


Abstract
The available reagents for the attachment of functional moieties to plasmid DNA are limiting. Most reagents bind plasmid DNA in a non-sequence- specific manner, with undefined stoichiometry, and affect DNA charge and delivery properties or involve chemical modifications that abolish gene expression. The design and ability of oligonucleotides (ODNs) containing locked nucleic acids (LNAs) to bind supercoiled, double-stranded plasmid DNA in a sequence-specific manner are described for the first time. The main mechanism for LNA ODNs binding plasmid DNA is demonstrated to be by strand displacement. LNA ODNs are more stably bound to plasmid DNA than similar peptide nucleic acid (PNA) ‘clamps’ for procedures such as particle-mediated DNA delivery (gene gun). It is shown that LNA ODNs remain associated with plasmid DNA after cationic lipid-mediated transfection into mammalian cells. LNA ODNs can bind to DNA in a sequence-specific manner so that binding does not interfere with plasmid conformation or gene expression. Attachment of CpG-based immune adjuvants to plasmid by ‘hybrid’ phosphorothioate–LNA ODNs induces tumour necrosis factor-α production in the macrophage cell line RAW264.7. This observation exemplifies an important new, controllable methodology for adding functionality to plasmids for gene delivery and DNA vaccination.
INTRODUCTION
The early promise of both DNA vaccination and gene therapy technologies has been tempered by a need to improve the efficiency of gene delivery to provide clinical benefit. Some of the major barriers to gene delivery, common to both technologies, are intracellular. Many attempts have been made to address these barriers, for example by attachment of functional moieties to DNA. However, effects of the coupling system can undermine such attachments either by altering parameters of the gene delivery system itself or by imposing inhibitory effects on gene expression (1,2). Positively charged peptides or moieties covalently linked to poly- or oligolysine can couple to plasmid DNA via electrostatic interactions, but do so in a non-sequence-specific manner and can cause random inhibitory effects upon gene expression. Such cationic peptides may also cause condensation or charge effects upon the plasmid DNA which would interfere with its properties, especially if delivered via a cationic gene delivery agent (2). Peptides have also been coupled to plasmid via direct chemical conjugation, but the position and numbers of copies of peptides attached to the plasmid in this way are difficult to control and the chemical modification usually inhibits gene expression (3,4). A few specifically engineered plasmid-derived gene expression vectors have been described which have unusual DNA sequences or attach linear hairpin loop structures to allow coupling of peptides or binding of oligonucleotides (ODNs) (5–7). However, these systems lose the simple growth and scale-up advantages of bacterial plasmids and have reduced gene expression because of loss of the supercoiled plasmid state (5–7).
Few coupling agents, if any, are robust enough to be applied to a broad range of gene delivery systems and conditions. One system described recently utilises ‘bis’ peptide nucleic acid (PNA) ODNs or ‘clamps’ for attachment of functional moieties to plasmid DNA, by hydrogen bonding with DNA bases, in a controllable and stoichiometric manner that does not interfere with gene expression (8,9). Although this has shown some promise for monitoring plasmid DNA distribution following gene delivery, in our hands the system is only useful for adding fluorophores to plasmid DNA under certain carefully defined binding conditions. The system is far from robust under some of the conditions required for delivery systems such as the gene gun, as described in this work. There is a clear need to design an improved controllable plasmid attachment system to add functional moieties to plasmid DNA in order to monitor and improve gene delivery and also for DNA vaccination to improve immunogenicity. We have devised an improved system based on ODNs of 100% locked nucleic acid (LNA) (10).
LNA is an analogue of RNA or DNA. The term LNA is used to describe both nucleotide monomers, in which the ribose ring is constrained by a methylene linkage between the 2′-oxygen and the 4′-carbon, and also ODNs that contain one or more monomers of LNA. The methylene bridge linkage can be through oxygen (oxy-LNA), sulfur (thio-LNA) or amine (amino-LNA). The conformation restriction increases binding affinity for complementary sequences (11). ODNs based upon LNA have extremely good hybridisation properties (i.e. high melting temperatures) and retention of sequence specificity of hybridisation/hydrogen bonding. The introduction of LNA monomers into DNA or RNA ODNs increases affinity for complementary DNA or RNA sequences, with melting temperature (Tm) increases in the range of 3–8°C, depending on the actual base, per LNA monomer present in the ODN (11).
The very high affinity and specificity for complementary nucleic acid targets of LNA-based ODNs has led to their successful use in a number of applications reliant on these properties, notably for antisense (12,13) and as probes for detecting DNA polymorphisms (14,15). The design and properties of LNA ODNs as antisense agents are now well described (16,17), and they have also been shown to be non-toxic for in vivo delivery at relatively high doses (12).
The aim of this work was to ascertain whether LNA ODNs could be used as agents to attach functional moieties to plasmid DNA and to compare their efficiency and robustness with PNA ODNs. Although LNA ODNs had been shown to bind and form triplexes with short double-stranded (ds) DNA sequences (18,19), this work is the first to show that LNA ODNs can bind to plasmid DNA and add a coupled functional moiety to that plasmid by stable binding (20). The sequence specificity and the mechanism of the binding of LNA ODNs to plasmid DNA is confirmed as being duplex invasion and strand displacement, by a modification of a single-stranded (ss)DNA sequencing procedure (21,22).
The science of DNA vaccination could be simplified if DNA and an immune adjuvant could be formulated for co-delivery. Synthetic ODNs containing core CpG motifs can mimic the immunostimulatory activity of bacterial DNA. Such signals can trigger B cells, natural killer (NK) cells and macrophages to proliferate, mature and secrete a variety of cytokines and chemokines (23). Previous work has shown that ODNs containing CpG motifs, particularly those based upon the more stable phosphorothioate (PTO) chemistry, can be useful vaccine adjuvants (24). It has been recently shown that the toll-like receptor 9 (TLR-9) is involved in the recognition of CpG motifs and that this interaction occurs via internalisation of CpGs into acidified endosomal vesicles (25–28). The recognition of CpGs and triggering of TLR-9 leads, in antigen-presenting cells (APCs) to the upregulation of co-receptor molecules and the secretion of a variety of cytokines including interleukin (IL)-12, IL-6, IL-1 and tumour necrosis factor-α (TNF-α) (24). RAW264.7 cells, which express high levels of TLR-9, have been used to confirm CpG immune adjuvant activity in vitro (29). In this work, CpG-based immune adjuvants have been attached to plasmid DNA as ‘hybrid’ PTO–LNA ODNs, and have been shown to induce high levels of TNF-α in RAW264.7 cells, similar to ‘free’ CpG ODNs (29).
MATERIALS AND METHODS
Oligonucleotides
The ODNs used in this study are listed in Table 1. All LNA ODNs were synthesised by Proligo LLC. Most were made with 5′-amino modifier C12 phosphoramidite spacers for labelling with Alexa Fluor dyes. Some were synthesised with rhodamine attached at the 5′ end. The PNAs described in Table 1 were purchased either from Oswel DNA Service or from GTS Inc. Most PNAs were purchased as 5′ rhodamine-labelled ‘bis’ PNA or PNA clamps (30,31); some were modified with a 5′-/N-terminal glycine residue and a 3′-/C-terminal lysine residue. LNA and PNA ODNs were based upon DNA sequences at the repeat binding sites 1 and 2, found on the GeneGrip plasmid series; see below (GTS). PNA/LNA ODNs containing either a (CT)n or a (GA)n repeat motif were designed to bind to GeneGrip site 1. LNA7 and LNA11 [(CCTT)n and (GGAA)n, respectively] were designed to bind to GeneGrip site 2 (Fig. 3A).
Table 1. List of oligonucleotides used in this study.
| Oligonucleotide | No. of plasmid-binding sites | Description | Sequence |
|---|---|---|---|
| LNA2 | 8 | 9mer 100% LNA | 5′-TAMRA-CTCTCTCTC-3′ |
| LNA4 | 6 | 13mer 100% LNA | 5′-TAMRA-CTCTCTCTCTCTC-3′ |
| LNA5 | 4 | 17mer 50% LNA | 5′-TAMRA-CtCtCtCtCtCtCtCtC-3′ |
| LNA6 | 6 | 13mer 100% LNA | 5′-NH2-CTCTCTCTCTCTC-3′ |
| LNA7 | 5 | 14mer 100% LNA | 5′-NH2-CCTTCCTTCCTTCC-3′ |
| LNA8 | 6 | 13mer 100% LNA | 5′-NH2-GAGAGAGAGAGAG-3′ |
| LNA9 | 6 | 11mer 100% LNA | 5′-NH2-CTCTCTCTCTC-3′ |
| LNA9R | 6 | 11mer 100% LNA | 5′-TAMRA-CTCTCTCTCTC-3′ |
| LNA10 | 8 | 9mer ‘bis’ 50% LNA, 50% DNA | 5′-NH2-CtCtCtCtC-XXX- CtCtCtCtC-3′ |
| LNA11 | 5 | 14mer 100% LNA | 5′-NH2-GGAAGGAAGGAAGG-3′ |
| PTOCpG–LNA | 6 | 20mer PTO, 1mer DNA, 13mer LNA | 5′-tccatgacgttcctgacgtttGAGAGAGAGAGAG-3′ |
| PTOGpC–LNA | 6 | 20mer PTO, 1mer DNA, 13mer LNA | 5′-tccatgagcttcctgagtcttGAGAGAGAGAGAG-3′ |
| CpG1826 | – | 20mer 100% PTO | 5′-tccatgacgttcctgacgtt-3′ |
| GpC1745 | – | 20mer 100% PTO | 5′-tccatgagcttcctgagtct-3′ |
| GTS PNA | 9–10 | 8mer ‘bis’ 100% PNA | 5′Rho-O-O-TCTCTCTC-O-O-O-JTJTJTJT-CONH2 |
| OsPNA2 | 6 | 13mer ‘bis’ 100% PNA | 5′ROX-O-O-gCTCTCTCTCTCTC-O-CTCTCTCTCTCTCk |
| OsPNA3 | 6 | 13mer 100% PNA | 5′ROX-O-O-gCTCTCTCTCTCTCk |
| OsPNA4 | 6 | 13mer ‘bis’ 100% PNA | 5′ROX-O-O-gCTCTCTCTCTCTC-O-O-O-CTCTCTCTCTCTCk |
| RevGG2B | 1 | 22mer 100% DNA | 5′(Cy5)-ggaaggaagttaggaaggaagg-3′ |
| KH2 | 4 | 19mer 100% DNA | 5′-Fl-gagagagagagagagagag- 3′ |
| KH3 | 3 | 18mer 100% DNA | 5′-Fl-ggaaggaaggaaggaagg- 3′ |
LNA residues are bold upper case; DNA residues are bold lower case; PTO residues are additionally italicised; PNA and amino acid residues are italicised, normal text, with PNA bases upper case. Numbers of binding sites refer to the maximum theoretical ODN-binding sites present on the GeneGrip plasmid series. O = 8-amino-3, 6-dioxaoctanoic acid linker; J = pseudoisocytosine; g = glycine; k = lysine; X = ‘PEG spacer’ -9-O-dimethoxytrityl-triethyleneglycol, 1-[(2-cyanoethyl)-(N,N-diisopropyl)]-phosphoramidite, spacer phosphoramidate 9; Fl = fluorescein, Rho = rhodamine, TAMRA = carboxytetramethylrhodamine, ROX = carboxy-X-rhodamine, NH2 = 5′-amino-modifier C12 phosphoramidite spacer.
Figure 3.


Sequencing strategy and results to show specific DNA strand displacement upon LNA ODN binding. (A) Sequencing strategy: GeneGrip site 2 on pGG2XGFP, plus bound LNA7 and DNA sequencing primer Cy5 RevGG2B (Table 1). Data displayed in (B–E) start from the marked T* and finish at the end of the arrow (A). (B) Sequence from pGG2XGFP by dsDNA sequencing. Further sequence data by ALF ssDNA sequencing kit, manually deciphered chromatograms: (C) 0.024 µM pGG2XGFP + 0.5 µM LNA7 (3 µg of template, primer annealed at 42°C); (D) 0.024 µM pGG2XGFP + 0.5 µM LNA7 (1 µg of template, primer annealed at 37°C); (E) pGG2XGFP alone (1 µg of template, primer annealed at 37°C).
The hybrid PTO/LNA ODNs, PTOCpG–LNA and PTOGpC–LNA, described in Table 1, were synthesised by Proligo LLC. Both ODNs contained twenty 5′ PTO residues, followed by a single deoxynucleoside phosphodiester residue, followed by thirteen 3′ LNA residues of the (GA)n motif as in LNA8 (Table 1). The PTO ODNs CpG1826 (24,32) and GpC1745 (32) (Table 1) and the remaining 100% DNA ODNs were synthesised by MWG-Biotech AG.
Plasmids
Plasmid DNA was prepared using the Qiagen MaxiPrep procedure or by the Qiagen Endofree Plasmid Maxi Kit and re-suspended in TE (10 mM Tris–HCl, 1 mM EDTA) pH 8.0 at 1 µg/µl. Plasmids were >95% supercoiled by agarose gel electrophoresis (33).
Most plasmids are based upon the pGeneGrip series, expressing either luciferase (gWiz) or green fluorescent protein (GFP; pGGGFP) [GTS; Zelphati et al. (8)]. Within the transcriptional terminator of plasmids gWiz and pGGGFP, enabling site-specific binding without interfering with gene expression, is GeneGrip site 1, a region in the plasmid of repeating sequences, based upon (CT)n with complementary repeat (GA)n on the opposite strand. Site 2 (Fig. 3A), which is located 5′ to the cytomegalovirus (CMV) promoter, is based upon (CCTT)n, with complementary strand (GGAA)n, and is found only in plasmids pGG2XGFP and pGG2XEMPTY, which additionally contain site 1 [GTS Catalogue 2002; Zephati et al. (8)].
Plasmid pGG2XEMPTY is derived from pGG2XGFP by deletion of the GFP gene. To construct plasmid pGG2XEMPTY, pGG2XGFP was digested with NheI and BamHI, and the remaining 5.1 kb plasmid fragment was gel purified, treated with Klenow DNA polymerase and re-circularised by ligation (33).
The plasmid pGL3CMV is a luciferase expression vector based upon pGL3 Basic (Promega) with the CMV immediate early promoter driving luciferase expression (10).
Endotoxin testing
Endotoxin levels were measured using either the Biowhittaker QCL-1000 LAL kit or the Pyrochrome LAL kit. Endotoxin levels for all plasmids and ODNs used in this study were <0.1 EU (endotoxin units)/µg DNA.
Fluorescent labelling of plasmid DNA
Plasmid DNA was labelled either with the Mirus Label IT, fluorescein or rhodamine labelling kits, or fluorescent labelled using TOTO-1 nucleic acid dye. Plasmid was usually labelled at the 100 µg scale and free dye was removed by ethanol precipitation (33). DNA was re-suspended at 1 µg/µl in water for LNA binding and transfection experiments.
Fluorescent labelling of oligonucleotides
LNA ODNs (25–100 µg) with 5′ primary amine groups were 5′ end labelled with Alexa Fluor dye, 568, 647 or 350. Labelled LNA ODNs were purified from free dye by ethanol precipitation and re-suspended in water at 20–200 µM.
ODNs without 5′ modifying groups were 5′ ARES fluorescent labelled. ODNs were fluorescent 3′ end labelled with fluorescein-12-ddUTP using terminal transferase or more efficiently with aminoallyl dUTP, followed by Alexa Fluor dye labelling as described above.
ODNs were also fluorescent labelled, by chemical modification, using the Alexa Fluor 488 Ulysis nucleic acid labelling kit. ODNs were generally labelled at the 5 µg scale and free dye was removed by ethanol precipitation (33). ODNs were re-suspended in water at 20–25 µM.
Binding of oligonucleotides to plasmid DNA and restriction enzyme analysis
Plasmid was usually incubated with PNA/LNA ODNs in 10 mM phosphate buffer, 1 mM EDTA, pH 5.8 for 16 h at 37°C, at a maximum of 4- to 40-fold molar excess of ODN to ODN-binding sites in the plasmid. Plasmid (0.023–0.027 µM) was incubated with ODNs at ‘low’ (0.5 µM) and ‘high’ (4 µM) concentrations. To visualise bound ODN, 2.5 µg of plasmid DNA was analysed by agarose–TAE gel electrophoresis without ethidium bromide (EtBr). High percentage (2%) gels were used to maximise separation of both plasmid-bound and free ODN, and resolution of the fluorescent signal. Conventional 0.8–1.0% gels resulted in diffusion of fluorescence intensity and overlap of plasmid-bound and free ODN signals [Zelphati et al. (9) and data not shown]. Any unbound ODNs were separated from plasmid and plasmid-bound ODN by gel exclusion chromatography using MicroSpin Sephacryl S400 HR columns.
For PTOCpG–LNA and PTOGpC–LNA ODNs binding to plasmid, 0.027 µM pGG2XEMPTY (2.5 µg) was incubated with 9 µM Ulysis Alexa Fluor 488-labelled PTOCpG–LNA or PTOGpC–LNA and the sample was split into two, one half being passed through an S400HR spin column (Fig. 6C and D). Prior to plasmid binding, the ODNs were heated at 80°C for 10 min and then plunged into ice, to disrupt any self-complementary interaction between the PTO and LNA bases within the ODN that might affect plasmid binding.
Figure 6.



‘Hybrid’ CpG PTO–LNA ODNs bind to plasmid and retain CpG activity in vitro. (A) Agarose gel (without EtBr) of 0.027 µM plasmid pGG2XEMPTY incubated with ODNs, labelled either by Ulysis Alexa Fluor 488 or by Alexa Fluor 568 at the 5′ end. (B) As (A) with EtBr. Lane M, 1 kb DNA ladder; lane 1, pGG2xEMPTY; lane 2, pGG2xEMPTY + 4 µM 5′ Alexa Fluor 568-labelled LNA8; lane 3, pGG2xEMPTY + 4 µM Ulysis-labelled LNA8; lane 4, pGG2xEMPTY + 9 µM Ulysis-labelled PTOCpG-LNA; lane 5, pGG2xEMPTY + 9 µM Ulysis-labelled PTOGpC-LNA. (C) Agarose gel (without EtBr) of plasmid incubated with Ulysis-labelled ‘hybrid’ PTO–LNA ODNs before and after S400HR gel exclusion chromatography. (D) As (C) with EtBr. Lane M, 1 kb DNA ladder; 0.027 µM pGG2xEMPTY + 9 µM PTOCpG-LNA before (lane 1) and after S400HR separation (lane 2). (E) TNF-α ELISA data for the dose response curve of gWiz plasmid ± bound PTOCpG–LNA or PTOGpC–LNA, transfected into RAW264.7.
Any additional binding of fluorescent DNA ODNs to plasmid DNA–LNA complexes was at 37°C for 45 min in 10 mM sodium phosphate pH 7.1, 1 mM EDTA at 4 µM DNA ODN.
Restriction enzyme digests of 2.5 µg of plasmid DNA were performed after overnight LNA or PNA ODN binding at 37°C (33). Plasmid gWiz was digested with BsaI and SphI, and plasmid pGG2XGFP was digested with NdeI. Samples were then analysed on 2% agarose–TAE gels without EtBr.
DNA sequencing reactions
Standard dsDNA sequencing was performed by ‘big dye’ PCR-based thermocycle sequencing using the fluorescent dideoxy terminator method, run on a PE-Biosystems Prism 3700 Capillary sequencer and visualised on an ABI 3700 DNA Analyser. To identify strand displacement from LNA ODNs binding to plasmid DNA, an ssDNA sequencing assay was established. This was based upon methods demonstrating PNA ODN strand displacement (21,22).
An optimal DNA sequencing primer (RevGG2B, Table 1) was designed (±5′ Cy5 label) and verified by good quality sequencing across the GeneGrip site 2 repeat region in pGG2XGFP (Fig. 3A) by standard ‘big dye’ sequencing (Fig. 3B). A 25 µg aliquot of plasmid pGG2XGFP (0.024 µM) was bound with ODN LNA7 (low concentration: 0.5 µM) and unbound LNA ODN was removed. Plasmid pGG2XGFP with and without bound LNA was then subject to a modified ssDNA sequencing protocol using the AutoRead Sequencing Kit (Amersham Pharmacia Biotech) with Cy5-labelled RevGG2B DNA primer and T7 DNA polymerase. The dose of template plasmid DNA was varied from 1 to 3 µg and the annealing temperature reduced to either 37 or 42°C, but the annealing time was extended to 30 min. This was to maximise sequence-specific binding of the DNA sequencing primer to any displaced ssDNA regions under conditions that should not disrupt the double-stranded nature of the plasmid. Sequencing reactions were then run on a Visible Genetics DNA Sequencer and modified using Chromas software. Using the known DNA sequence of the region, the DNA sequence obtained for plasmid with LNA bound was interpreted by eye.
Preparation and gene gun delivery of plasmid DNA
Plasmid DNA (1 µg/µl) and 2 µm gold particles were re-suspended in 0.05 M spermidine. DNA was precipitated on to gold particles (‘gold slurry’) by addition of 1 M CaCl2. The DNA–gold complex was incubated for 10 min at room temperature and washed in moisture-free absolute ethanol. Samples were re-suspended in absolute ethanol containing 0.05 mg/ml polyvinylpyrrolidone (PVP). DNA was eluted from the ‘gold slurry’ by shaking in TE for 15 min at 37°C. Eluted plasmid samples, ‘gold slurry’ samples and 2.5 µg plasmid samples with bound fluorescent PNA or LNA ODNs (‘pre-gold slurry preparations’) were analysed on 2% agarose–TAE gels without EtBr.
For gene gun cartridge preparation, plasmid DNA-coated ‘gold slurry’ was re-suspended in ethanol/PVP at a sufficient volume to allow a DNA loading rate of 2 µg DNA/mg gold. DNA-coated gold was applied to the inner surface of the Tefzel tubing by centrifugal force and was cut into cassettes.
Gene gun delivery of plasmid DNA to MC57 cells was performed using the plate method (34). Cells were transfected using an Accell XR1 gene gun at 250 p.s.i. (PowderJect). Cells were incubated for 24 h at 37°C and 5% CO2 before harvesting for luciferase and total protein assays.
Cell lines, growth and transfection conditions
Cells were cultured at 37°C in a 5% CO2 humidified incubator. HeLa cells and MC57, a murine fibrosarcoma cell line (35), were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 2 mM glutamine. RAW264.7, a murine macrophage cell line, was grown in RPMI 1640 with 10% low IgG FCS and 4 mM glutamine. Chinese hamster ovary (CHO) K1 cells were maintained in Iscove’s modified Dulbecco’s medium with 10% FCS, 2 mM glutamine with non-essential amino acids [1% (v/v)] and Hypoxanthine/Thymidine (HT) supplement (Invitrogen). All media contained 100 U/ml penicillin and 100 µg/ml streptomycin.
HeLa cells were transfected using DMRIE-C. Cells were harvested 24 h post-transfection and assayed for luciferase and total protein. CHO K1 cells were transfected using Transfast™ (Promega). For DNA transfection and ODN incubation, RAW264.7 cells were grown to confluence (96-well plates), washed once with phosphate-buffered saline (PBS) and incubated in Optimem. A transfection mixture of 0.01–10 µM PTO ODNs ± FuGENE6, at a ratio of 1 µM ODN:0.5 µl FuGENE6, in Optimem was added to RAW264.7 cells and incubated for 14 h at 37°C. Controls included solely FuGENE 6 and 10–500 ng of gWiz plasmid at 1 µg of DNA:6 µl of FuGENE 6.
Luciferase and protein assays
Cells, washed with PBS, were lysed in passive lysis buffer (Promega) or Magnus lysis buffer [10 mM EDTA, 0.25% Triton X-100, 10 mM dithiothreitol (DTT), 250 mM HEPES pH 7.5] and, for Magnus lysis buffer, frozen at –20°C and thawed at room temperature. Lysate was assayed together with luciferase assay reagent (Promega) in 96-well plates. Luciferase activity was measured as counts per second in the TopCountNXT HTS scintillation and luminescence counter, or as counts per minute on the Victor II 1420 multilabel HTS counter on the luminescence program. Total protein concentration was calculated by Coomassie Plus protein assay reagent kit. The absorbance was measured at 595 nm on a Molecular Devices Spectra Max 340. Results were expressed as the mean of triplicate samples (µg/ml). Luciferase activity was expressed as relative light units (RLU)/mg of total protein.
Confocal microscopy
CHO K1 cells, grown on 8-well glass chamber slides, washed with PBS, were fixed with 4% paraformaldehyde. Cells were mounted in Vectorshield containing 4′,6-diamidino-2-phenylindole (DAPI) for analysis by confocal fluorescence microscopy on a Leica TCS NT microscope, utilising argon (UV, 351 and 364 nm), argon (visible, 476 and 488 nm), krypton (visible, 568 nm) and helium/neon (visible, 633 nm) lasers. Samples were viewed under a 63× water immersion lens. Images were analysed by the TCS image analysis package.
TNF-α ELISA
RAW264.7 cell culture supernatants were taken to detect murine TNF-α levels using the R&D systems Duoset enzyme-linked immunosorbent assay (ELISA) kit. Samples were harvested 14 h post-incubation with CpG stimulatory sequences. Fresh medium was added to RAW264.7 cells, for harvesting 24 h post-transfection, for luciferase assays upon the lysed cells. Supernatant samples were diluted in 1% bovine serum albumin (in PBS) in 96-well ELISA plates. Absorbance was measured at 450 nm on a Molecular Devices Spectra Max 190, and TNF-α values were calculated using a 4-PL curve fit on the Softmax Pro 3.1.2 software. Results were expressed as the mean of duplicate samples (ng/ml).
Conversion of plasmid dose to equivalent ‘bound’ CpG oligonucleotide concentration
Doses of plasmid gWiz (µg), transfected into RAW264.7 cells, were converted to doses of CpG ODN (µM) that would bind to plasmid if the CpG ODN was attached to an LNA ODN with binding sites in the DNA (e.g. for PTOCpG–LNA; Table 1). The conversion was based upon the following: 1 µg of a 5 kb plasmid is 0.3 pmol (GTS ‘PNA clamp’, manufacturer’s instructions), gWiz is 6.7 kb, so 1 µg of gWiz binds 0.23 pmol of PTOCpG–LNA at each of the six binding sites on the plasmid (Table 1). ‘Hybrid’ PTO–LNA ODNs, such as PTOCpG–LNA, were normalised to PTO ODN dose. These values were used to normalise TNF-α levels between gWiz plasmid and free CpG-containing ODNs in RAW264.7 transfections.
RESULTS AND DISCUSSION
LNA oligonucleotides bind to supercoiled plasmid DNA
Fluorescent labelled LNA ODNs bind to plasmids of the pGeneGrip series (GTS). The 11–14mer, 100% LNAs based on the (CT)n-, (GA)n-, (CCTT)n- and (GGAA)n-binding motifs for GeneGrip sites 1 and 2, including ODNs LNA6, LNA7, LNA8, LNA9, LNA9R, LNA10 and LNA11 (Table 1), all bind to plasmid. An example is shown in Figure 1, where binding of 5′ fluorescent labelled LNAs to plasmid gWiz is demonstrated. Agarose gel electrophoresis of LNA ODN binding to plasmid can be visualised under UV illumination in the absence of EtBr staining [Fig. 1A; Zelphati et al. (9)]. Plasmid not incubated with a fluorescent LNA ODN shows no such fluorescence (Fig. 1A, lane 1). LNA ODNs are able to accommodate fluorophores at the 5′ end (Fig. 1 and fig. 1 of the Supplementary Material), at the 3′ end (data not shown) and internally (Fig. 6A and B, lanes 3–5) without interfering with plasmid binding. Plasmid binding was not seen if LNA ODNs were incubated with plasmids that did not contain their binding site (data not shown).
Figure 1.

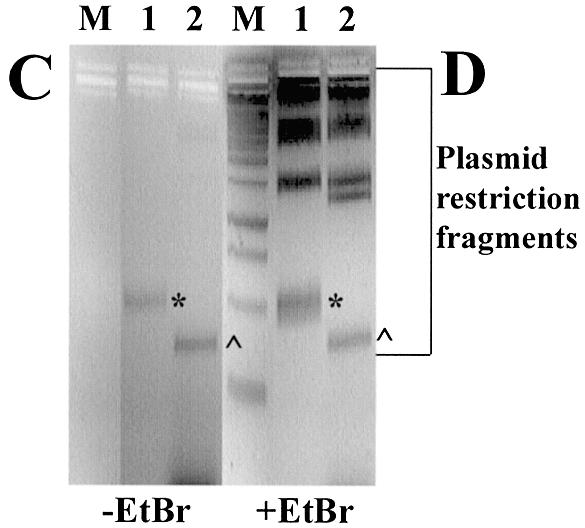
Binding of ODNs to plasmid and restriction mapping of the binding site. (A) Agarose gel (without EtBr) of 0.023 µM plasmid gWiz incubated with Alexa Fluor 568- (or rhodamine for LNA4) labelled LNA ODNs at high, (H, 4 µM) or low (L, 0.5 µM) ODN concentrations. (B) As (A) with EtBr. Lane M, 1 kb DNA ladder; lane 1, gWiz without ODN. (C) Agarose gel (without EtBr) of plasmids bound with Alexa Fluor 568- labelled LNA ODNs digested with restriction enzymes. (D) As (C) with EtBr. Lane M, 1 kb DNA ladder; lane 1, 0.024 µM pGG2XGFP + 4 µM LNA7 digested with NdeI; lane 2, 0.023 µM gWiz + 4 µM LNA8 digested with BsaI and SphI.
The rhodamine-labelled LNA ODNs LNA2, LNA4 and LNA5 (Table 1) were designed based upon the (CT)n GeneGrip site 1 motif on plasmid gWiz, to compare their ability to bind plasmid DNA with that of similar PNA ODNs: GTS PNA (8), OsPNA2, OsPNA3 and OsPNA4 (Table 1). The data demonstrated that (CT)n-based LNA ODNs, from 9mer 100% LNAs to 17mer 50% LNA–DNA ODNs, bind to plasmid at least as well as similar ‘bis’ PNA ODNs (8,21,22,30,31; Supp. Fig. 1).
A range of LNA–plasmid binding conditions were evaluated for LNA2, LNA4, LNA5 and gWiz, including pH range, from 5.8 to 7.0, addition of sodium chloride to 100 mM and the presence of 3 M tetramethyl ammonium chloride (TMAC). Incubation time at 37°C was also reduced to 1–3 h, and incubation temperatures of 20 or 4°C for overnight incubation were also evaluated. For all the conditions described above, LNAs could be detected as bound to plasmid by the agarose gel electrophoresis, UV visualisation assay (Supp. Fig. 2). From this analysis, the most efficient binding appeared to be to LNA4 (100% LNA 13mer) in 10 mM phosphate buffer, 1 mM EDTA pH 5.8 for 16 h at 37°C. Similar studies with PNA ODNs (Table 1) showed that OsPNA3, the only non-‘bis’ PNA, did not bind plasmid DNA under conditions which allowed LNA ODNs to bind. Even OsPNA4, the longest ‘bis’ PNA, only bound plasmid after incubation for 16 h at 37°C in 10 mM phosphate, 1 mM EDTA pH 5.8 (data not shown).
Confirmation of LNA ODN binding to the expected complementary sequence within plasmid DNA was obtained by restriction enzyme analysis of plasmid DNA from LNA binding experiments. An example is shown in Figure 1C and D for Alexa Fluor 568-labelled LNAs LNA7 and LNA8. Binding was only to the small restriction fragment containing the cognate binding site: GeneGrip 1 on a 300 bp BsaI–SphI fragment of gWiz for LNA8 (lane 2 ^) and GeneGrip 2 on a 491 bp NdeI fragment of pGG2XGFP for LNA7 (lane 1*). All LNAs with (CT)n and (GA)n motifs only showed binding to the expected GeneGrip 1-containing fragment, whereas LNAs with (CCTT)n and (GGAA)n motifs only showed binding to the expected GeneGrip 2-containing fragment (Table 1, data not shown).
LNA oligonucleotide binding is primarily by strand displacement
Differences in the binding of LNA ODNs to plasmid DNA at low (0.5 µM) and high (4 µM) ODN concentrations were also studied. Analyses of LNAs based upon the (CT)n sequence (LNA2, LNA4 and LNA5) for binding to gWiz plasmid DNA are shown in Figure 2A and B. All LNA ODNs show increased binding to plasmid DNA at high compared with low ODN concentrations (Fig. 2A). The numbers of theoretical binding sites for the ODNs range from four to eight for longer to shorter ODNs. The increase in plasmid-bound LNA ODNs at the higher concentration suggests that two mechanisms for LNA ODN binding may be in operation, perhaps doubling the amount of plasmid-bound LNA at high compared with low ODN concentrations. This could be achieved by both Watson–Crick and Hoogsteen base pairing at high LNA ODN concentrations and led to triplexes of LNA:DNA:LNA, similar to those described for ‘bis’ PNAs (8,30). At low LNA ODN concentrations, only one binding mechanism may be occurring, either Watson–Crick or Hoogsteen.
Figure 2.
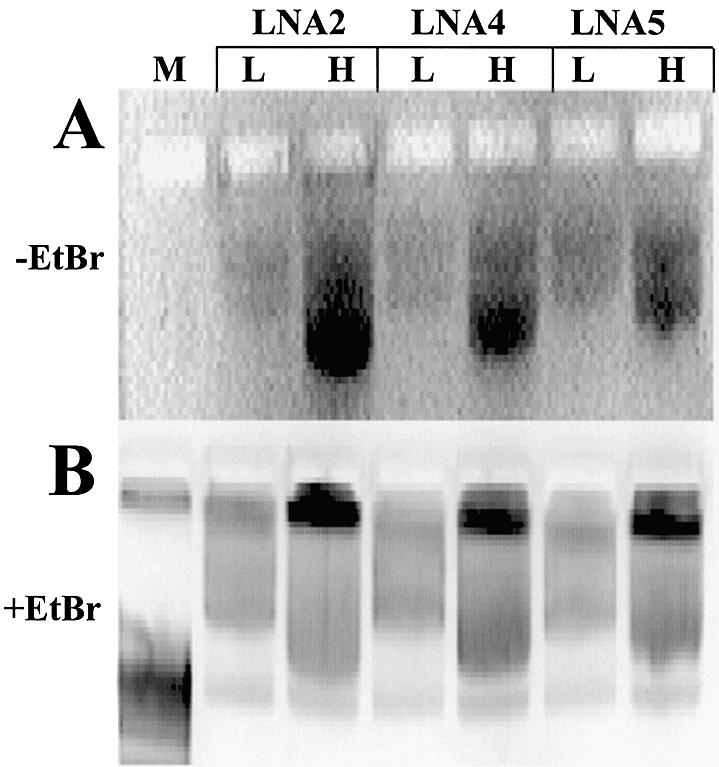
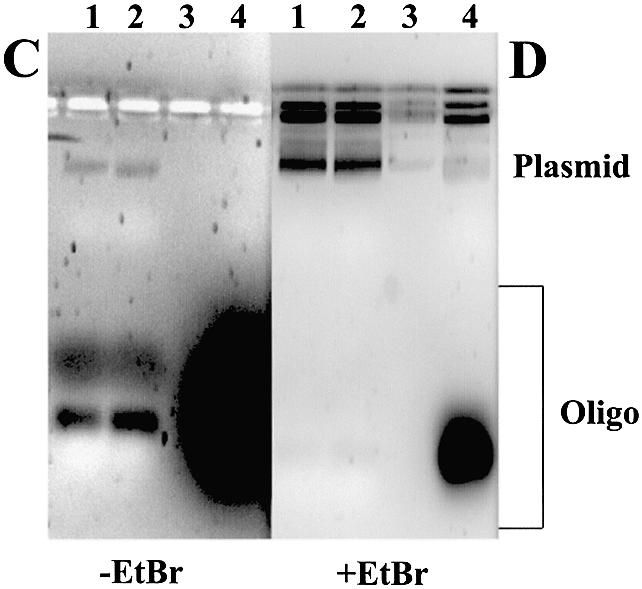
Binding of LNA ODNs to plasmid allows complementary DNA ODNs to bind. (A) Agarose gel (without EtBr) of 0.023 µM plasmid gWiz incubated with rhodamine-labelled LNA ODNs at high (H, 4 µM) or low (L, 0.5 µM) ODN concentrations. (B) As (A) with EtBr. Lane M, 1 kb DNA ladder. (C) Agarose gel (without EtBr) of 0.023 µM plasmid gWiz incubated with LNA ODNs at high (H, 4 µM) or low (L, 0.5 µM) ODN concentrations. Unbound LNA ODN was removed, the plasmid–LNA ODN complex was incubated with the fluorescein-labelled DNA primer KH2 (4 µM) and free ODN was again removed prior to loading on a gel (except lane 4). (D) As (C) with EtBr. Lane 1, gWiz + LNA6 H + KH2; lane 2, gWiz + LNA6 L + KH2; lane 3, empty; lane 4, gWiz + KH2.
An assay to show strand displacement of the complementary DNA strand upon LNA ODN binding to plasmid was designed from published methods (36). LNA ODNs were bound to plasmid at low and high concentrations and any unbound LNA was removed. A fluorescein-labelled DNA ODN of complementary sequence to the LNA, capable of binding to the displaced DNA strand, was incubated with the plasmid–LNA complex and free DNA ODN was removed. The LNA ODN–plasmid–DNA ODN complex was analysed by agarose gel electrophoresis. An example is shown in Figure 2C and D (lanes 1 and 2), showing detection of LNA6-mediated strand displacement at GeneGrip site 1 of plasmid gWiz with the DNA ODN KH2 (Table 1). In the absence of bound LNA ODN on the opposite plasmid DNA strand, the fluorescent DNA ODN is unable to bind (Fig. 2C and D, lane 4). Very similar data were obtained using plasmid pGG2XGFP as template and LNA7 for binding and DNA strand displacement, at GeneGrip site 2, so that DNA ODN KH3 (Table 1) could bind the displaced strand (data not shown).
To demonstrate that sequence-specific LNA ODN binding to plasmid DNA, at its cognate binding site, causes strand displacement of the unbound DNA strand, an ssDNA sequencing assay was established. This was based upon a modification of similar methods that have been described to demonstrate PNA ODN-based strand invasion and displacement (21,22). The modified procedure was to use double-stranded supercoiled plasmid DNA (dsDNA) in the presence or absence of a bound LNA ODN as a template for a DNA sequencing procedure that is optimal only for ssDNA. The plasmid employed for this analysis was pGG2XGFP, and LNA7 was used as a ‘strand-displacing agent’ (Table 1).
The data are shown in Figure 3C and D, with the known DNA sequence of the region in Figure 3A and B. The DNA sequence obtained in Figure 3C and D for plasmid with LNA bound is clearly the correct DNA sequence for specific binding of the DNA sequencing primer to its cognate binding site. The trace data displayed in Figure 3B–E represent DNA sequence primed from the marked T*, and finish at the end of the arrow (Fig. 3A). Control DNA sequencing reactions run in parallel under identical conditions, containing plasmid without LNA bound, generated only non-specific signal (Fig. 3E). These data suggest that, at low LNA ODN concentrations (0.5 µM), LNAs bind to the correct binding sites, by strand invasion and Watson–Crick base pairing, leading to strand displacement and allowing ssDNA sequencing of the displaced strand.
Ulysis labelling adds fluorescent molecules to N7 atoms of guanine (G) and adenine (A) purine rings. For ODNs based upon LNA8, LNA binding to plasmid is only via G and A residues (Table 1). Hoogsteen base pairing via the N7 atoms would be blocked by the large fluorescent label (19). Watson–Crick base pairing through purines should be unaffected by the labelling. Ulysis-labelled LNA8 binds to plasmid (Fig. 6A, lane 3). This provides further evidence that the primary mechanism for LNA ODN binding to plasmid is strand displacement by Watson–Crick base pairing. From solution structure studies of LNA:DNA duplexes, high stability of binding is favoured by local changes in the phosphate backbone geometry that favour a higher degree of base stacking with LNA:DNA Watson–Crick base pairing (37).
The DNA sequence data (Fig. 3), the DNA ODN binding data to the displaced strand (Fig. 2C and D) and the Ulysis-labelled LNA ODN binding data (Fig. 6A, lane 3), taken together, suggest that the major mechanism of binding of LNA ODNs, even at the low ODN concentration, is by Watson–Crick-based strand invasion and strand displacement.
For LNA ODNs based upon the (CT)n motif, there is some evidence for additional Hoogsteen base pairing with DNA at high ODN concentrations (Fig. 2A and B). Similar behaviour could not be demonstrated for other LNA ODNs that were not based upon the sequence (CT)n (data not shown), so this may be a feature of ODNs based upon this repeating polypyrimidine sequence which are thought to Hoogsteen base-pair more readily (8,30).
Gene gun preparations show enhanced stability of LNA over PNA oligonucleotides bound to plasmid
Although GTS PNA [Table 1; Zelphati et al. (8)] binds to gWiz plasmid DNA (Supplementary fig. 1), the binding is unstable when bound plasmid is used to coat gold beads for gene gun delivery (Fig. 4C and D). Plasmid binding stability in this assay was studied for LNA ODNs based upon (CT)n-, (GA)n- and (CCTT)n-binding motifs (LNA7, LNA8, LNA9R and LNA10), and for PNA ODNs based on the (CT)n motif (GTS PNA, OsPNA2 and OsPNA4) (Table 1). An example of these data, showing plasmid-bound ODNs before and after ‘gold slurry’ preparation, can be seen in Figure 4A and B. All the LNA ODNs described above and those shown in Figure 4A and B remained plasmid bound upon ‘gold’ slurry’ preparation, the shortest being LNA9R, a (CT)n motif, 11mer 100% LNA.
Figure 4.

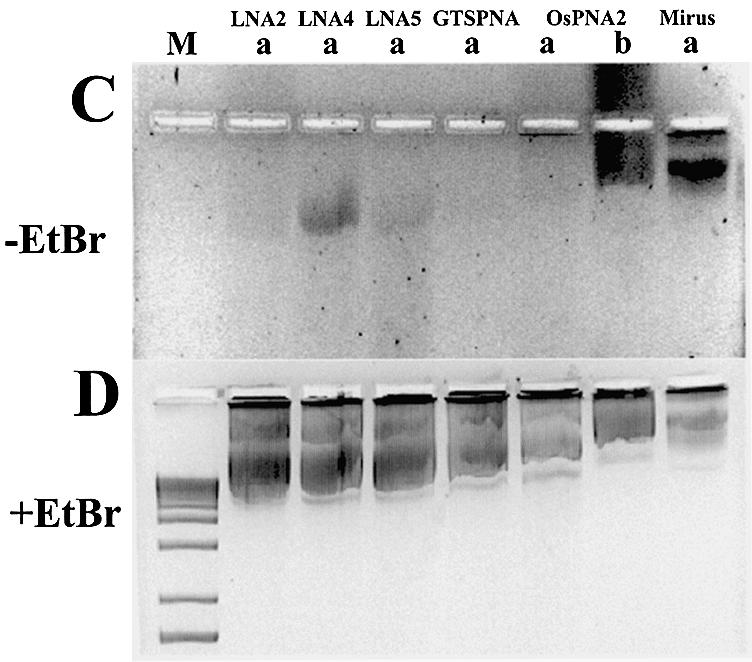
Binding stability of LNA and PNA ODNs to plasmid in gene gun preparations. (A) Agarose gel (without EtBr) of 0.024 µM plasmid pGG2XGFP incubated with 4 µM Alexa Fluor 568 (LNA) or rhodamine-labelled (PNA) ODNs before (b) and after (a) plasmid binding to gold beads. (B) As (A) with EtBr. Lane M, 100 bp DNA ladder. (C) Agarose gel (without EtBr) of 0.023 µM plasmid gWiz incubated with 4 µM rhodamine-labelled LNA or PNA ODNs after (a) plasmid binding to gold beads, unless stated as before (b). (D) As (C) with EtBr. Lane M, 1 kb DNA ladder; Mirus, pGL3CMV rhodamine labelled with Mirus Label IT kit.
A comparison was made between LNA and PNA ODNs based on the (CT)n motif, for stability of plasmid-bound ODN upon coating gold beads. Figure 4C and D shows ODN-bound plasmid samples eluted from ‘gold slurry’ and analysed on agarose gels. ODN LNA4 remains attached to plasmid (Fig. 4C and D), whereas LNA2 and LNA5 show reduced levels of plasmid-bound ODN (cf. Supplementary fig. 1). Little or no PNA remains attached to plasmid, post-‘gold slurry’ preparation (Fig. 4A–D). Attached PNA ODNs are mainly removed by this procedure.
For all LNA ODNs studied, binding of LNA8 [(GA)n motif] seems least disrupted by the ‘gold slurry’ procedure (Fig. 4A and B). Comparisons between purified rhodamine-labelled ODNs and non-purified Alexa Fluor-labelled LNA ODNs may, however, not be strictly quantitative. PNA ODNs based on polypurines are not well described and may only be able to bind by Hoogsteen base pairing. PNA ODNs offer less sequence flexibility for binding stably to plasmid DNA compared with LNA ODNs (38).
A 9mer ‘bis’ 50% LNA–DNA ODN (LNA10) was designed, comparable with ‘bis’ PNA ODNs (8,22,31). Binding of LNA10 to plasmid should allow both Watson–Crick and Hoogsteen base pairing to enable triplex formation (30). However, LNA10 did not demonstrate improved binding (Table 1; Fig. 1), and only showed marginally improved stability over the 9mer LNA2 when bound to plasmid DNA in ‘gene gun’ preparations (see Fig. 4A and B, compared with C and D). Most simple 100% LNA ODNs bind to DNA more stably than the ‘bis’ LNA10 (Table 1, Fig. 1 and Supplementary fig. 1). LNA10 is 50% LNA–50% DNA and might be expected to have intermediate hybridisation properties between those of 100% LNA and 100% DNA ODNs. LNA10 was difficult to synthesise (Proligo LLC, personal communication) and offers no advantage over simple linear 13–14mer 100% LNA ODNs.
The longest ‘bis’ PNA ODN (OsPNA4) showed very little plasmid binding after ‘gold slurry’ preparation (Fig. 4A and B). The ‘O’ residues (Table 1) separate PNAs into two; the region 3′ to the ‘O’ residues Hoogsteen base-pairs with DNA. Hoogsteen base pairing is more robust for PNAs containing pseudoisocytosine, not cytosine, residues (e.g. GTS PNA, Table 1), enabling Hoogsteen base pairing at high pH >5–6, whereas PNAs containing cytosine only bind at low pH <5–6 (30). The addition of certain amino acids improves the stability of ‘bis’ PNAs when bound to DNA (21). However, the 13mer ‘bis’ PNA with additional amino acids to improve stability (OsPNA4, Table 1) was outperformed by most 100% LNA ODNs tested in the ‘gold slurry’ preparation assay (Fig. 4).
Of those tested, the LNA ODNs appear superior to the PNA ODNs in withstanding the conditions required for ‘gene gun’-mediated delivery when bound to plasmid DNA. LNA:DNA hybridisation properties appear more robust than PNA:DNA for direct Watson–Crick base pairing, whereas PNA:DNA interactions require additional, weaker Hoogsteen base pairing. One or a combination of the excipients (i.e. spermidine, CaCl2 and PVP) used to condense and coat plasmid DNA on to gold particles interferes with the PNA:DNA hybridisation and removes even sophisticated PNA ODNs such as PNA clamps or ‘bis’ PNA. From these data, LNA ODNs may represent a simpler and more robust alternative to PNA ODNs for adding functionality to plasmid DNA, at least for gene gun delivery applications.
LNA oligonucleotides remain associated with plasmid DNA in mammalian cells and allow plasmid-derived gene expression
To demonstrate that LNA ODNs, when bound to plasmid DNA, remain associated with the plasmid after transfection into mammalian cells, experiments were performed in CHO cells, with fluorescent plasmids bound with differentially fluorescent LNA ODNs, and analysed by confocal microscopy. Fluorescent LNA ODNs were bound to plasmids gWiz or pGG2XEMPTY, which had been previously labelled with either a Mirus fluorescein labelling kit or with bound TOTO-1 dye. Mirus-labelled plasmid gWiz was bound with LNA9R (Table 1), free LNA was removed and then the plasmid–LNA conjugate was transfected into CHO cells. The fixed CHO cells were analysed by confocal microscopy and the data are shown in Figure 5A. This shows an association or co-localisation of the plasmid-derived fluorescein signal and the LNA9R-derived rhodamine signal (Fig. 5A, panels 1, 3 and 4). The data suggest that bound LNAs remain associated with plasmid following lipid-mediated transfection into CHO cells. In this transfection, plasmid and ODN are largely endosomally located. Similar experiments showed for three different LNA ODNs, binding to two different sites within a plasmid, co-localisation of all labelled LNAs with plasmid DNA after transfection into CHO cells (data not shown).
Figure 5.



Co-localisation of plasmid and bound LNA ODNs when transfected into CHO cells and demonstration that plasmid with bound LNA ODNs shows unmodified gene expression. (A) Co-localisation in CHO cells, 36 h post-transfection, of LNA bound to plasmid, visualised by confocal microscopy. 1, gWiz labelled with fluorescein Mirus Label IT kit, FITC channel; 2, DAPI-stained nuclei, UV light; 3, LNA9R, TRITC channel; 4, overlay of 1, 2 and 3. (B) As (A), but control of gWiz mixed with Alexa Fluor 568-labelled, unbound LNA ODN. 1, gWiz labelled with fluorescein Mirus Label IT kit, FITC channel; 2, DAPI-stained nuclei, UV light; 3, LNA11, TRITC channel; 4, overlay of 1, 2 and 3. (C) Luciferase activity, 24 h post-transfection, in MC5-7 cells transfected by gene gun with gWiz ± LNA or PNA ODNs, or pGL3CMV ± rhodamine Mirus Label IT. Values are a mean of six pooled, independent gene gun transfections. (D) Luciferase activity, 24 h post-transfection, in HeLa cells lipofected by DMRIE-C with gWiz ± LNA or PNA ODNs. Values are a mean of four independent transfections, with standard deviations shown.
Control experiments were performed to show that co-localisation did not occur due to co-uptake of unlinked plasmid and LNA ODNs into the same endosomes. Plasmid gWiz was mixed with Alexa Fluor 568-labelled LNA11 (unable to bind to gWiz as GeneGrip site 2 is absent; Table 1). The plasmid and LNA mixture was similarly transfected into CHO cells, which were fixed for confocal analysis (Fig. 5B). No co-localisation of plasmid and LNA signal can be seen (Fig. 5B, panels 1, 3 and 4).
No ‘cross-talk’ between any of the individual signals within any of the channels could be identified from separate, parallel transfections of individual fluorescent DNAs or ODNs into the same cells (data not shown).
For LNA ODNs to be used for attachment of functional moieties to plasmid DNA, LNA binding should not interfere with plasmid-derived gene expression. The ODN-binding sites present on the GeneGrip plasmids are within regions where binding does not obstruct RNA polymerase progression (8). LNA ODNs only bind to the cognate ODN-binding sites on these plasmids (8), but the direct effect of LNA ODN binding upon plasmid gene expression had not been determined. This was assayed by transfecting mammalian cells with gWiz, with or without bound LNA or PNA ODNs. Figure 5C shows comparative luciferase expression from in vitro gene gun delivery of LNA and PNA ODN-bound gWiz plasmid in MC57 cells. Plasmid gWiz with bound LNA4 ODN is as transcriptionally active as gWiz bound with GTS PNA. The PNA is removed in the ‘gold slurry’ preparation procedure (Fig. 4C and D, lane 4), so this is equivalent to plasmid without bound ODN. Data are compared with unlabelled versus Mirus-labelled pGL3CMV, where, for the latter chemically modified plasmid, gene expression is abolished (Fig. 5C). Results similar to those in Figure 5C were obtained in HeLa cells. Comparative luciferase expression data are shown in Figure 5D. Similarly, gWiz plasmid, with or without bound LNA or PNA ODNs, showed no significant difference in gene expression (Fig. 5D). The binding of LNA, similarly to PNA ODNs, does not interfere with gene expression in this system.
LNA oligonucleotides can add CpG adjuvant activity to plasmid DNA
Synthetic ODNs containing core CpG motifs can act as immune adjuvants (24). An in vitro assay was established based upon transfection of RAW264.7 cells. RAW264.7 express high levels of TLR-9, and can be used to measure CpG activity as induction of TNF-α (29,39–42). Experiments were performed with PTO CpG ODNs to determine if the assay was sensitive enough to detect CpG activity from doses of PTO CpG ODNs equivalent to those that could be coupled to plasmid, via LNA ODNs, against any background TNF-α levels induced by plasmid alone. An example of this assay is shown in Supp. Fig. 4. The PTO ODN CpG1826 shows an adjuvant effect, whereas the negative control GpC1745 does not (Table 1, Supp. Fig. 4). The conversion of plasmid to CpG dose suggested that if PTOCpG–LNA (Table 1) was bound to gWiz at all cognate ODN-binding sites, a sufficient (6-fold) increase in TNF-α signal over gWiz alone would be detected (Supp. Fig. 4). Plasmid gWiz has one murine 6mer ‘core’ CpG sequence which may be responsible for its baseline TNF-α production (26).
‘Hybrid’ PTO–LNA ODNs were designed to combine CpG adjuvant activity with the plasmid binding of the LNA. Two ‘hybrid’ PTO–LNA ODNs were synthesised: PTOCpG–LNA and PTOGpC–LNA, based upon CpG1826 and its negative control GpC1745, respectively (24,32; Table 1). PTOCpG–LNA and PTOGpC–LNA ODNs were separately fluorescent labelled and bound to plasmid, and the resulting products were analysed on agarose gels (Fig. 6A and B). In both cases, ‘hybrid’ PTO–LNA ODN bound to plasmid, as did a 100% LNA ODN control (Fig. 6A and B, Table 1). A 5′ extension of a 13mer 100% LNA ODN by 21 nucleotides does not prevent plasmid binding (Fig. 6A and B).
Binding of PTOCpG–LNA or PTOGpC–LNA ODNs to plasmid left some free ODN (labelled ‘Oligo’, Fig. 6A and B). To be sure that the effect of adding CpG to plasmid, via LNA binding of PTO CpG ODNs, is being measured, free CpG ODN must be removed from plasmid–PTOCpG–LNA. S400HR spin column separation is sufficient to remove all unbound PTOCpG–LNA (Fig. 6C and D); bands of free ODN (labelled ‘Oligo’, Fig. 6C and D), present on the gel in the non-S400HR-treated sample, have been completely removed.
For proof of concept studies to show that LNA ODNs could be used to add CpG adjuvant activity to plasmid, unlabelled PTOCpG–LNA and PTOGpC–LNA ODNs were bound separately to plasmid gWiz, and any free ODN was removed. The ODN and plasmid samples used were endotoxin free. The plasmid–PTOCpG–LNA conjugates were used in RAW264.7 transfection experiments to show TNF-α induction. Data demonstrating the adjuvant effect from PTOCpG–LNA ODN bound to plasmid gWiz after transfection into RAW264.7 cells is shown in Figure 6E. The difference in levels of TNF-α induced by gWiz, with or without bound PTOCpG–LNA or PTOGpC–LNA, is shown. If plasmid gWiz has PTOCpG–LNA ODN bound, there is a 6- to 8-fold increase in TNF-α levels over those induced by plasmid alone. This compares well with that predicted from Supplementary figure 4 (6-fold). It is unclear whether free ODNs or plasmid-derived CpG motifs can interact equally well with TLR-9 (23). PTOCpG–LNA (Table 1) contains a phosphodiester link for base 21 which could allow the PTO CpG portion of the ODN to be cleaved from the plasmid in the endosome by DNases. This may also help to contribute to the high level of TNF-α induction demonstrated in this assay. The data demonstrate that an immune adjuvant, CpG ODN, can be added to plasmid coupled by LNA ODNs.
This is a significant finding, as previous attempts to enhance the immunogenicity of plasmid DNA by simply directly mixing PTO CpG ODNs with plasmid resulted in reduced immunogenicity of the plasmid-encoded antigen (43,44). This is due to a PTO ODN dose-dependent reduction in antigen expression, which is thought to be due to competition between ODN and plasmid DNA for cellular uptake (43). In the coupled LNA system, CpG ODN is bound to plasmid and so should not compete for uptake with plasmid. Plasmid gWiz encodes luciferase, enabling luminescence activity to be measured from cells used for the TNF-α ELISA (Fig. 6E), receiving plasmid with and without bound PTOCpG–LNA or PTOGpC–LNA (Supp. Fig. 5A). Luciferase levels were comparable, so bound CpG ODN on gWiz DNA does not reduce plasmid-encoded gene expression. RAW264.7 cells were transfected with gWiz plasmid in the presence of increasing amounts of PTO ODN to confirm that, as reported in vivo (43,44), this results in reduced gene expression. A dramatic ODN dose-dependent inhibition of luciferase expression was observed both for CpG1826, (data not shown) and for GpC1745 (Supp. Fig. 5B).
These data also exclude the possibility that induction of Th1 cytokines by either plasmid-bound or free CpG ODNs might lead to reduced gene expression by downregulation of the CMV promoter driving the luciferase expression (43). With the PTO CpG ODN and the plasmid, encoding the antigen, unlinked, the benefit of the immune adjuvant is lost because of the inhibitory effect upon antigen expression (43,44). The coupled LNA system may help resolve the problem of co-formulating PTO CpG ODN adjuvants and DNA vaccines, since using LNA to bind PTO CpG ODNs to plasmid encoding an antigen can lead to an immune adjuvant effect without inhibiting high-level antigen expression.
Conclusions
In this work, LNA ODNs were used to bind fluorophores to plasmid DNA. Data described in this study demonstrate that LNA ODNs are strand displacement agents of supercoiled plasmid DNA. Simple LNA ODNs seem more robustly attached to plasmid than similar ‘bis’ PNA ODNs for plasmid delivery by gene gun (10). However, ‘bis’ PNA ODNs with the addition of a few, positively charged amino acids are also excellent strand displacement agents and were not tested alongside the LNA ODNs in this work (21). LNA ODNs can remain associated with plasmid DNA after cationic lipid-mediated transfection into mammalian cells. The mechanism of the binding of LNA ODNs to plasmid DNA was confirmed as being duplex invasion and strand displacement. LNA ODNs can add CpG adjuvant activity to plasmid DNA without inhibiting high-level antigen expression.
These results suggest that coupling to LNA ODNs represents a novel and powerful methodology for adding functional moieties to plasmid DNA to improve and monitor gene delivery and DNA vaccination. This system offers real promise of being able to controllably attach numbers of functional peptides to plasmid DNA, without interfering with gene expression, and also to be compatible with a wide range of gene delivery systems. Work is ongoing to demonstrate how flexible this system is in attaching numbers of functional moieties to plasmid DNA.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Chin, Cheryl Hite, Alex Amiet and colleagues at Proligo LLC for discussions on LNA ODN production, Dwaine Braasch for discussions on PNA ODN design, Brian Hayes for training and help with confocal microscopy, Nalini Mehta and Chris Christodoulou for help establishing the fluorescent strand displacement DNA sequencing assay, Peter Topley and Patrick Gilboy for help and advice on gene gun cartridge preparation and in vitro gene gun delivery, and Jennie Wolstenholme and Claire Ashman for endotoxin tests.
REFERENCES
- 1.Scott E., Wiseman,J., Evans,M. and Colledge,W.H. (2001) Enhanced gene delivery to human airway epithelial cells using an integrin-targeting lipoplex. J. Gene Med., 3, 125–134. [DOI] [PubMed] [Google Scholar]
- 2.Cartier R. and Reszka,R. (2002) Utilization of synthetic peptides containing nuclear localization signals for nonviral gene transfer systems. Gene Ther., 9, 157–167. [DOI] [PubMed] [Google Scholar]
- 3.Ciolina C., Byk,G., Blanche,F., Thuiller,V., Scherman,D. and Wils,P. (1999) Coupling of nuclear localization signals to plasmid DNA and specific interaction of the conjugates with importin α. Bioconj. Chem., 10, 49–55. [DOI] [PubMed] [Google Scholar]
- 4.Sebestyen M., Ludtke,J., Bassik,M., Zhang,G., Budker,V., Lukhtanov,E., Hagstrom,J. and Wolff,J. (1997) DNA vector chemistry: the covalent attachment of signal peptides to plasmid DNA. Nat. Biotechnol., 16, 80–85. [DOI] [PubMed] [Google Scholar]
- 5.Neves C., Byk,G., Scherman,D. and Wils,P. (1999) Coupling of a targeting peptide to plasmid DNA by covalent triple helix formation. FEBS Lett., 453, 41–45. [DOI] [PubMed] [Google Scholar]
- 6.Zanta M., Belguise-Valladier,P. and Behr,J.P. (1999) Gene delivery: a single nuclear localization signal peptide is sufficient to carry DNA to the cell nucleus. Proc. Natl Acad. Sci. USA, 96, 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schirmbeck R., Konig-Merediz,S., Riedl,P., Kwissa,M., Sack,F., Schroff,M., Junghans,C., Reimann,J. and Wittig,B. (2001) Priming of immune responses to hepatitis B surface antigen with minimal DNA expression constructs modified with a nuclear localization signal peptide. J. Mol. Med., 79, 343–350. [DOI] [PubMed] [Google Scholar]
- 8.Zelphati O., Liang,X., Hobart,P. and Felgner,P. (1999) Gene chemistry: functionally and conformationally intact fluorescent plasmid DNA. Hum. Gene Ther., 10, 15–24. [DOI] [PubMed] [Google Scholar]
- 9.Zelphati O., Liang,X., Nguyen,C., Barlow,S., Sheng,S., Shao,Z. and Felgner,P. (2000) PNA-dependent gene chemistry: stable coupling of peptides and oligonucleotides to plasmid DNA. Biotechniques, 28, 304–316. [DOI] [PubMed] [Google Scholar]
- 10.Catchpole I. (2002) Patent WO 02/102825 A2; PCT/GB02/02728.
- 11.Braasch D. and Corey,D. (2001) Locked nucleic acid (LNA), fine tuning the recognition of DNA and RNA. Chem. Biol., 8, 1–7. [DOI] [PubMed] [Google Scholar]
- 12.Wahlestedt C., Salmi,P., Good,L., Kela,J., Johnsson,T., Hokfelt,T., Broberger,C., Porreca,F., Lai,J., Ren,K., Ossipov,M., Koshkin,A., Jakobsen,N., Skouv,J., Oerum,H., Jacobsen,M. and Wengel,J. (2000) Potent and non-toxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl Acad. Sci. USA, 97, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braasch D., Liu,Y. and Corey,D. (2002) Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design. Nucleic Acids Res., 30, 5160–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simeonov A. and Nikiforov,T. (2002) Single nucleotide polymorphism genotyping using short, fluorescently labeled locked nucleic acid (LNA) probes and fluorescence polarization detection. Nucleic Acids Res., 30, e91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demidov V. (2002) PNA and LNA throw light on DNA. Trends Biotechnol., 21, 4–7. [DOI] [PubMed] [Google Scholar]
- 16.Kurreck J., Wyszko,E., Gillen,C. and Erdmann,V. (2002) Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res., 30, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crinelli R., Bianchi,M., Gentilini,L. and Magnani,M. (2002) Design and characterization of decoy oligonucleotides containing locked nucleic acids. Nucleic Acids Res., 30, 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Torigue H., Hari,Y., Sekiguchi,M., Obika,S. and Imanishi,T. (2001) 2′-O,4′-C-Methylene bridged nucleic acid modification promotes pyrimidine motif triplex formation at physiological pH. J. Biol. Chem., 276, 2354–2360. [DOI] [PubMed] [Google Scholar]
- 19.Obika S., Hari,Y., Morio,K. and Imanishi,T. (2000) Triplex formation by an oligonucleotide containing conformationally locked C-nucleoside, 5-(2′-O,4′-C-methylene-β-D-ribofuranosyl) oxazole. Tetrahedron Lett., 41, 221–224. [Google Scholar]
- 20.Chernolovskaya E., Koshkin,A. and Vlassov,V. (2001) Interaction of LNA oligonucleotides with MDR1 promoter. Nucleosides Nucleotides Nucleic Acids, 20, 847–850. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X., Ishihara,T. and Corey,D. (2000) Strand invasion by mixed base PNAs and a PNA–peptide chimera. Nucleic Acids Res., 28, 3332–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smulevitch S., Simmons,C., Norton,J., Wise,T. and Corey,D. (1996) Enhancement of strand invasion by oligonucleotides through manipulation of backbone charge. Nat. Biotechnol., 14, 1700–1704. [DOI] [PubMed] [Google Scholar]
- 23.Kreig A. (2001) Immune effects and mechanisms of action of CpG motifs. Vaccine, 19, 618–622. [DOI] [PubMed] [Google Scholar]
- 24.Weeratna R., McCluskie,M., Xu,Y. and Davis,H. (2000) CpG DNA induces stronger immune responses with less toxicity than other adjuvants. Vaccine, 18, 1755–1762. [DOI] [PubMed] [Google Scholar]
- 25.Hemmi H., Takeuchi,O., Kawai,T., Kalsho,T., Sato,S., Sanjo,H., Matsumuto,M., Hoshino,K., Wagner,H., Takeda,K. and Akira,S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature, 408, 740–745. [DOI] [PubMed] [Google Scholar]
- 26.Bauer S., Kirschning,C., Hacker,H., Redecke,V., Hausmann,S., Akira,S., Wagner,H. and Lipford,G. (2001) Human TLR-9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl Acad. Sci. USA, 98, 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hacker H., Mischak,H., Miethke,T., Liptay,S., Schmid,R., Sparwasser,T., Heeg,K., Lipford,G. and Wagner,H. (1998) CpG-DNA-specific activation of antigen-presenting cells requires stress kinase activity and is preceded by non-specific endocytosis and endosomal maturation. EMBO J., 17, 6230–6240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad-Nejad P., Hacker,H., Rutz,M., Bauer,S., Vabulas,R. and Wagner,H. (2002) Bacterial CpG-DNA and lipopolysaccharides activate Toll-like receptors at distinct cellular compartments. Eur. J. Immunol., 32, 1958–1968. [DOI] [PubMed] [Google Scholar]
- 29.Crabtree T., Jin,L., Raymond,D., Pelletier,S., Houlgrave,C., Gleason,T., Pruett,T. and Sawyer,R. (2001) Preexposure of murine macrophages to CpG oligonucleotide results in a biphasic tumour necrosis factor alpha response to subsequent lipopolysaccharide challenge. Infect. Immun., 69, 2123–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Egholm M., Christensen,L., Dueholm,K., Buchardt,O., Coull,J. and Nielsen,P. (1995) Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res., 23, 217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Griffith M., Risen,L., Grieg,M., Lesnik,E., Sprankle,K., Griffey,R., Kiely,J. and Freier,S. (1995) Single and bis peptide nucleic acids as triplexing agents, binding and stoichiometry. J. Am. Chem. Soc., 117, 831–832. [Google Scholar]
- 32.Shirota H., Sano,K., Hirasawa,N., Terui,T., Ohuchi,O., Hattori,T., Shirato,K. and Tamura,G. (2001) Novel roles of CpG oligodeoxynucleotides as a leader for the sampling and presentation of CpG-tagged antigen by dendritic cells. J. Immunol., 167, 66–74. [DOI] [PubMed] [Google Scholar]
- 33.Maniatis T., Fritsch,E. and Sambrook,J. (1992) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 34.Tuting T., Wilson,C., Martin,D., Kasamon,Y., Rowles,J., Ma,D., Slingluff,C., Wagner,S., van der Bruggen,P., Baar,J., Lotze,M. and Storkus,W. (1998) Autologous human monocyte-derived dendritic cells genetically modified to express melanoma antigens elicit primary cytotoxic T cell responses in vitro, enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12 and IFN-α. J. Immunol., 160, 1139–1147. [PubMed] [Google Scholar]
- 35.Butz E. and Bevan,M. (1998) Differential presentation of the same MHC class I epitopes by fibroblasts and dendritic cells. J. Immunol., 160, 2139–2144. [PubMed] [Google Scholar]
- 36.Bukanov N., Demidov,V., Nielsen,P. and Frank-Kamenetskii,M. (1998) PD-loop: a complex of duplex DNA with an oligonucleotide. Proc. Natl Acad. Sci. USA, 95, 5516–5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen K., Singh,S., Wengel,J. and Jacobsen,J. (2000) Solution structure of an LNA hybridised to DNA: NMR study of the d(CTLGCTLTLCTLGC):d(GCAGAAGCAG) duplex containing four locked nucleotides. Bioconj. Chem., 11, 228–238. [DOI] [PubMed] [Google Scholar]
- 38.Nielsen P. and Christensen,L. (1996) Strand displacement binding of a duplex forming homopurine PNA to a homopyrimidine duplex DNA target. J. Am. Chem. Soc., 118, 2287–2288. [Google Scholar]
- 39.Jin L., Raymond,D., Crabtree,T., Pelletier,S., Houlgrave,C., Pruett,T. and Sawyer,R. (2000) Enhanced murine macrophage TNF receptor shedding by cytosine–guanine sequences in oligodeoxynucleotides. J. Immunol., 165, 5153–5160. [DOI] [PubMed] [Google Scholar]
- 40.Dalpke A., Opper,S., Zimmermann,S. and Heeg,K. (2001) Suppressors of cytokine signalling (SOCS)-1 and SOCS-3 are induced by CpG-DNA and modulate cytokine responses in APCs. J. Immunol., 166, 7082–7089. [DOI] [PubMed] [Google Scholar]
- 41.Gao J., Xue,Q., Papasian,J. and Morrison,D. (2001) Bacterial DNA and lipopolysaccharide induce synergistic production of TNF-α through a post-transcriptional mechanism. J. Immunol., 166, 6855–6860. [DOI] [PubMed] [Google Scholar]
- 42.Yi A., Yoon,J., Hong,S., Redford,T. and Kreig,A. (2001) Lipopolysaccharide and CpG DNA synergize for tumor necrosis factor-α production through activation of NF-κB. Int. Immunol., 13, 1391–1404. [DOI] [PubMed] [Google Scholar]
- 43.Weernata R., Brazalot,M.C., Krieg,A. and Davis,H. (1998) Reduction of antigen expression from DNA vaccines by coadministered oligodeoxynucleotides. Antisense Nucleic Acid Drug Dev., 8, 351–356. [DOI] [PubMed] [Google Scholar]
- 44.Schirmbeck R. and Reimann,J. (2001) Modulation of gene-gun-mediated Th2 immunity to hepatitis B surface antigen by bacterial CpG motifs or IL-12. Intervirology, 44, 115–123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.