC is produced (lane 9). Finally, heparin treatment strips IHF from the IHF-shifted DNA fragment and ffbPEC is converted to sfbPEC (lane 10). Note that no tPEC is produced in this experiment because IHF remains locked onto at least one of the two binding sites in the absence of divalent metal ion.
Quantitative hydroxyl radical footprinting was used to estimate the fractional occupancy of the IHF sites in the various PECs (Fig. 4A). The results support our interpretation of the unfolding experiments: the tPEC (no IHF) has no protection, the ffbPEC (2 × IHF) has the strongest protection and the sfbPEC (1 × IHF) has an intermediate level of protection.
Figure 4.

Quantitative footprinting and mobility assay of the ffbPEC and the sfbPEC. (A) Hydroxyl radical footprints of the indicated complexes were performed as described in Materials and Methods. Fractional occupancy of the IHF site can be estimated from the strength of the footprint between bp+33 and +50 because this region is free of non-specific perturbations. (B) A mobility shift assay of the indicated complexes was performed as in Figure 3. An autoradiogram is shown. Abbreviations are as in Figures 2 and 3.
If our interpretation of the mobility shift data is correct, it means that the ffbPEC and the sfbPEC co-migrate in the gel, explaining why the complexes have not been detected previously. To prove this interpretation is correct, we assembled a sfbPEC directly by mixing an outside end with the even-end which lacks an IHF binding site. The even-end fails to form PEC on its own, but is recruited into a mixed PEC when an outside end partner is provided. Hence, if the radioactive label is present only on the even-end fragment, only the mixed PEC will be detected.
Isogenic outside end and even-end DNA fragments were prepared and used to form PEC (Fig. 4B). These fragments have identical flanking DNA and transposon sequences up to bp+19. However, the DNA between bp+19 and +47 of the even-end was replaced by a tandem repeat of the bases 5′-CTGA, as described above. As expected, both fragments have the same mobility when uncomplexed with proteins and the even-end does not change position in the presence of IHF (compare lanes 1 and 3). Crucially, the mixed PEC, incorporating the unfolded even-end, has exactly the same mobility as the standard bPEC with two outside ends (compare lanes 2 and 3). This shows unequivocally that ffbPEC and sfbPEC co-migrate in the standard gel mobility assay. Taken together, these results further confirm the fundamental asymmetry of the PEC.
Target interactions in the presence of IHF
Target interactions in Tn10 require cleavage of the flanking DNA and dissociation of IHF (19,25). One mechanism to explain the requirement for IHF release is that the folded arm of the transposon occludes the target binding site. If this is true, the model in Figure 1D predicts that a transposon end less than about 60 bp in length should allow target interactions in the presence of IHF.
An insertion assay was performed with transposon ends of 94 or 56 bp and pBluescript as a target (Fig. 5). PECs were assembled in the presence of IHF and then supplemented with Mg2+ and target. Mg2+ initiates cleavage of the transposon ends and the target plasmid acts as a competitor to titrate the IHF from the transposon ends. Both of these events are required for the strand transfer step later in the reaction. Insertion of the transposon ends into the closed circular target yields a linear product slightly larger than the target plasmid (Fig. 5). When additional IHF is titrated into the reaction, insertion of the 94 bp transposon end into the target is abolished. In contrast, insertion of the 56 bp transposon end is not affected by the presence of IHF. This result is in perfect agreement with the prediction of the molecular model. IHF therefore inhibits target interactions by a simple steric hindrance mechanism, and not by promoting a conformational change in transposase as mentioned as an alternative model above.
Figure 5.
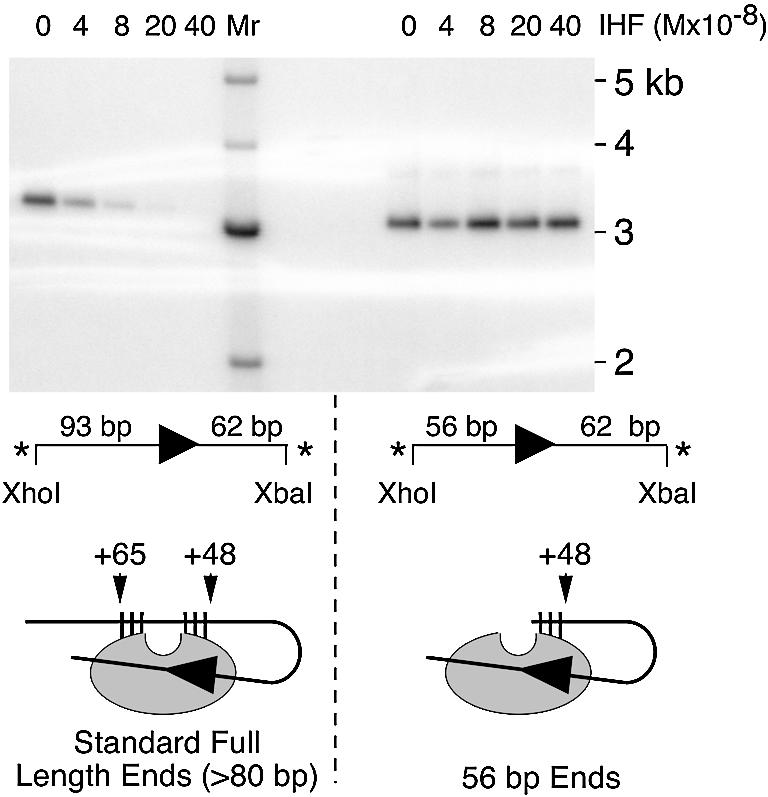
A short transposon end allows target interaction in the presence of IHF. PECs were assembled in the standard buffer which contains only enough IHF to bind the linear transposon end fragments (Materials and Methods). After 1 h, the reaction was supplemented with 4 mM MgCl2, 1 µg pBluescript as target and the indicated additional amounts of IHF. After 80 min incubation at 37°C, the reactions were extracted with phenol and run on a 1.1% agarose/TAE gel at 65 V overnight. The gel was dried and recorded on a phosphoimager. The linear insertion product is slightly larger than the 3 kb target plasmid. Molecular weight markers (Mr) were end labeled by treatment with polynucleotide kinase. The transposon ends, shown below each panel, were generated by the indicated restriction digestion of pRC98 (left panel) and pRC351 (right panel). Other symbols and abbreviations are as in Figures 2 and 3. Below the gel is an illustration of the two different transposon ends as they would appear in the molecular model. In this schematic representation, the target binding groove is at the top as illustrated in the molecular model on the left of Figure 1C. The large arrowhead is at the junction between the transposon and the flanking DNA. The vertical ticks are the subterminal transposase contacts determined by hydroxyl radical footprinting. The 56 bp transposon ends lack all of the contacts distal to the target binding groove.
DISCUSSION
The locked status of the IHF site imposes asymmetry and defines α and β sides of the PEC
Quantitative hydroxyl radical footprinting of IS10 and Tn10 PECs showed that only one transposon arm can engage the subterminal transposase contacts (Fig. 2). Together with the unfolding experiments (Fig. 3), these results support a model for the IHF-dependent assembly and unfolding of an asymmetric Tn10 transpososome (Fig. 6). The model shows how treatment of the ffbPEC with heparin produced sfbPEC by dissociating one bound IHF. The second IHF remains locked in position until released by divalent metal ion. The different behavior of the IHF sites introduces a structural and functional asymmetry in the complex. Here we define the locked IHF binding site as the α side of the complex. The opposite IHF binding site, which can be dissociated in the absence of divalent metal ion, is defined as the β side of the complex (Fig. 6).
Figure 6.

Model for the metal-ion dependent unfolding of the asymmetric Tn10 transpososome. IHF binds specifically to the outside end of IS10 and activates transpososome assembly by transposase (21). Synapsis of two IHF-folded transposon ends produces the ffbPEC. The Tn10 and Tn5 transpososomes contain a dimer of transposase (43,45). The relative geometry of the transposon ends are not known for Tn10 but are likely to be antiparallel as shown here and in the crystal structure of the related Tn5 transpososome. In the absence of divalent metal ion, competitor DNA or heparin strips the IHF from one of the transposon arms to produce the sfbPEC. IHF bound to the second transposon arm remains locked into the sfbPEC. Since IHF freely associates and dissociates from DNA in the presence or absence of metal ion, this behavior is dictated by the transpososome and is probably mediated by the subterminal transposase contacts on one transposon arm. Addition of metal ion unlocks the remaining IHF which dissociates in the presence of competitor DNA or heparin to produce the tPEC. The asymmetric behavior defines two sides of the PEC. The α side of the complex is defined as that which requires divalent metal ion to unlock the IHF binding site. IHF can dissociate from the β side in the absence of divalent metal ion. Arrowhead, transposon end; hatched oval, IHF; white oval, transposase; ffbPEC, fully folded bottom PEC; sfbPEC, semi folded bottom PEC; tPEC, top PEC; Me++, divalent metal ion.
Divalent metal ion-induced conformational changes
Release of the α IHF from the synaptic complex by divalent metal ion suggests that a conformational change has taken place in transposase. The conformational change is necessary to release IHF from the complex and also to activate later steps in the reaction. If the complex is deprived of the subterminal contacts by using a truncated transposon end, some intermolecular transposition is achieved (Fig. 5), but only about 50% of the complexes complete the cleavage step of the reaction (18). The phenotype of truncated arm complexes can be fully complemented with a single full-length transposon arm that can fold and unfold normally (to be presented elsewhere). Thus, it appears that unfolding of the α transposon arm promotes a conformational change that is coupled between each side of the complex and is required for efficient cleavage.
Previously, Ca2+ ions were shown to promote a conformational change that rendered bp +1 and –1 of the transferred stand of the transposon end hyperreactive to hydroxyl radicals (18). This change is likely to be important to activate catalysis as the hyperreactivity is located on either side of the first phosphodiester bond to be cleaved in the reaction. These observations raise the question of whether the PEC prepared in the absence of divalent metal ion has any biological significance. However, there are several good arguments that PEC prepared without metal ion is a natural intermediate of the transposition reaction (18). In the transition state model proposed for the two metal ion catalytic mechanism used by Tn10, the metal ions have as many points of coordination with the phosphate oxygens on the DNA as with the DDE residues (27–29). It is therefore highly unlikely that both metal ions can be properly located in the active site before the substrate. Thus, although metal ion is always present in vivo, there will be discrete points during PEC assembly before which it cannot exert its effects because the structures on which it acts have not been established.
Divalent metal ions cause conformational changes in other transposition systems as well. Many transpososomes, such as in phage Mu and V(D)J recombination, are stabilized by Ca2+ (23,26). There is also a conformational change in the DNA near the end of phage Mu in response to divalent metal ion (30). This was detected by an increase in 2-aminopurine fluorescence which indicates the loss of base stacking in the DNA. Since metal ions do not affect 2-aminopurine fluorescence in uncomplexed DNA, the divalent metal ion must induce the conformational change in the transposase.
In HIV integrase, divalent metal ions have several profound effects. Recognition of the viral ends is promoted by catalytic metal ions (31). Metal ions also induce two separate conformational changes in HIV integrase per se (32–34). The first is due to metal ion binding to the active site, but the second is due to metal ion interacting with the small N-terminal domain. Interestingly, in the related ASV integrase, divalent metal ions do not induce a conformational change. It was suggested that divalent metal ions are needed to stabilize a structural conformation of HIV that is native to the ASV integrase. This is reminiscent of the way in which IHF and negative supercoiling are required for Tn10 transposition but not for Tn5. Perhaps these factors are required to promote a conformational change in Tn10 transposase that is achieved directly by the larger Tn5 transposase.
The mechanism of IHF activation
Tn10 transposase belongs to a superfamily of proteins that includes all of the classical DNA transposons, HIV integrase and the V(D)J recombinases Rag1/2. IHF is not absolutely essential for Tn10 transposition but stimulates assembly of the PEC about 200-fold in the absence of negative supercoiling. Likewise, HMG proteins activate the cleavage step of V(D)J recombination and probably promote synapsis, particularly for the physiologically relevant pairing of the 12/23 RSS (4). Thus, Tn10 and V(D)J recombinases both bind in the major groove of DNA and utilize as cofactors specialized proteins that bend DNA by interactions with the minor groove. Despite the huge evolutionary distance between these proteins, HMG proteins can substitute for IHF and the related protein HU in phage λ recombination and the HIN invertasome (35,36). Even so, some functional constraints remain because although IHF and HU promote synapsis in V(D)J, they do not support catalysis.
In many of the systems in which IHF and other DNA bending proteins act as accessory proteins the DNA helix is melted or underwound. Examples include initiation of transcription and DNA replication, and the processing of hairpin intermediates in transposition and V(D)J recombination. The precise mechanism of IHF action in these systems is not known, but IHF is unlikely to promote DNA unwinding directly as distortions cannot be transmitted more than a few base pairs through the helix.
More than 150 E.coli promoters are up- or down-regulated by IHF (10). IHF stimulates transcription via DNA loops that recruit a regulator bound to a distant site or loops that promote interaction of RNA polymerase with an upstream activating sequence (37,38). IHF also appears capable of activating transcription by partitioning the energy of supercoiling in different DNA domains (39). In Tn10, an IHF loop is required to establish the subterminal transposase contacts (Figs 1 and 6). Furthermore, there are close parallels between the role of IHF in Tn10 transposition and the events required for open complex formation at the tyrT promoter in E.coli (40). The tyrT promoter has a binding site for the DNA bending protein FIS (factor for inversion stimulation). Time resolved UV laser footprinting of tyrT shows that FIS protein is part of the higher order complex during the first 35 ms of assembly. FIS next completes a cycle of dissociation and reassociation before the isomerization event melts the DNA to produce open complex (40). This is analogous to the cycle of IHF association and dissociation in Tn10 transposition. Transient DNA looping is therefore a recurrent theme in DNA metabolism. The looping events generally appear to have kinetic effects, accelerating the reaction by activating a rate limiting step.
IHF inhibition of target interactions
It was reported previously that IHF has a higher affinity for the transposon end in a tPEC than when it is free in solution (19). This raised the possibility of a direct interaction between IHF and transposase that could potentially alter the conformation of the target binding groove. However, co-migration of the ffbPEC and sfbPEC suggests that the apparent high affinity of IHF for the tPEC is a simple consequence of the fact that the tPEC has two IHF binding sites, but that the free transposon end has only one.
In vivo, IHF appears to regulate the rate of transposition both positively and negatively, depending on the length of the transposon and whether it is located on the chromosome or a multi-copy plasmid (41). These properties probably reflect a mechanism to couple the rate of transposition to the physiology of the cell and to limit the accumulation of transposons in vivo (19,41,42). In vitro, low concentrations of IHF positively regulate transposition by stimulating assembly of the synaptic complex. In contrast, high IHF concentrations negatively regulate transposition by inhibiting intermolecular target interactions and channeling insertions to an intramolecular target site only 120 bp from the transposon end (42). The molecular model of the transpososome (Fig. 1A) suggests that the mechanism responsible for inhibiting target interactions is the occlusion of the target binding groove by the IHF-folded transposon arm. The predictions of the model were confirmed by intermolecular transposition of the 56 bp transposon ends in the presence of a saturating concentration of IHF (Fig. 5). However, the channeling of insertions into sites close to the transposon end in vitro remains to be explained.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank: Phoebe Rice for rendering the space filling model of the Tn5 transpososome in Figure 1C; Greg Van Duyne for first pointing out that the crystal packing in the Tn5 transpososome structure provided evidence for the location of the target binding groove and Mick Chandler for suggesting inclusion of the explanatory diagram at the bottom of Figure 5. This work was funded by The Wellcome Trust grant 065119 to R.C. R.C. is a Royal Society University Research Fellow.
REFERENCES
- 1.Katinka M.D., Duprat,S., Cornillot,E., Metenier,G., Thomarat,F., Prensier,G., Barbe,V., Peyretaillade,E., Brottier,P., Wincker,P. et al. (2001) Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature, 414, 450–453. [DOI] [PubMed] [Google Scholar]
- 2.Buisine N., Tang,C.M. and Chalmers,R. (2002) Transposon-like Correia elements: structure, distribution and genetic exchange between pathogenic Neisseria sp. FEBS Lett., 522, 52–58. [DOI] [PubMed] [Google Scholar]
- 3.Rice P.A. and Baker,T.A. (2001) Comparative architecture of transposase and integrase complexes. Nature Struct. Biol., 8, 302–307. [DOI] [PubMed] [Google Scholar]
- 4.Gellert M. (2002) V(D)J recombination: rag proteins, repair factors and regulation. Annu. Rev. Biochem., 71, 101–132. [DOI] [PubMed] [Google Scholar]
- 5.Chalmers R., Sewitz,S., Lipkow,K. and Crellin,P. (2000) Complete nucleotide sequence of Tn10. J. Bacteriol., 182, 2970–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haniford D.B. (2002) Transposon Tn10. In Craig,N.L., Craigie,R., Gellert,M. and Lambowitz,A.M. (eds), Mobile DNA II. American Society for Microbiology, Washington, DC, USA, pp. 457–483.
- 7.Rice P.A., Yang,S., Mizuuchi,K. and Nash,H.A. (1996) Crystal structure of an IHF-DNA complex: a protein-induced DNA U-turn. Cell, 87, 1295–1306. [DOI] [PubMed] [Google Scholar]
- 8.Nash H.A. (1996) The E. coli HU and IHF proteins: accessory factors for complex protein–DNA assemblies. In Lin,C.C. and Lynch,A.S. (eds), Regulation of Gene Expression in Escherichia coli. Landes Co., Austin, TX, USA, pp. 149–166. [Google Scholar]
- 9.Friedman D.I. (1988) Integration host factor: a protein for all reasons. Cell, 55, 545–554. [DOI] [PubMed] [Google Scholar]
- 10.Arfin S.M., Long,A.D., Ito,E.T., Tolleri,L., Riehle,M.M., Paegle,E.S. and Hatfield,G.W. (2000) Global gene expression profiling in Escherichia coli K12. The effects of integration host factor. J. Biol. Chem., 275, 29672–29684. [DOI] [PubMed] [Google Scholar]
- 11.Nash H.A. and Granston,A.E. (1991) Similarity between the DNA-binding domains of IHF protein and TFIID protein [letter]. Cell, 67, 1037–1038. [DOI] [PubMed] [Google Scholar]
- 12.Borca M.V., Irusta,P.M., Kutish,G.F., Carillo,C., Afonso,C.L., Burrage,A.T., Neilan,J.G. and Rock,D.L. (1996) A structural DNA binding protein of African swine fever virus with similarity to bacterial histone-like proteins. Arch. Virol., 141, 301–313. [DOI] [PubMed] [Google Scholar]
- 13.Azaro M.A. and Landy,A. (2002) Lambda integrase and the lambda Int family. In Craig,N.L., Craigie,R., Gellert,M. and Lambowitz,A.M. (eds), Mobile DNA II. American Society for Microbiology, Washington, DC, USA, pp. 118–148. [Google Scholar]
- 14.Nash H.A. and Robertson,C.A. (1981) Purification and properties of the Escherichia coli protein factor required for lambda integrative recombination. J. Biol. Chem., 256, 9246–9253. [PubMed] [Google Scholar]
- 15.Goryshin I.Y. and Reznikoff,W.S. (1998) Tn5 in vitro transposition. J. Biol. Chem., 273, 7367–7374. [DOI] [PubMed] [Google Scholar]
- 16.Surette M.G., Lavoie,B.D. and Chaconas,G. (1989) Action at a distance in Mu DNA transposition: an enhancer-like element is the site of action of supercoiling relief activity by integration host factor (IHF). EMBO J., 8, 3483–3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Surette M.G. and Chaconas,G. (1989) A protein factor which reduces the negative supercoiling requirement in the Mu DNA strand transfer reaction is Escherichia coli integration host factor. J. Biol. Chem., 264, 3028–3034. [PubMed] [Google Scholar]
- 18.Crellin P. and Chalmers,R. (2001) Protein–DNA contacts and conformational changes in the Tn10 transpososome during assembly and activation for cleavage. EMBO J., 20, 3882–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakai J.S., Kleckner,N., Yang,X. and Guhathakurta,A. (2000) Tn10 transpososome assembly involves a folded intermediate that must be unfolded for target capture and strand transfer. EMBO J., 19, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chalmers R.M. and Kleckner,N. (1994) Tn10/IS10 transposase purification, activation and in vitro reaction. J. Biol. Chem., 269, 8029–8035. [PubMed] [Google Scholar]
- 21.Sakai J., Chalmers,R.M. and Kleckner,N. (1995) Identification and characterization of a pre-cleavage synaptic complex that is an early intermediate in Tn10 transposition. EMBO J., 14, 4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lynch T.W., Read,E.K., Mattis,A.N., Gardner,J.F. and Rice,P.A. (2003) Integration host factor: putting a twist on protein–DNA recognition. J. Mol. Biol., 330, 493–502. [DOI] [PubMed] [Google Scholar]
- 23.Savilahti H., Rice,P.A. and Mizuuchi,K. (1995) The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J., 14, 4893–4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiom K., Melek,M. and Gellert,M. (1998) DNA transposition by the RAG1 and RAG2 proteins: a possible source of oncogenic translocations. Cell, 94, 463–470. [DOI] [PubMed] [Google Scholar]
- 25.Sakai J. and Kleckner,N. (1997) The Tn10 synaptic complex can capture a target DNA only after transposon excision. Cell, 89, 205–214. [DOI] [PubMed] [Google Scholar]
- 26.Hiom K. and Gellert,M. (1998) Assembly of a 12/23 paired signal complex: a critical control point in V(D)J recombination. Mol. Cell, 1, 1011–1019. [DOI] [PubMed] [Google Scholar]
- 27.Beese L.S. and Steitz,T.A. (1991) Structural basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J., 10, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joyce C.M. and Steitz,T.A. (1995) Polymerase structures and function: variations on a theme? J. Bacteriol., 177, 6321–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy A.K., Haniford,D.B. and Mizuuchi,K. (2000) Single active site catalysis of the successive phosphoryl transfer steps by DNA transposases: insights from phosphorothioate stereoselectivity. Cell, 101, 295–305. [DOI] [PubMed] [Google Scholar]
- 30.Yanagihara K. and Mizuuchi,K. (2003) Progressive structural transitions within Mu transpositional complexes. Mol. Cell, 11, 215–224. [DOI] [PubMed] [Google Scholar]
- 31.Yi J., Asante-Appiah,E. and Skalka,A.M. (1999) Divalent cations stimulate preferential recognition of a viral DNA end by HIV-1 integrase. Biochemistry, 38, 8458–8468. [DOI] [PubMed] [Google Scholar]
- 32.Asante-Appiah E. and Skalka,A.M. (1997) A metal-induced conformational change and activation of HIV-1 integrase. J. Biol. Chem., 272, 16196–16205. [DOI] [PubMed] [Google Scholar]
- 33.Asante-Appiah E., Seeholzer,S.H. and Skalka,A.M. (1998) Structural determinants of metal-induced conformational changes in HIV-1 integrase. J. Biol. Chem., 273, 35078–35087. [DOI] [PubMed] [Google Scholar]
- 34.Asante-Appiah E. and Skalka,A.M. (1999) HIV-1 integrase: structural organization, conformational changes and catalysis. Adv. Virus Res., 52, 351–369. [DOI] [PubMed] [Google Scholar]
- 35.Paull T.T., Haykinson,M.J. and Johnson,R.C. (1993) The nonspecific DNA-binding and -bending proteins HMG1 and HMG2 promote the assembly of complex nucleoprotein structures. Genes Dev., 7, 1521–1534. [DOI] [PubMed] [Google Scholar]
- 36.Segall A.M., Goodman,S.D. and Nash,H.A. (1994) Architectural elements in nucleoprotein complexes: interchangeability of specific and non-specific DNA binding proteins. EMBO J., 13, 4536–4548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dworkin J., Ninfa,A.J. and Model,P. (1998) A protein-induced DNA bend increases the specificity of a prokaryotic enhancer-binding protein. Genes Dev., 12, 894–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goosen N. and van de Putte,P. (1995) The regulation of transcription initiation by integration host factor. Mol. Microbiol., 16, 1–7. [DOI] [PubMed] [Google Scholar]
- 39.Sheridan S.D., Benham,C.J. and Hatfield,G.W. (1998) Activation of gene expression by a novel DNA structural transmission mechanism that requires supercoiling-induced DNA duplex destabilization in an upstream activating sequence. J. Biol. Chem., 273, 21298–21308. [DOI] [PubMed] [Google Scholar]
- 40.Pemberton I.K., Muskhelishvili,G., Travers,A.A. and Buckle,M. (2002) FIS modulates the kinetics of successive interactions of RNA polymerase with the core and upstream regions of the tyrT promoter. J. Mol. Biol., 318, 651–663. [DOI] [PubMed] [Google Scholar]
- 41.Signon L. and Kleckner,N. (1995) Negative and positive regulation of Tn10/IS10-promoted recombination by IHF: two distinguishable processes inhibit transposition off of multicopy plasmid replicons and activate chromosomal events that favor evolution of new transposons. Genes Dev., 9, 1123–1136. [DOI] [PubMed] [Google Scholar]
- 42.Chalmers R., Guhathakurta,A., Benjamin,H. and Kleckner,N. (1998) IHF modulation of Tn10 transposition: sensory transduction of supercoiling status via a proposed protein/DNA molecular spring. Cell, 93, 897–908. [DOI] [PubMed] [Google Scholar]
- 43.Davies D.R., Goryshin,I.Y., Reznikoff,W.S. and Rayment,I. (2000) Three-dimensional structure of the Tn5 synaptic complex transposition intermediate [see comments]. Science, 289, 77–85. [DOI] [PubMed] [Google Scholar]
- 44.Stewart B.J., Wardle,S.J. and Haniford,D.B. (2002) IHF-independent assembly of the Tn10 strand transfer transpososome: implications for inhibition of disintegration. EMBO J., 21, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kennedy A.K. (1999) Mechanistic aspects of Tn10 transposition. PhD Thesis, University of Western Ontario, London, Ontario, Canada. [Google Scholar]