

Abstract
α-L-LNA (α-L-ribo configured locked nucleic acid) is a nucleotide analogue that raises the thermostability of nucleic acid duplexes by up to ∼4°C per inclusion. We have determined the NMR structure of a nonamer α-L-LNA:RNA hybrid with three α-L-LNA modifications. The geometry of this hybrid is intermediate between A- and B-type, all nucleobases partake in Watson–Crick base pairing and base stacking, and the global structure is very similar to that of the corresponding unmodified hybrid. The sugar–phosphate backbone is rearranged in the vicinity of the modified nucleotides. As a consequence, the phosphate groups following the modified nucleotides are rotated into the minor groove. It is interesting that the α-L-LNA:RNA hybrid, which has an elevation in melting temperature of 17°C relative to the corresponding DNA:RNA hybrid, retains the global structure of this hybrid. To our knowledge, this is the first example of such a substantial increase in melting temperature of a nucleic acid analogue that does not act as an N-type (RNA) mimic. α-L-LNA:RNA hybrids are recognised by RNase H with subsequent cleavage of the RNA strand, albeit with slow rates. We attempt to rationalise this impaired enzyme activity from the rearrangement of the sugar–phosphate backbone of the α-L-LNA:RNA hybrid.
INTRODUCTION
Drug therapy at the genetic level is an appealing strategy for treatment of diseases due to the generality and rationale of this approach. One of the possible methods is the antisense approach, a method applied by Nature for the control of gene regulation and shown by Stephenson and Zamecnik to be applicable with synthetic oligonucleotides (1,2). In the antisense approach, a synthetic antisense oligonucleotide (AO) is transfected into the cell where it binds to its target mRNA, thus causing silencing of this given mRNA (3). This silencing can be brought about in various ways, e.g. by pure steric blocking of the mRNA or by induced degradation of the mRNA, possibly by RNase H (4). The RNase H degradation mechanism is appealing as this enzyme is ubiquitous in all cells. However, very strict rules have to be adhered to with the AO in order for the AO:RNA hybrid to be amenable to degradation by RNase H. As such, only very few, of the many, synthetic oligonucleotides tested for RNase H activity possess this feature; prominent amongst these are phosphorothioate DNA (5), 2′F-ANA (6) and CeNA (7,8), although the latter two have cleavage rates substantially lower than for native nucleic acids.
RNase H binds dsRNA duplexes and DNA:RNA hybrids, but only the latter are recognised with ensuing cleavage of the RNA strand (9). As RNase H recognises its putative substrate in the minor groove and as dsRNA duplexes and DNA:RNA hybrids have differing minor groove widths (the latter hybrids generally have minor grooves ∼2–3 Å narrower than the dsRNA duplexes), the width of the minor groove has been proposed as a key element in RNase H recognition (9,10). Other features might also play an important role in RNase H recognition, as will be discussed below in light of our results.
In general, nucleotide modifications which promote the N-like character of the ribose or deoxyribose ring, such as 2′-OMe RNA (11), phosphoramidates (12) or locked nucleic acid (LNA) (13,14), yield increased thermostability when hybridised with RNA, and thus possess one of the prerequisites of the ideal AO; however, the hybrids which these analogues form with RNA are not recognised by RNase H (12,15,16). This might be due to an A-like (dsRNA-like) geometry adopted by these hybrids (17). In contrast, phosphorothioates and 2′F-ANA form hybrids with RNA which retain the intermediate geometry of DNA:RNA hybrids (18,19).
Within the last few years, LNA (see Fig. 1) has been introduced (13,14). LNA oligonucleotides [we have defined LNA and α-L-LNA, respectively, as oligonucleotides containing one or more 2′-O,4′-C-methylene-β-d-ribofuranosyl, 2′-O,4′-C-methylene-α-L-ribofuranosyl, respectively, nucleotide monomer(s)] display substantial increases in thermostability when hybridised with cognate DNAs and RNAs, with increases in melting temperatures of up to 10°C per LNA modification. This is possibly the largest increase in thermostability observed for a nucleic acid analogue based on phosphodiester linkages and Watson–Crick base pairing. The eight stereoisomers of LNA have also been evaluated and, of these, α-L-LNA (see Fig. 1) caught the attention because it showed thermostability properties only slightly inferior to those of LNA (20). In contrast to LNA, it was shown that α-L-LNA:RNA hybrids could elicit RNase H cleavage of the RNA strand, albeit that the cleavage rate was significantly lower than for unmodified DNA:RNA hybrids (16). Further more, α-L-LNA nucleotides have been incorporated into an RNA-cleaving DNAzyme, thereby increasing the DNAzyme’s cleavage rate and improving its access to highly structured RNA targets (21).
Figure 1.

The chemical structure of LNA and α-L-LNA.
We have previously studied α-L-LNA:RNA hybrids by NMR and CD spectroscopy and by molecular dynamics (MD) simulations (22), and we now extend these studies with a high-resolution NMR structure of a nonamer hybrid (base composition and numbering is shown in Scheme 1). The structure was determined by applying nuclear Overhauser effect (NOE)-derived distance bounds in a simulated annealing (SA) scheme. To investigate the dynamic behaviour of the α-L-LNA:RNA hybrid, the distance bounds derived by NMR spectroscopy were included in rMD-tar (restrained MD with time-averaged restraints) calculations. These calculations were conducted using explicit solvent, periodic boundary conditions and Ewald treatment of electrostatics. Finally, we compare the structural implications of α-L-LNA and LNA in DNA:RNA hybrids, and discuss the α-L-LNA:RNA structure in the context of RNase H recognition.

Scheme 1. The numbering scheme for the α-L-LNA:RNA hybrid studied. αLTL denotes α-L-LNA-modified thymidines.
MATERIALS AND METHODS
Sample preparation
The NMR sample was prepared by mixing equimolar amounts of the two strands, each purified by site exclusion on a Sephadex G15 column. The final concentration was ∼3 mM hybrid, 10 mM sodium phosphate buffer (pH 7), 100 mM NaCl and 0.05 mM EDTA. The numbering scheme used for the hybrid is shown in Scheme 1. Further details on the sample preparation can be found in Petersen et al. (22).
NMR experiments
NMR experiments were performed on either Varian UNITY 500 or Varian INOVA 800 spectrometers at 25°C. NOESY spectra with mixing times of 60, 150, 200 and 300 ms were acquired in D2O at 800 MHz using 2048 complex points in t2 and a spectral width of 8000 Hz. A total of 400 t1 experiments, each with 32 scans and a dwell-time of 3.75 s between scans, were recorded using the States phase cycling scheme. The residual signal from HOD was removed by low-power pre-saturation. In addition, an inversion recovery experiment was recorded at 800 MHz to extract T1 relaxation rates. A NOESY spectrum in H2O was acquired at 800 MHz using the WATERGATE NOESY pulse sequence with a spectral width of 16 000 Hz using 4096 complex points in t2, 770 t1 experiments, each with 24 scans, and a dwell-time of 3.30 s between scans. In addition to the NOESY spectra, TOCSY and DQF-COSY spectra were recorded at 500 MHz, and a 1H,13C HSQC spectrum at 800 MHz using standard parameters. 1H,31P correlated experiments were recorded in D2O at 500 MHz, with spectral widths of 5000 and 600 Hz in the 1H and 31P dimensions, respectively.
The acquired data were processed using FELIX (version 97.2, MSI, San Diego, CA). All spectra were apodised by skewed sinebell squared functions in F1 and F2. The NOESY spectra recorded in D2O were linear predicted from 400 to 800 points in F1, and the NOESY spectrum recorded in H2O was linear predicted from 770 to 1540 points in F1. All NOESY spectra were baseline corrected by the FLATT procedure in F1 (23) and by the automatic baseline correction procedure as implemented in FELIX in F2.
Distance restraints
A total of 484 distance restraints were obtained from 2D NOE cross-peak intensities using the method of Wijmenga et al. (24); in this variation of the isolated spin pair approximation, spin diffusion is accounted for in an average manner. NOESY cross-peak intensities from the four spectra recorded in D2O were corrected for saturation effects using T1 relaxation times obtained from an inversion recovery experiment and were subsequently transformed to distance restraints by calibrating against known distances. The final upper and lower distance bounds used in the structure determination were determined from the standard deviations calculated by performing the procedure 100 times with slightly perturbed NOESY volumes. This resulted in the bounds being on average ±11% of the average distance obtained from the 100 calculations. A total of 412 restraints were derived from the NOESY spectra recorded in D2O. From the NOESY spectrum recorded in H2O, a further 72 restraints including exchangeable protons were derived by a procedure as described above; the upper and lower distance bounds were set to the distances calculated ±20%. The distribution of NOE restraints for the α-L-LNA:RNA hybrid is shown in Figure 2. In total, we had 178 intra-nucleotide, 276 inter-nucleotide and 30 cross-strand restraints. The average width of the distance restraints was 1.24 Å and the average restraint length 4.96 Å.
Figure 2.

The distribution of NOE restraints employed in structure calculations. The numbers of intra-nucleotide, sequential and cross-strand restraints are indicated. Indicated in red are restraints between non-neighbour residues, i.e. a red 1 above T2 indicates one restraint between C1 and G3, and so forth.
Normal Watson–Crick base pairing was inferred from the NOESY spectrum acquired in H2O and, consequently, 22 hydrogen bond distance restraints were included in the calculations. Target values for these restraints were taken from crystallographic data (25).
All distance restraints were incorporated into the AMBER potential energy by a flat-well pseudo potential with the form:

where the force constants, KNOE, are in units of kcal/(mol Å) or kcal/(mol Å2), and r1 and r2 are the lower and upper distance bounds, respectively.
Structure calculations
All calculations were performed with the AMBER5 suite of programs (26). An SA protocol was utilised to obtain the structure of the hybrid. The starting structures (A- and B-form duplexes) were obtained with the nucgen module of AMBER5. The appropriate nucleotides were modified to α-L-LNA nucleotides, and atomic charges for the modified nucleotides were calculated using the RESP procedure (27). A table giving the atomic charges is included in the Supplementary Material (Table S1).
Initially the starting structure was restrained energy minimised before being subjected to 50 ps of restrained molecular dynamics (rMD) in time steps of 1 fs: during the initial 4 ps, the temperature was raised from 600 to 800 K and the force constants from 50 to 175 kcal/(mol Å2). For 14 ps, these values were kept constant, and subsequently the temperature was decreased to 300 K and the force constants to 50 kcal/(mol Å2) over the final 32 ps of rMD. Finally, a further restrained energy minimisation was carried out. A distance-dependent dielectric constant, ε = 4r, was used and the non-bonded cut-off was 30 Å.
MD calculations with time-averaged restraints
A representative structure from the ensemble calculated using the SA protocol was chosen for further calculations. Sodium counterions were added at a distance of 5 Å from the phosphorus atoms on the OPO-bisectors and the structure was solvated with a periodic box of TIP3P waters extending ∼10 Å from the solute. The SHAKE algorithm was used to keep X–H bond lengths fixed during the simulation, and the time step was 1 fs. The simulations were run at a constant pressure of 1 atmosphere and at 300 K. The non-bonded cut-off was 9 Å. To accommodate the water molecules around the solute, 75 ps MD was carried out with the atoms of the duplex fixed. In this equilibration process, the force constants restraining the duplex atoms were gradually lowered from 500 to 10 kcal/(mol Å2) and the temperature and pressure couplings were gradually loosened. At the start of the production runs, the restraint force constants were gradually ramped up from 0.5 to 5.0 kcal/(mol Å2) over the first 20 ps to prevent heating of the systems. The rMD-tar calculations were carried out using a third-root averaging for distances and a memory decay constant of 20 ps. Periodic boundary conditions and Ewald treatment of electrostatics were employed during the calculations with a direct sum cut-off of 9 Å. The temperature and pressure couplings both were 1 ps. In the calculations, the Watson–Crick hydrogen bond restraints, included with force constants of 5 kcal/(mol Å2), were not time-averaged. The trajectories were extended to 1 ns, with coordinates being saved every 0.5 ps.
RESULTS
Spectral analysis
The 1D 1H-NMR spectrum of the α-L-LNA:RNA hybrid showed exclusively lines from the expected hybrid at 25°C. No signs of alternative hybridisation were observed. The NOESY spectra of the hybrid displayed the characteristic connectivities of a right-handed nucleic acid duplex with all the nucleobases in the anti conformation. The assignment of the non-exchangeable protons was performed using standard methods (28–31). The NOESY spectrum with the shortest mixing time (60 ms) allowed unambiguous assignments of the H2′, H2′′, H6′ and H6′′ resonances. The H6′ and H6′′ resonances in the 2′-O,4′-C-methylene bridge of the α-L-LNA nucleotides were assigned by their intra-nucleotide cross-peaks to the methyl protons of the modified nucleotides, with H6′ being assigned as the proton closer to the methyl group. The NOESY spectrum acquired in H2O exhibited normal Watson–Crick connectivities and was employed in the assignment of the exchangeable protons. A selection of chemical shift values for the α-L-LNA:RNA hybrid is included in the Supplementary Material (Table S2).
The average in vacuo structure
The final ensemble of average NOE structures was obtained using the SA protocol as described above. Two sets of 20 structures each were generated from either canonical A- or B-type duplex starting geometries by randomly varying initial atomic velocities. The two families of structures converged well with an average pairwise root mean square deviation (r.m.s.d.) of the 40 structures of 0.82 Å for non-terminal base pairs. However, the structures generated from A-type starting geometry possessed a lower force field energy than those derived from B-type geometry. This difference in energy originates from slightly different conformations for the terminal base pairs. Consequently, we have chosen only to include the 20 structures generated from A-type starting geometry in further analyses.
We chose only to include NOE-derived restraints in the calculations and did not include any torsion angle restraints which could have been derived from COSY and 1H,31P correlated spectra by analysis of coupling constants. Our rationale for this approach was to use the complementary information on torsion angles for validation of the structure and the structural ensembles calculated with the rMD-tar approach.
The final average structures feature low distance restraint energies and low distance violations when compared with the starting structure (see Table 1). The largest violation is 0.34 Å, and just 11 violations in excess of 0.2 Å were observed.
Table 1. Structural parametersa for the in vacuo structure and for the rMD and rMD-tar calculations.
| Structure | EAMBER (kcal/mol) | ENOE (kcal/mol) | Δdav (Å) | Jr.m.s.d. (Hz) | R.m.s.d versus NMR (Å) |
|---|---|---|---|---|---|
| A-type | 2.5 | 4029 | 0.218 | 4.97 | 2.1 |
| B-type | 36 | 7037 | 0.343 | 2.58 | 2.9 |
| In vacuo NMR | 120 | 81.8 | 0.021 | 1.76 | 0.58 (0.17)b |
| rMD | –43 556 | 67.1 | 0.063 | 1.94 | 0.97 (0.19)c |
| rMD-tar | –43 458 | 41.0 | 0.047 | 0.49 | 1.70 (0.36)c |
aForce field (EAMBER) and restraint (ENOE) energies, average restraint violations (Δdav), coupling constant r.m.s.ds (Jr.m.s.d.) and pairwise atomic r.m.s.ds for the starting structures, in vacuo NMR structure and rMD and rMD-tar ensembles. All-atomic r.m.s.ds were calculated for all atoms of the seven internal base pairs. Where applicable, standard deviations are included in parentheses. Comparisons between in vacuo calculations and calculations with solvent included cannot be made.
bAtomic r.m.s.ds for the 20 structures calculated from A-form starting geometry.
cAtomic r.m.s.ds relative to the in vacuo structure along the trajectories of the calculations.
General description of the average structure
The α-L-LNA:RNA hybrid structure is a regular right-handed helix with all nucleobases in the anti conformation and Watson–Crick base pairing maintained in all base pairs as was indicated by the NOESY spectra. The hybrid possesses an overall geometry intermediate between A- and B-type, with r.m.s.ds of 2.1 and 2.9 Å to canonical A- and B-type structures, respectively. The global helix axis, as calculated with CURVES 5.2 (32,33), is almost straight, with a slight kink at the T2:A17 base pair. The above observations show that the α-L-LNA nucleotides can be incorporated into a Watson–Crick duplex framework without any global disruptions being introduced. The 2′-O,4′-C-methylene linkers of the α-L-LNA nucleotides are positioned at the rim of the major groove, where they pose no steric hindrances for duplex formation with complementary nucleic acids. Stereo views of the α-L-LNA:RNA hybrid are presented in Figure 3.
Figure 3.

Stereo views of the in vacuo structure of the α-L-LNA:RNA hybrid. For clarity, only hydrogens on the modifications are shown. The colouring scheme is: nucleobases, blue; sugar–phosphate backbone, red; and the α-L-LNA 2′-O,4′-C bridge, yellow. (a) Superposition of the 20 structures calculated and (b) a single structure.
The intermediate geometry of the α-L-LNA:RNA hybrid is also mirrored in the minor groove width with an average value of 8.4 Å; this value lies between those observed for A-type duplexes (∼11.0 Å) and for B-type duplexes (∼5.2 Å). Minor groove widths between 8 and 10 Å are usually found in DNA:RNA hybrids (10,17,18,34–36). The intra-strand phosphorus distances in the RNA strand of the α-L-LNA:RNA hybrid are ∼6.0 Å, while across the α-L-LNA nucleotides ∼7.3 Å is measured and across the deoxyriboses values of ∼6.9 Å are observed. An intra-strand phosphorus distance of ∼6 Å is found in A-type duplexes, whilst in B-type duplexes values of ∼7 Å are observed. As such, the RNA strand of the hybrid is A-like, while the α-L-LNA strand is more B-like.
The stacking pattern in the hybrid structure agrees perfectly well with the pattern observed in the unmodified reference DNA:RNA hybrid and in isosequential LNA:RNA hybrids (17), i.e. no intra-strand stacking between the thymine nucleobases and the 3′-flanking nucleobases but purine–purine inter-strand stacking between G3 and A17, A6 and A14, and G8 and A12. This purine–purine stacking alleviates the inherent weaknesses of the pyrimidine–purine base steps. Thus the overall stacking pattern is not altered by the incorporation of α-L-LNA nucleotides. Changes in chemical shift values for adenine H2 protons are indicators of changes in the stacking arrangement as these protons are located in the centre of the duplex. When compared with the unmodified hybrid, we observe upfield shifts of 0.20, 0.15 and 0.39 p.p.m. for the H2 protons of A12, A14 and A17 (the adenines base paired with α-L-LNA nucleotides). This indicates that subtle changes in the stacking pattern indeed are occurring upon introduction of the modified nucleotides. However, whether these changes correspond to more or less favourable stacking interactions between the nucleobases cannot be deduced from changes in chemical shift values alone in a simple manner.
Helix parameters
The global helix axis and the helix parameters were determined with CURVES 5.2. X-displacement, Y-displacement, inclination and tip are the helix parameters with which the global helix axis are determined. Consequently, these parameters allow a direct comparison between different duplex types and the structure determined. X-displacement and inclination are both parameters discriminative of A- and B-type duplexes, and values intermediate between those of canonical A- and B-form geometries are observed, although somewhat closer to the A-type parameters. Y-displacement and tip offer no discrimination of different helix types, but serve to describe the regularity of the duplex, and only slight variations are observed along the duplex (values of 0.0 ± 0.3 Å and 4.2 ± 1.8°, respectively). A table with a selection of helix parameters is included in the Supplementary Material (Table S3). In general, the helix parameters determined are much like those of the corresponding unmodified hybrid in terms of values and variations along the duplex, and as such these variations are more likely to be attributable to sequence dependence rather than to the inclusion of the α-L-LNA nucleotides. For example, we observe a roll of 21° at the T2pG3:C16pA17 base step; however, high values of roll in TpG steps is a common feature of dsDNA duplexes (37). Worth noting is that at base pair steps 2, 5 and 7, where inter-strand nucleobase stacking is occurring, high values of twist (37–40°) and positive values of roll (5–21°) are observed. At the remaining base pair steps, fairly low values of twist (23–36°) and negative values of roll (–6 to –4°) are observed. The average rise in the duplex is 2.8 Å, which is intermediate between that of A- and B-type duplex forms, although closest to the A-type value. The average values for twist and propeller twist are 33 and –15°, respectively; these values are rather close to the values observed in A-type duplexes.
Dynamics in the α-L-LNA:RNA hybrid
We have previously established that the deoxyribo sugar puckers in the hybrid are best described by a two-state N/S model (22), and as such the single sugar pucker obtained for each furanose in the in vacuo average is an NOE averaged conformation which may not be a representative picture of the real sugar conformation. In addition, it has been found for DNA duplexes that multiple backbone conformations are energetically allowed (38,39). To investigate the sugar–phosphate backbone conformation, which is dramatically altered to accommodate the modified α-L-LNA nucleotides, and to investigate the general dynamics of the α-L-LNA:RNA hybrid, we have carried out rMD-tar calculations. In the time-averaged approach, the NOE restraints have to be satisfied over a period of time rather than instantaneously. Consequently, the dynamics encoded in the NOE restraints are explored in rMD-tar calculations. For completeness, we have included a table with sugar–phosphate backbone angles and sugar puckers of the in vacuo structure in the Supplementary Material (Table S4).
rMD-tar calculations
We have calculated a 1 ns trajectory for the α-L-LNA:RNA hybrid using time-averaged NOE restraints. For comparison, we calculated a 1 ns trajectory using ordinary rMD, i.e. without time averaging of the restraints. Both simulations are stable as indicated by average r.m.s.ds of 1.7 and 1.0 Å, respectively, relative to the in vacuo structure along the trajectories. The average distance violations decrease when employing time-averaged restraints as compared with the control rMD simulation; thus the enhanced conformational sampling afforded by rMD-tar results in an improved fit to the NOE-based distance restraints (see Table 1). In addition, the error between the sugar coupling constants calculated from the rMD-tar trajectory and the experimental ones is very low (0.5 Hz, see Table 1).
The global structural features remain as discussed above for the in vacuo structure throughout both simulations. The main difference between the rMD-tar simulation and the in vacuo structure is unwinding of the duplex in the simulation as judged by lower values of twist (average 28°) and higher values for rise (average 3.2 Å), whilst in the rMD calculation the unwinding is less pronounced. At the local level, dynamics are observed in the rMD-tar calculation as evidenced by repuckering of the furanose sugars and by motions of the sugar–phosphate backbone. Consequently, this will be our focus in the following sections.
Sugar puckers dynamics. The pseudorotation angles and puckering amplitudes were calculated for the last 950 ps of the simulation (1900 frames). All the riboses in the RNA strand display little flexibility, with almost pure N-type puckers and average pseudorotation angles ranging from 12 to 26°. The locked α-L-LNA furanoses, as expected, display very narrow sugar pucker distributions throughout the simulation, with their average pseudorotation angle being 13° (taking into account that they are l-sugars) and puckering amplitudes of ∼57°. The deoxyriboses, on the other hand, show frequent repuckering between N- and S-type conformations resulting in bimodal distributions of the pseudorotation angles (see Table 2). In the control rMD calculation, we observe a unimodal distribution of sugar puckers for the deoxyriboses. We have previously determined the ratio of N- and S-type sugar pucker for the deoxyriboses in the α-L-LNA strand by analysis of COSY-type NMR spectra, and these results are included in Table 2. As can be observed, strikingly good agreement is obtained between the rMD-tar calculations and the experimental results. As no restraints were included to explicitly govern the sugar conformations in the rMD-tar calculations, this shows that the dynamics encoded in the NOE restraints can reproduce, in an average manner at least, the sugar conformations determined in a complementary manner. This, in turn, validates that the information on dynamics derived from the rMD-tar calculation is reflecting the real dynamics of the hybrid.
Table 2. The sugar conformations and the JH3′P coupling constants of the α-L-LNA strand in the α-L-LNA:RNA hybrid.
| Sugar conformations | JH3′P (Hz) | |||
|---|---|---|---|---|
| rMD-tar | Experimental | rMD-tar | Experimental | |
| C1 | 3 | 25 (13) | 6.5 | 12.0 |
| αLTL2 | –a | –a | 4.3 | 6.0 |
| G3 | 41 | 37 (10) | 5.9 | 8.0 |
| A4 | 21 | 24 (9) | 7.7 | 7.2 |
| αLTL5 | –a | –a | 5.0 | 5.1 |
| A6 | 34 | 30 (8) | 7.4 | 8.3 |
| αLTL7 | –a | –a | 5.5 | 5.6 |
| G8 | 53 | 46 (8) | 5.7 | 5.7 |
| C9 | 33 | 43 (12) | ||
The sugar conformations are given as the percentage of N-type sugar conformation. The N-type sugar conformation percentages for the rMD-tar calculation were obtained by summation over conformations in the entire N-type range (–90° ≤ P ≤ 90°). The experimental values are taken from Petersen et al. (22) and standard deviations are included in parentheses.
aThe modified nucleotides were not included in the analysis owing to the locked furanose conformation.
The deoxyriboses in the α-L-LNA strand possess between 20 and 40% N-type conformation, except C1 (3%) and G8 (53%), which is in agreement with the ratios of N-type sugar puckers generally obtained for DNA:RNA hybrids (17,18,34–36). Thus, on the whole, the α-L-LNA nucleotides do not perturb the deoxyribose sugar equilibrium significantly from an unmodified hybrid. The high percentage of N-type pucker for G8 is intriguing. In our investigation of the corresponding α-L-LNA:DNA hybrid (40), a similar trend was observed.
Sugar–phosphate backbone conformation and dynamics. From inspection of the backbone torsion angles of the α-L-LNA:RNA hybrid, it is evident that the α-L-LNA nucleotides are accommodated in a geometry suitable for Watson–Crick base pairing and stacking through rearrangement of the sugar–phosphate backbone. The rMD-tar calculation allows us to investigate the conformation and dynamics of the backbone. The unnatural stereochemistry of the α-L-LNA nucleotides imposes changes of the δ (∼120° to gauche–) and ζ (gauche– to gauche+) torsion angles of the modified nucleotides. The most pronounced changes observed in response to these changes are for the α and γ angles of the modified and 3′-flanking residues. In native nucleic acids, these angles are usually found in (α, γ: gauche–, gauche+) conformations although concerted transitions to (trans, trans) conformations can be observed. The former conformation is lowest in energy and consequently most populated (39). The behaviour of the α and γ angles of the α-L-LNA:RNA hybrid is summarised in Table 3. In the modified nucleotides, the γ angle is shifted to the trans conformation while the α angle assumes two major conformations: approximately –90° (∼60–70%) and trans (∼15–20%). For the nucleotides 3′-flanking the modified ones, two major and coupled α, γ conformations are observed: (gauche+, trans) and (approximately –120, gauche+). The population of the trans conformation for the γ angle is corroborated by vanishing JH4′P coupling constants for these nucleotides. Non-vanishing JH4′P coupling constants (JH4′P ≈ 2 Hz) can only be observed if the P-O5′-C5′-C4′-H4′-fragment adopts a planar W-shaped conformation, i.e. β, γ: trans, gauche+ (24). As the β angles adopt trans conformations, the disappearances of the JH4′P coupling constants imply that the γ angles cannot be exclusively in gauche+ conformations. The conformation of the γ angle can also be gauged from the JH4′H5′ and JH4′H5′′ coupling constants; however, extensive spectral overlap precluded this analysis. In the RNA strand, the nucleotides possess entirely (α, γ: gauche–, gauche+) conformation, except C11, U13 and U15, which have 3, 24 and 60% (trans, trans) conformation, respectively. There appears to be no correlation between the different conformations in the two strands. The α, γ conformations found in the in vacuo structure and in the rMD calculations correspond for all nucleotides to one of the major conformations observed in the rMD-tar calculation.
Table 3. α and γ backbone torsion angles in the rMD-tar calculations given as population percentages.
| (α, γ) | (–90, trans) | (trans, trans) | (gauche+, trans) | |
|---|---|---|---|---|
| α-L-LNA | 2 | 76 | 14 | |
| 5 | 66 | 15 | ||
| 7 | 56 | 19 | 14 | |
| (α, γ) | (gauche+, trans) | (–120, gauche+) | (trans, trans) | |
| 3′-flankers | 3 | 25 | 38 | 25 |
| 6 | 64 | 31 | ||
| 8 | 33 | 46 | ||
| (α, γ) | (gauche–, gauche+) | (–120, gauche+) | ||
| Other | 4 | 80 | ||
| 9 | 79 |
aConformations with populations of <10% are not included.
As for the remaining backbone angles, β and ε are found in trans conformations and ζ in gauche– conformations in the unmodified nucleotides, although a high degree of flexibility is observed for the nucleotides 3′-flanking α-L-LNA modifications, with the ε and ζ backbone angles adopting multiple (three or four) concerted conformations. The α-L-LNA nucleotides have β = trans and ζ = gauche+, the latter owing to the change in chirality of C3′, and they display flexibility for their ε angles, with values ranging from gauche– to gauche+.
The JH3′P coupling constant can give information on the ε backbone angle and, as a validation of the rMD-tar calculations, we calculated the average values of these coupling constants from the trajectory and compared them with the values determined experimentally (Table 2). We generally obtain good agreement between the calculated and experimental values. It is observed that the JH3′P coupling constants are smaller for the modified nucleotides than for the deoxyribonucleotides.
The glycosidic angles adopt anti conformations for all nucleotides, including the modified ones, throughout the simulation. Riboses and α-L-LNA show average values of –159 and –150°, respectively, and deoxyriboses average values of –155 and –119° in the N- and S-type domains, respectively.
DISCUSSION
Nucleic acid duplex structure
The α-L-LNA-modified hybrid adopts a right-handed duplex geometry, with all the nucleobases in anti conformations and forming normal Watson–Crick base pairing. It is evident from the minor groove width (average value 8.4 Å), intra-strand phosphorus distances and several helical parameters that the α-L-LNA:RNA hybrid adopts a geometry intermediate between that of A- and B-type duplex forms. The RNA strand is A-like, with the riboses in N-type sugar conformations, while the α-L-LNA strand is more B-like, with the deoxyriboses repuckering between N- and S-type conformations. This picture of an A-like RNA strand and a B-like DNA strand is corroborated by the intra-strand phosphorus distances, as described above. Thus, the α-L-LNA:RNA, in the overall structure, resembles a DNA:RNA hybrid, and indeed the r.m.s.d. between this hybrid and the corresponding unmodified one is only 1.2 Å (17). This close structural resemblance between the modified and unmodified hybrid is corroborated by the CD spectra of the two hybrids as they display almost identical curves (22).
We have previously determined the solution structure of the corresponding LNA:RNA hybrid, i.e. the isosequential hybrid with LNA modifications at nucleotides 2, 5 and 7 (17,41). This hybrid adopts an almost canonical A-type duplex geometry, with all furanoses adopting N-type sugar puckers, i.e. the LNA nucleotides perturb the remaining deoxyribonucleotides in the LNA strand so as to shift their sugar conformations to N-type. This is in stark contrast to what is observed in the present study of the α-L-LNA:RNA hybrid, where the deoxyriboses in the α-L-LNA strand repucker between N- and S-type conformations.
According to our results, we can consider α-L-LNA nucleotides as deoxyribose mimics at the global structural level in the context of DNA:RNA hybrids. We have previously shown that α-L-LNA nucleotides act as DNA mimics in the context of dsDNA (40). However, at the local level of geometry, the α-L-LNA nucleotides introduce rather dramatic perturbations in the sugar–phosphate backbone of the chimeric strand. These changes are caused by the geometric constraints introduced by the participation of the α-L-LNA nucleobases in Watson–Crick base pairing and nucleobase stacking, and the unnatural stereochemistry of the modified nucleotides. The sugar–phosphate backbone responds to the changes in the δ and ζ backbone angles imposed by the stereochemistry of the α-L-LNA nucleotides by a slight adjustment of the α torsion angle (major conformation –90°) and a rotamer shift of the γ angle to trans conformation in the modified nucleotides. In addition, the backbone in the 3′ direction appears rather flexible, with flexible ε angles in the α-L-LNA nucleotides themselves and frequent α, γ transitions in their 3′-flanking neighbours. As a consequence of the backbone rearrangement imposed by the α-L-LNA nucleotides, the phosphates located in their 3′ direction are rotated ∼70° into the minor groove (see Fig. 4).
Figure 4.
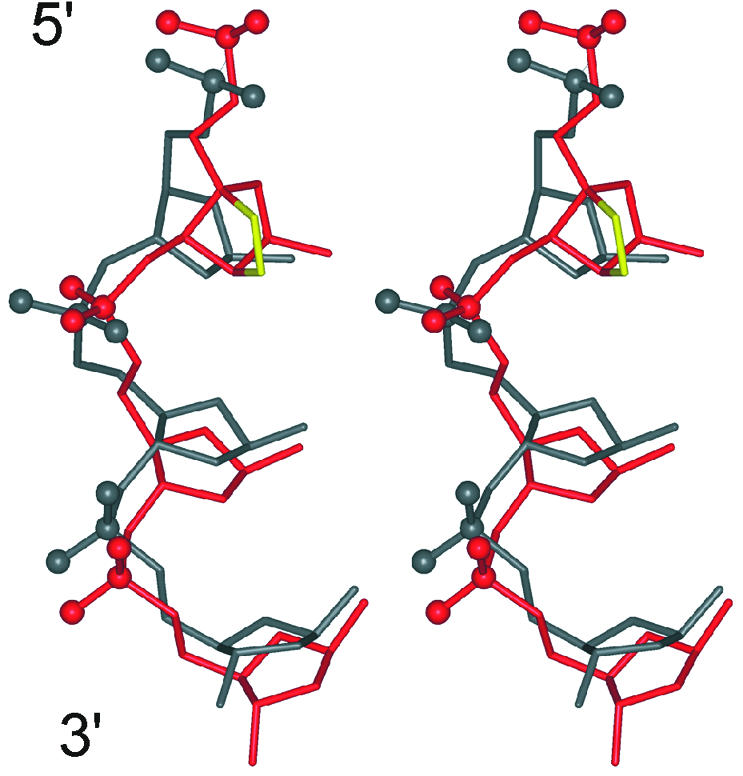
Stereo view of an overlay of an excerpt of the α-L-LNA sugar–phosphate backbone (shown in red) and the backbone of the unmodified DNA:RNA hybrid (shown in grey) viewed into the major groove. The phosphorus and non-bridging oxygen atoms of the phosphate groups are shown as balls. The overlay was prepared by r.m.s.d. fitting the RNA strands of the two hybrids.
In the rMD-tar calculations, multiple backbone conformations were populated for the modified and neighbouring nucleotides. This suggests that the α-L-LNA sugar–phosphate backbone retains the plasticity of the native DNA backbone, where multiple backbone conformations are energetically allowed. We observe an increased dynamic behaviour of the backbone in nucleotides flanking the α-L-LNA modifications relative to the α-L-LNA nucleotides themselves; this may suggest either that the modifications introduce increased dynamics in neighbouring nucleotides or that the modifications introduce a decrease of dynamics in these nucleotides. rMD-tar calculations of an unmodified DNA:RNA hybrid are required to distinguish between these possibilities.
Thermal stability
α-L-LNA nucleotides introduce a significant increase in helical thermostability when introduced into DNA:RNA hybrids or dsDNA duplexes (16,20). In the hybrid used in this study, the three α-L-LNAs give an increase in melting temperature of 17°C (5.6°C per modification) relative to the unmodified hybrid. For comparison, the increase in melting temperature of the corresponding LNA:RNA hybrid is 24°C, which corresponds to a staggering increase of 8°C per modification (41). However, the structural implications of introduction of LNA and α-L-LNA are entirely different, as discussed above. Whereas the LNA nucleotides propel the structure of the LNA:RNA hybrid into an almost canonical A-type geometry, the α-L-LNA nucleotides leave the global structure of a hybrid unaltered, i.e. intermediate between A- and B-type geometries. To our knowledge, all known nucleic acid analogues based on Watson–Crick hybridisation increasing the helical stability as much as α-L-LNA do are RNA mimics [e.g. LNA, N3′–P5′ phosphoramidates (42,43) and 2′-F-ribo-DNA (44)], which consequently drives the structure of modified hybrids towards A-type duplex geometry. In this light, the substantial increase in thermostability of α-L-LNA-modified nucleic acids is quite remarkable.
The structure, as it is, offers few clues as to why α-L-LNA nucleotides introduce this increase in thermostability; however, some possible explanations are forthcoming. The 2′-oxygen is located in the major groove, where it may provide an additional anchoring point for hydration. A change in the stacking of the nucleobases is indicated by the change in chemical shift values as discussed above; this change may entail a more efficient stacking in the α-L-LNA-modified hybrid than in the unmodified one. With the locked nature of the α-L-LNA furanose, a decrease in the entropic gain upon denaturation of the two strands is achieved as single-stranded α-L-LNA is made less advantageous than single-stranded DNA.
RNase H recognition of α-L-LNA-modified nucleic acids
It has been shown with Escherichia coli RNase H assays that RNase H recognises and cleaves α-L-LNA:RNA hybrids, albeit with an efficiency of cleavage that is substantially reduced compared with unmodified DNA:RNA hybrids (16).
It is generally assumed that one of the key elements of RNase H recognition of DNA:RNA substrates is the intermediate minor groove width of these hybrids. Weight has been added to this hypothesis with the recent X-ray crystallography structure of human immunodeficiency virus type 1 reverse transcriptase (HIV-1 RT) complexed with a polypurine tract (PPT) DNA:RNA hybrid (45). HIV-1 RT contains an RNase H domain which is highly homologous with E.coli RNase H1, and the structure reveals numerous contacts between this domain and the nucleic acid hybrid. The PPT DNA:RNA hybrid is not cleaved by the RNase H domain of HIV-1 RT, and in the structure it is evident that this hybrid has a minor groove width smaller than usual for DNA:RNA hybrids in the part bound to the RNase H domain.
From the X-ray structure, information on the mode of action of RNase H is deducible. The primer grip of the RNase H domain in HIV-1 RT makes numerous contacts to the nucleic acid hybrid, mainly to the phosphate groups and sugars in the DNA strand half a turn away from the potentially scissile RNA phosphate group. The distance between the primer grip and the active site is ∼10–11 Å, which perfectly matches the minor groove width in DNA:RNA hybrids. As such, two demands must be fulfilled for a nucleic acid hybrid to be a substrate, the minor groove must have the right width (∼10–11 Å) and the trajectory of the nucleic acid must be right when bound to the enzyme (see Fig. 5).
Figure 5.

Models of interactions between RNase H and nucleic acids. RNase H is shown as spheres with coordinates taken from pdb entry 2rn2. Residues 43–46 and 73 are coloured blue; these residues are homologous to the primer grip of the RNase H domain of HIV-1 RT; the active site is coloured red. Shown as ribbons is part of a nucleic acid duplex, with the putative scissile RNA strand coloured green. The double-headed arrow indicates the minor groove width. (a) A duplex with the right trajectory and minor groove width, leading to cleavage of the RNA strand; this could be the case for a DNA:RNA hybrid. (b) The right trajectory but with the minor groove too wide, leading to abolishment of cleavage; this could be the case for an LNA:RNA hybrid. (c) Right minor groove width but wrong trajectory, leading to diminished cleavage of the RNA strand; this could be the case for an α-L-LNA:RNA hybrid.
The α-L-LNA:RNA hybrid has a minor groove width compliant with RNase H cleavage; however, inspection of the features in the NMR structure reveals that the phosphate groups adjoining α-L-LNA nucleotides are rotated ∼70° into the minor groove relative to the unmodified DNA:RNA hybrid (see Fig. 4). Thus it is very likely that α-L-LNA:RNA hybrids are recognised by the RNase H enzyme, with the α-L-LNA strand interacting with the residues of E.coli RNase H that correspond to the primer grip in HIV-1 RT. However, the rotated phosphate groups would alter the trajectory of an α-L-LNA:RNA hybrid relative to that of a native DNA:RNA hybrid, hence positioning the phosphate groups of the RNA strand in non-optimum geometries for cleavage, thus impairing the cleaving efficiency (see Fig. 5).
It is noteworthy that the locked α-L-LNA sugars do not abolish RNase H activity altogether, and this shows that a locked furanose conformation is not incompatible with RNase H activity. Recently, it was demonstrated by Damha and co-workers that the incorporation of an acyclic inter-residue insert restored full RNase H activity of 2′F-ANA oligonucleotides (46). It is possible that such a construct could be applied to increase the RNase H activity of α-L-LNA oligonucleotides by introducing appropriate flexibility in the modified strand.
Conclusion
We have determined the high-resolution structure of a nonamer α-L-LNA:RNA hybrid. This hybrid adopts a duplex geometry intermediate between A- and B-type, and the global structure is very similar to that of the corresponding unmodified DNA:RNA hybrid. Although the α-L-LNA-modified hybrid retains the native intermediate geometry, we have shown that local rearrangements of the sugar–phosphate backbone are necessary to accommodate the modified nucleotides. From our calculations, it appears that the backbone linkage between the α-L-LNA nucleotides and their 3′-flanking neighbours is very flexible, with multiple conformations of the ε, α and γ backbone torsion angles allowed. Introduction of α-L-LNA modifications in DNA:RNA hybrids yields substantial increases in helical thermostability, which is quite remarkable as α-L-LNA is not moulded on an N-type furanose skeleton. α-L-LNA:RNA hybrids are recognised and cleaved by E.coli RNase H, albeit much more slowly than native hybrids. In our structure, we observe a distinct rotation into the minor groove of the phosphate groups neighbouring the modifications; this might be the rationale for the impaired RNase H activity of α-L-LNA modified hybrids.
Summarising, α-L-LNA acts as a DNA (B-type) mimic in the contexts of both DNA:RNA hybrids and dsDNA duplexes, leaving the global duplex structure unperturbed, yet yield substantial elevation in duplex stability. As such, α-L-LNA-modified nucleic acids could prove useful as decoys for proteins recognising B-type (or intermediate type) duplex geometries.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank the Instrument Centre for NMR Spectroscopy of Biological Macromolecules at The Carlsberg Laboratory, Copenhagen granted by The Danish Natural Science Research Council for providing spectrometer time at the 800 MHz spectrometer. The Danish National Research Foundation is thanked for financial support, and Britta M. Dahl (Department of Chemistry, University of Copenhagen) is thanked for oligonucleotide synthesis. The Nucleic Acid Center is a research centre funded by the Danish National Research Foundation for studies on the chemical biology of nucleic acids. Coordinates and restraints employed in calculations have been deposited in the Protein Data Bank (accession code: 1okf).
REFERENCES
- 1.Stephenson M.L. and Zamecnik,P.C. (1978) Inhibition of rous-sarcoma viral-RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl Acad. Sci. USA, 75, 285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zamecnik P.C. and Stephenson,M.L. (1978) Inhibition of rous-sarcoma virus-replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl Acad. Sci. USA, 75, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crooke S.T. (1999) Molecular mechanisms of action of antisense drugs. Biochim. Biophys Acta, 1489, 31–44. [DOI] [PubMed] [Google Scholar]
- 4.Kurreck J. (2003) Antisense technologies: improvement through novel chemical modifications. Eur. J. Biochem., 270, 1628–1644. [DOI] [PubMed] [Google Scholar]
- 5.Levin A.A. (1999) A review of issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim. Biophys Acta, 1489, 69–84. [DOI] [PubMed] [Google Scholar]
- 6.Damha M.J., Wilds,C.J., Noronha,A., Brukner,I., Borkow,G., Arion,D. and Parniak,M.A. (1998) Hybrids of RNA and arabinonucleic acids (ANA and 2′F-ANA) are substrates of ribonuclease H. J. Am. Chem. Soc., 120, 12976–12977. [Google Scholar]
- 7.Wang J., Verbeure,B., Luyten,I., Lescrinier,E., Froeyen,M., Hendrix,C., Rosemeyer,H., Seela,F., Van Aerschot,A. and Herdewijn,P. (2000) Cyclohexene nucleic acids (CeNA): serum stable oligonucleotides that activate RNase H and increase duplex stability with complementary RNA. J. Am. Chem. Soc., 122, 8595–8602. [Google Scholar]
- 8.Verbeure B., Lescrinier,E., Wang,J. and Herdewijn,P. (2001) RNase H mediated cleavage of RNA by cyclohexene nucleic acid (CeNA). Nucleic Acids Res., 29, 4941–4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toulmé J.-J. and Tidd,D. (1998) The role of ribonuclease H in antisense oligonucleotide-mediated effects. In Crouch,R.J. and Toulmé,J.-J. (eds), Ribonucleases H. INSERM, Paris, pp. 225–250. [Google Scholar]
- 10.Fedoroff O.Y., Salazar,M. and Reid,B.R. (1993) Structure of a DNA:RNA hybrid duplex. Why RNase H does not cleave pure DNA. J. Mol. Biol., 233, 509–523. [DOI] [PubMed] [Google Scholar]
- 11.Manoharan M. (1999) 2′-Carbohydrate modifications in antisense oligonucleotide therapy: importance of conformation, configuration and conjugation. Biochim. Biophys Acta, 1489, 117–130. [DOI] [PubMed] [Google Scholar]
- 12.Gryaznov S.M. (1999) Oligonucleotide N3′→P5′ phosphoramidates as potential therapeutic agents. Biochim. Biophys Acta, 1489, 131–140. [DOI] [PubMed] [Google Scholar]
- 13.Wengel J. (1999) Synthesis of 3′-C- and 4′-C-branched oligonucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res., 32, 301–310. [Google Scholar]
- 14.Petersen M. and Wengel,J. (2003) LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol., 21, 74–81. [DOI] [PubMed] [Google Scholar]
- 15.Inoue H., Hayase,Y., Iwai,S. and Ohtsuka,E. (1987) Sequence-dependent hydrolysis of RNA using modified oligonucleotide splints and RNase H. FEBS Lett., 215, 327–330. [DOI] [PubMed] [Google Scholar]
- 16.Sørensen M.D., Kværnø,L., Bryld,T., Håkansson,A.E., Verbeure,B., Gaubert,G., Herdewijn,P. and Wengel,J. (2002) α-L-ribo-configured locked nucleic acid (α-L-LNA): synthesis and properties. J. Am. Chem. Soc., 124, 2164–2176. [DOI] [PubMed] [Google Scholar]
- 17.Petersen M., Bondensgaard,K., Wengel,J. and Jacobsen,J.P. (2002) Locked nucleic acid (LNA) recognition of RNA: NMR solution structures of LNA:RNA hybrids. J. Am. Chem. Soc., 124, 5974–5982. [DOI] [PubMed] [Google Scholar]
- 18.Bachelin M., Hessler,G., Kurz,G., Hacia,J.G., Dervan,P.B. and Kessler,H. (1998) Structure of a stereoselective phosphorothioate DNA/RNA duplex. Nature Struct. Biol., 5, 271–276. [DOI] [PubMed] [Google Scholar]
- 19.Trempe J.-F., Wilds,C.J., Denisov,A.Y., Pon,R.T., Damha,M.J. and Gehring,K. (2001) NMR solution structure of an oligonucleotide hairpin with a 2′F-ANA/RNA stem: implications for RNase H specificity toward DNA/RNA hybrid duplexes. J. Am. Chem. Soc., 123, 4896–4903. [DOI] [PubMed] [Google Scholar]
- 20.Rajwanshi V.K., Håkansson,A.E., Sørensen,M.D., Pitsch,S., Singh,S.K., Kumar,R., Nielsen,P. and Wengel,J. (2000) The eight stereoisomers of LNA (locked nucleic acid): a remarkable family of strong RNA binding molecules. Angew Chem. Int. Ed., 39, 1656–1659. [DOI] [PubMed] [Google Scholar]
- 21.Vester B., Lundberg,L.B., Sørensen,M.D., Babu,B.R., Douthwaite,S. and Wengel,J. (2002) LNAzymes: incorporation of LNA-type monomers into DNAzymes markedly increases RNA cleavage. J. Am. Chem. Soc., 124, 13682–13683. [DOI] [PubMed] [Google Scholar]
- 22.Petersen M., Håkansson,A.E., Wengel,J. and Jacobsen,J.P. (2001) α-L-LNA (α-L-ribo configured locked nucleic acid) recognition of RNA. A study by NMR spectroscopy and molecular dynamics simulations. J. Am. Chem. Soc., 123, 7431–7432. [DOI] [PubMed] [Google Scholar]
- 23.Güntert P. and Wüthrich,K. (1992) FLATT—new prodedure for high-quality baseline correction of multidimensional NMR spectra. J. Magn. Reson., 96, 403–407. [Google Scholar]
- 24.Wijmenga S.S. and van Buuren,B.N.M. (1998) The use of NMR methods for conformational studies of nucleic acids. Prog. Nucl. Magn. Reson. Spectrosc., 32, 287–387. [Google Scholar]
- 25.Saenger W. (1984) Principles of Nucleic Acid Structure. Springer Verlag, New York. [Google Scholar]
- 26.Case D.A., Pearlman,D.A., Caldwell,J.W., Cheatham,T.E.,III, Ross,W.S., Simmerling,C.L., Darden,T.A., Merz,K.M., Stanton,R.V., Cheng,A.L. et al. (1997) AMBER 5. University of California, San Francisco. [Google Scholar]
- 27.Bayly C.I., Cieplak,P., Cornell,W.D. and Kollman,P.A. (1993) A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem., 97, 10269–10280. [Google Scholar]
- 28.Hare D.R., Wemmer,D.E., Chou,S.-H., Drobny,G. and Reid,B.R. (1983) Assignment of the non-exchangeable proton resonances of d(C-G-C-G-A-A-T-T-C-G-C-G) using two-dimensional nuclear magnetic resonance methods. J. Mol. Biol., 171, 319–336. [DOI] [PubMed] [Google Scholar]
- 29.Wüthrich K. (1986) NMR of Proteins and Nucleic Acids. John Wiley & Sons, New York. [Google Scholar]
- 30.Feigon J., Leupin,W., Denny,W.A. and Kearns,D.R. (1983) Two dimensional proton nuclear magnetic resonance investigation of the synthetic deoxyribonucleic acid decamer d(ATATCGATAT)2. Biochemistry, 22, 5943–5951. [DOI] [PubMed] [Google Scholar]
- 31.Scheek R.M., Russo,N., Boelens,R. and Kaptein,R. (1983) Sequential resonance assignments in DNA 1H NMR spectra by two-dimensional NOE spectroscopy. J. Am. Chem. Soc., 105, 2914–2916. [DOI] [PubMed] [Google Scholar]
- 32.Lavery R. and Sklenar,H. (1988) The definition of generalized helicoidal parameters and of axis curvature of irregular nucleic acids. J. Biomol. Struct. Dyn., 6, 63–91. [DOI] [PubMed] [Google Scholar]
- 33.Lavery R. and Sklenar,H. (1989) Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn., 7, 655–667. [DOI] [PubMed] [Google Scholar]
- 34.Gonzáles C., Stec,W., Reynolds,M. and James,T.L. (1995) Structure and dynamics of a DNA·RNA hybrid duplex with a chiral phosphorothioate moiety: NMR and molecular dynamics with conventional and time-averaged restraints. Biochemistry, 34, 4969–4982. [DOI] [PubMed] [Google Scholar]
- 35.Gyi J.I., Lane,A.N., Conn,G.L. and Brown,T. (1998) Solution structures of DNA:RNA hybrids with purine-rich and pyrimidine-rich strands: comparison with the homologous DNA and RNA duplexes. Biochemistry, 37, 73–80. [DOI] [PubMed] [Google Scholar]
- 36.Aramini J.M. and Germann,M.W. (1999) Solution structure of a DNA·RNA hybrid containing an α-anomeric thymidine and polarity reversals: d(ATGG-3′-3′-αT-5′-5′-GCTC)·r(gagcaccau). Biochemistry, 38, 15448–15458. [DOI] [PubMed] [Google Scholar]
- 37.Ulyanov N.A. and James,T.L. (1995) Statistical analysis of DNA duplex structural features. Methods Enzymol., 261, 90–115. [DOI] [PubMed] [Google Scholar]
- 38.Issacs R.J. and Spielmann,H.P. (2001) NMR evidence for mechanical coupling of phosphate BI–BII transitions with deoxyribose conformational exchange in DNA. J. Mol. Biol., 311, 149–160. [DOI] [PubMed] [Google Scholar]
- 39.Varnai P., Djuranovic,D., Lavery,R. and Hartmann,B. (2002) α/γ transitions in the B-DNA backbone. Nucleic Acids Res., 30, 5398–5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen K.M.E., Petersen,M., Håkansson,A.E., Wengel,J. and Jacobsen,J.P. (2002) α-L-LNA (α-L-ribo configured locked nucleic acid) recognition of DNA: an NMR spectroscopic study. Chem. Eur. J., 8, 3001–3009. [DOI] [PubMed] [Google Scholar]
- 41.Bondensgaard K., Petersen,M., Singh,S.K., Rajwanshi,V.K., Wengel,J. and Jacobsen,J.P. (2000) Structural studies of LNA:RNA duplexes by NMR: conformations and implications for RNase H activity. Chem. Eur. J., 6, 2687–2695. [DOI] [PubMed] [Google Scholar]
- 42.Ding D., Gryaznov,S.M., Lloyd,D.H., Chandrasekaran,S., Yao,S., Ratmeyer,L., Pan,Y. and Wilson,W.D. (1996) An oligodeoxyribonucleotide N3′→P5′ phosphoramidate duplex forms an A-type helix in solution. Nucleic Acids Res., 24, 354–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tereshko V., Gryaznov,S. and Egli,M. (1998) Consequences of replacing the DNA 3′-oxygen by an amino group: high-resolution crystal structure of a fully modified N3′→P5′ phosphoramidate DNA dodecamer duplex. J. Am. Chem. Soc., 120, 269–283. [Google Scholar]
- 44.Kawasaki A.M., Casper,M.D., Freier,S.M., Lesnik,E.A., Zounes,M.C., Cummins,L.L., Gonzalez,C. and Cook,P.D. (1993) Uniformly modified 2′-deoxy-2′-fluoro phosphorothioate oligonucleotides as nuclease-resistant antisense compounds with high affinity and specificity for RNA targets. J. Med. Chem., 36, 831–841. [DOI] [PubMed] [Google Scholar]
- 45.Sarafianos S.G., Das,K., Tantillo,C., Clark,A.D.,Jr, Ding,J., Whitcomb,J.M., Boyer,P.L., Hughes,S.H. and Arnold,E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J., 20, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mangos M.M., Min,K.-L., Viazovkina,E., Galarneau,A., Elzagheid,M.I., Parniak,M.A. and Damha,M.J. (2003) Efficient RNase H-directed cleavage of RNA promoted by antisense DNA or 2′F-ANA constructs containing acyclic nucleotide inserts. J. Am. Chem. Soc., 125, 654–661. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.