

Abstract
A Drosophila forward genetic screen for mutants with defective synaptic development identified bad reception (brec). Homozygous brec mutants are embryonic lethal, paralyzed, and show no detectable synaptic transmission at the glutamatergic neuromuscular junction (NMJ). Genetic mapping, complementation tests, and genomic sequencing show that brec mutations disrupt a previously uncharacterized ionotropic glutamate receptor subunit, named here “GluRIID.” GluRIID is expressed in the postsynaptic domain of the NMJ, as well as widely throughout the synaptic neuropil of the CNS. In the NMJ of null brec mutants, all known glutamate receptor subunits are undetectable by immunocytochemistry, and all functional glutamate receptors are eliminated. Thus, we conclude that GluRIID is essential for the assembly and/or stabilization of glutamate receptors in the NMJ. In null brec mutant embryos, the frequency of periodic excitatory currents in motor neurons is significantly reduced, demonstrating that CNS motor pattern activity is regulated by GluRIID. Although synaptic development and molecular differentiation appear otherwise unperturbed in null mutants, viable hypomorphic brec mutants display dramatically undergrown NMJs by the end of larval development, suggesting that GluRIID-dependent central pattern activity regulates peripheral synaptic growth. These studies reveal GluRIID as a newly identified glutamate receptor subunit that is essential for glutamate receptor assembly/stabilization in the peripheral NMJ and required for properly patterned motor output in the CNS.
Keywords: synapse, postsynaptic, synaptogenesis, glutamatergic, glutamate receptor, subunit, embryo, pattern generator
Introduction
Ionotropic glutamate receptors (GluRs) are heteromeric glutamate-gated cation channels that mediate most excitatory synaptic transmission in mammalian brains (Dingledine et al., 1999; Kubo and Ito, 2004; McFeeters and Oswald, 2004; Simeone et al., 2004; Wollmuth and Sobolevsky, 2004). Changes in GluR subunit composition have important functional consequences; ligand binding, pore gating, and receptor trafficking are all altered by subunit substitution (Monaghan and Wenthold, 1997; Dingledine et al., 1999). However, the mechanisms controlling subunit composition remain poorly understood. For example, it is not clear how cells “choose” which GluR subunits to express under specific conditions, or whether rules worked out using heterologous expression systems are followed in vivo. An attractive approach to answering these questions is genetic manipulation of the well defined Drosophila glutamatergic neuromuscular junction (NMJ). However, despite decades of characterization, the subunit composition of glutamate receptors in the Drosophila NMJ remains incompletely described.
The Drosophila genome encodes 30 putative ionotropic glutamate receptor subunits (Littleton and Ganetzky, 2000), but only 21 genes contain amino acid sequences thought to be required for pore formation (Sprengel et al., 2001). Three genes, called “GluRIIA,” “GluRIIB,” and “GluRIII” (also known as “GluRIIC”) (Sprengel et al., 2001), have been shown to encode functional ionotropic glutamate receptor subunits localized to the NMJ (Schuster et al., 1991; Petersen et al., 1997; Marrus et al., 2004). GluRIIC null mutants are embryonic lethal, and strong hypomorphs have many fewer GluRs at the larval NMJ (Marrus and DiAntonio, 2004; Marrus et al., 2004). GluRIIA null mutants are viable but display reduced receptor channel open time, smaller miniature excitatory junction potentials, and reduced sensitivity to the antagonist argiotoxin 636 (Petersen et al., 1997; DiAntonio et al., 1999). GluRIIB null mutants are also viable but show no significant change in receptor function (DiAntonio et al., 1999), suggesting that GluRIIB is less important for channel function or that most native receptors lack GluRIIB. Simultaneous deletion of both GluRIIA and GluRIIB causes embryonic lethality (Petersen et al., 1997; DiAntonio et al., 1999) and a presumed complete loss of functional glutamate receptors. Antibody staining suggests that GluRIIA and GluRIIB occupy adjacent partially overlapping domains (Marrus et al., 2004; Chen and Featherstone, 2005), indicating that at least some receptors contain either GluRIIA or GluRIIB but not both. Thus, it has been proposed that glutamate receptors at the Drosophila NMJ are composed of GluRIIC plus either GluRIIA or GluRIIB (Marrus et al., 2004). However, the possibility of other subunits has not been ruled out. The identification of essential subunits is particularly important, because these proteins, by definition, contain properties critical for formation and/or stabilization of glutamate receptors.
Here, we describe a new Drosophila NMJ GluR subunit: “GluRIID.” Electrophysiology, immunohistochemistry, and confocal microscopy show that Drosophila NMJs lack all postsynaptic glutamate receptors in the absence of GluRIID. GluRIID is also expressed in the CNS synaptic neuropil, in which it plays a role in central motor pattern generation. Together, our results demonstrate that GluRIID is essential at the Drosophila NMJ and the first glutamate receptor subunit known to function within the CNS.
Materials and Methods
Genetics. Two ethyl methanesulfonate (EMS) (brec1 and brec2) and three P-element transposon (brecP1, brecP2 and brecP5) mutant alleles of bad reception (brec) were generated. brec1 was generated from EMS mutagenesis of a rucuca marked third chromosome. Mutants were outcrossed to Oregon R (OR) several times and screened with a combination of anatomical, behavioral, and electrophysiological assays to identify NMJ dysfunction mutants, as described previously (Featherstone et al., 2000). brec2 was identified as a “second-site hit” on a chromosome containing a previously isolated lethal glutamic acid decarboxylase (gad) mutation (gada30) (Featherstone et al., 2000). To separate the brec2 and gada30 mutations, the brec2, gada30 double-mutant flies were outcrossed to rucuca and then Oregon R several times, subsequently tested for complementation to another lethal gad allele, and then retested for embryonic lethality and paralysis after genetic tests confirmed that the portion of the chromosome containing the gada30 mutation was replaced with that of Oregon R. Both brec1 and brec2 still carry portions of the rucuca chromosomes; the genotypes of the brec mutant chromosomes are brec1, e, ca, and sr, brec2. Rescue of homozygous brec mutants by the CG18039 genomic transgene (see Results) was confirmed in brec1 by the presence of homozygous e (non-TM3 Sb) flies and in brec2 by the presence of sr (non-TM3 Sb) flies after crossing the transgene into w; brec/TM3 Sb mutant stocks. Homozygous rucuca flies (which are homozygous for e, ca, and sr, among other markers) do not differ significantly from OR wild-type (WT) controls in any parameters measured (Featherstone et al., 2002 and data not shown). The P-element alleles (brecP1, brecP2, and brecP5) were generated by Δ2-3-induced mobilization of a P{w[+mC] = lacW}bon[S048706] element previously mapped to the bonus gene in 92F (Beckstead et al., 2001) and screened for failure to complement the embryonic lethal EMS mutant allele brec2. Rescue of brecP5 homozygotes by the CG18039 transgene was confirmed by observing an increase (from ∼5% in unrescued flies to 100% of the expected Mendelian ratio in those carrying the transgene) of non-TM3 adult flies after crossing in the wild-type transgene. In all cases (brec1, brec2, brecP1, brecP2, and brecP5), homozygous mutants were selected before experiments using green fluorescent protein (GFP)-marked TM3 or TM6b balancer chromosomes.
Df(2L)SP22 is a deletion that removes genes encoding both GluRIIA and GluRIIB (Petersen et al., 1997; DiAntonio et al., 1999). Musclespecific overexpression of myc-tagged GluRIIA was accomplished using a myc-tagged GluRIIA transgene driven by the myosin heavy chain (MHC) promoter (Petersen et al., 1997; DiAntonio et al., 1999). Overexpression flies were constructed by crossing +/+;brec/TM3-GFP × MHC-mycGluRIIA/MHC-mycGluRIIA:+/+ to yield MHC-mycGluRIIA/MHC-mycGluRIIA;brec/brec offspring in the F2 generation.
Molecular biology. The entire 3.8 kb genomic region of CG18039 was amplified for sequencing using the primers CCAGAGGACGGAAATGAAAA and TGAGTTTCCCGAACAGAACA. To identify the EMS mutations, this entire region was redundantly (approximately four times) sequenced in Oregon R, rucuca, brec1/brec1, and brec2/brec2 animals. Sequence fragments were assembled using AssemblyLign software (Oxford Molecular Group, San Diego, CA), and consensus sequences for each genotype were determined after visual inspection. Consensus sequences were subsequently aligned for presentation using the ClustalW algorithm in MacVector (Accelrys). For quantitative real-time reverse transcription (RT)-PCR, total RNA was isolated from 22-24 h after egg laying (AEL) embryos using TriZol extraction and reverse transcribed using oligo-dT primers. Real-time RT-PCR was then performed using subunit cDNA-specific primers in an Opticon 2 real-time cycler (MJ Research, Waltham MA), using SYBR green (Molecular Probes, Eugene, OR) for fluorescent measurement of amplicon quantity. As a loading control, actin 5C RNA levels were also measured for every extraction using actin 5C-specific primers, and GluRII RNA levels were normalized using this measurement. Each measurement represents the actin 5C c(t) divided by a GluRII c(t), in which both measurements were made from the same RNA isolation and RT reaction.
Immunocytochemistry. Temporally and morphologically staged embryos were dechorionated in bleach, manually devitellinated and dissected, and then fixed in either Bouin's fixative (for GluRIIA staining) or 4% paraformaldehyde for 30-60 min (for all other antibodies). Mouse monoclonal anti-GluRIIA (“8B4D2”; Developmental Studies Hybridoma Bank, Iowa City, IA) was used at 1:10 to 1:100. Rabbit polyclonal anti-GluRIIB and anti-GluRIII (GluRIIC) were kind gifts from Dr. Aaron DiAntonio (Washington University, St. Louis, MO) and used at 1:1000 (Marrus and DiAntonio, 2004; Marrus et al., 2004). Fluorescently conjugated anti-HRP and anti-myc (Jackson ImmunoResearch, West Grove, PA) were used at 1:100; mouse monoclonal anti-cysteine string protein (CSP) was a gift from Dr. Konrad Zinsmeier (University of Arizona, Tucson, AZ) and was used at 1:200 (Zinsmaier et al., 1994). Mouse monoclonal anti-Discs large (anti-DLG “4F3”; Developmental Studies Hybridoma Bank) was used at 1:1000 (Parnas et al., 2001). Rabbit polyclonal anti-GluRIID antibody was a generous gift from Dr. Stephan Sigrist (European Neuroscience Institute, Göttingen, Germany) and was used at 1:500 [see companion paper by Qin et al. (2005) in this issue]. For visualization of immunoreactivity, goat anti-rabbit or goat anti-mouse fluorescent (FITC or rhodamine) secondaries (Jackson ImmunoResearch) were used. Images were obtained using either a Zeiss (Oberkochen, Germany) LSM 510 or Olympus Optical (Tokyo, Japan) FV500 laser-scanning confocal microscope. Quantitative image analysis was performed using NIH ImageJ software.
NMJ electrophysiology. Temporally and morphologically staged embryos were dechorionated in bleach, manually devitellinated and dissected, and then treated with 1 mg/ml collagenase type IV (Sigma, St. Louis, MO) for 30-60 s. Muscle 6 was whole-cell voltage clamped (-60 mV) in standard Drosophila saline using standard patch-clamp techniques. Muscle 6 whole-cell capacitance was typically 25-30 pF, and input resistances were 15-40 MΩ. For evoked currents, the segmental nerve leading to the patch-clamped muscle was stimulated via a suction electrode, using a 0.5-ms-long, 5-20 V pulse from a Grass Instruments (Quincy, MA) stimulator (Grass-Telefactor). To assay glutamate-gated currents, 1 mm glutamate in extracellular saline was pressure ejected from a small (∼10 μm tip opening) glass pipette using a 100 ms pressure pulse from a transistor-transistor logic-gated valve connected to the building air supply (Featherstone et al., 2002). Data were acquired and subsequently analyzed using an Axopatch 1D amplifier and pClamp 9 (Axon Instruments, Union City, CA). Spontaneous synaptic events were identified and analyzed using the template-matching algorithm of Clampfit 9.
CNS electrophysiology. Temporally staged embryos and early first-instar larvae (26-29 h AEL) were collected from agar plates or dechorionated with bleach and manually removed from the vitelline membrane in recording saline. Animals were secured at the head and tail to sylgard-coated coverslips with surgical histoacryl glue (histoacryl blue; B. Braun, Emmenbrucke, Switzerland), dissected open dorsally, and glued flat as described previously to expose the dorsal surface of the ventral ganglion (Baines et al., 1999; Rohrbough and Broadie, 2002). Dorsal neuronal cell bodies were exposed using a large-diameter (∼20 μm) patch pipette containing 0.5-1% protease [type XIV (Sigma) in recording saline]. The CNS sheath material was drawn by suction into the pipette tip for 1-2 min until the sheath ruptured. Standard whole-cell voltage-clamp recordings were made at 18-20°C at a holding potential of -60 mV. The patch saline contained the following (in mm): 140 K-acetate, 2 MgCl2, 0.1 CaCl2, 10 HEPES, 1.1 EGTA, 2 Na2-ATP, and ∼6 KOH, pH 7.2. Lucifer yellow (1 mg/ml; Molecular Probes) was included in the patch pipette saline to confirm motor neuron or interneuron morphology. Recorded neurons included the identified dorsal motor neurons aCC and RP2 (Baines et al., 1999, 2001), as well as excitatory interneurons. All recorded cells displayed sustained neuronal properties, including action potential firing, fast and sustained synaptic currents, and responses to acetylcholine (ACh) application. ACh was applied iontophoretically to the neuronal soma via a sharp microelectrode containing100 mm ACh in dH2O, at pH 4-5 to favor agonist iontophoresis by positive current.
Statistics. Statistics were computed using Instat or Prism software (Graphpad Software, San Diego, CA). All values are mean ± SEM; n always refers to the number of animals. Asterisks in figures represent statistical significance compared with appropriate controls: *p < 0.05; **p < 0.01; ***p < 0.001. Individual animal values for spontaneous excitatory junctional currents (sEJCs) are computed from average measurements obtained over at least 5 min of recording.
Results
Isolation of bad reception mutants
To identify genes required for glutamatergic synaptic development, we screened ∼6300 Drosophila third-chromosome EMS mutant lines for embryonic/early larval lethality and paralysis (Featherstone et al., 2000). Paralyzed mutants were assayed directly for NMJ synaptic transmission defects with whole-cell patch-clamp recording of the muscle coupled to suction-electrode stimulation of the motor nerve. brec1 mutants were identified based on the absence of detectable NMJ neurotransmission but normal excitation-contraction coupling in response to direct muscle stimulation. A second member of this complementation group, brec2, was originally identified on a chromosome containing a lethal EMS gad allele (gada30) (Featherstone et al., 2000) and then separated from the gad mutation by recombination. Homozygous brec1 and brec2 mutants, and transheterozygous brec1/brec2 mutants, display normal gross morphology and normally patterned neuromusculature but always fail to hatch (100% embryonic lethality) and appear paralyzed within the eggshell. Manually dechorionated and devittelinized brec mutant embryos fail to produce any detectable peristaltic waves typical of locomotory movements in wild-type animals of the same age. The name of this paralyzed mutant class (bad reception) derives from the subsequent finding that paralysis is caused by lack of NMJ postsynaptic glutamate receptors (see below).
Using recombination mapping, brec1 and brec2 were independently mapped to region 92E-F on the right arm of chromosome 3. To identify the mutant gene, we conducted a P-element mutagenesis and screened for inserts that failed to complement the brec embryonic lethality. A local P-element was mobilized by crossing y[1] w[67c23]; P{w[+mC] = lacW}bon[S048706]/TM3, Sb[1] Ser[1] to T(2;3)ap[Xa], ap[Xa]/CyO, H{PDelta2-3}HoP2.1; Sb[1], and ∼800 of the resulting “local hop” mutant lines were screened for failure to complement brec1 and brec2. Three brec P-element alleles were isolated (brecP1, brecP2, and brecP5) that completely fail to complement the viability of both brec1 and brec2. Some homozygous brecP2 mutants hatch, but none survive to adulthood. Homozygous brecP1 and brecP5 mutants die throughout larval and pupal development, with a minority (5-10% for brecP5 and 20-30% for brecP1) surviving into adulthood. Based on sequence data and phenotype (see below), brec1 and brec2 likely represent two null alleles, whereas the three P-element alleles are partial loss-of-function hypomorphic mutants. The five brec mutant alleles, in order of severity, are as follows: brec1 = brec2 > brecP2 > brecP5 > brecP1.
Unfortunately, use of P-element-specific primers and inverse PCR suggested that none of the P-element alleles (P1, P2, or P5) generated by the local hop contains an intact lacW P-element insertion within 92E-F, so the gene could not be identified using these P-element alleles. However, subsequent recombination mapping of brec1 and brec2 using P-element w+ markers (Zhai et al., 2003) narrowed the gene region to 92F, and complementation tests to candidate gene mutants demonstrated that brec1 and brec2 fail to complement Df(3R)E3, a small deficiency that removes all or part of predicted genes CG18039, CG31201, and CG4058 [see companion paper by Qin et al. (2005) in this issue]. Each brec allele, however, also complements another small Df e01443 that removes CG31201, suggesting that brec mutants specifically disrupt CG18039. To confirm that the brec mutations were in CG18039, we sequenced the entire genomic region of CG18039 in homozygous brec1 and brec2 mutants, as well as the genetic controls rucuca and Oregon R (GenBank accession numbers AY768536, AY768537, AY768538, and AY768539 for Oregon R, rucuca, brec1, and brec2, respectively). CG18039 contains 14 predicted exons (Fig. 1). In brec1 mutants, exon 4 contains a 50 bp deletion and frame shift. In brec2 mutants, exon 11 contains a 7 bp duplication and frame shift (Fig. 1). Genescan analysis predicts that both mutations, assuming the gene product is translated and stable, would lead to abnormal protein. Specifically, brec1 would lead to a deletion and then large insertion in the N terminus near the putative ligand-binding region, and brec2 would lead to a large deletion between predicted transmembrane segments 3 and 4. However, both mutations appear to destabilize the CG18039 protein, because no detectable protein is observed with immunocytochemistry confocal imaging in either brec1 or brec2 mutants (see below). Homozygous brec1 and brec2 mutants are rescued to full viability using a transgene containing only the CG18039 genomic sequence.
Figure 1.

brec mutations disrupt the CG18039 gene encoding an ionotropic glutamate receptor subunit GluRIID. Gray boxes represent predicted exons in CG18039, a putative ionotropic glutamate receptor subunit most similar in sequence to mammalian kainite receptor subunits. The entire 3.8 kb genomic region was sequenced in brec1 and brec2 mutants, as well as the control genotypes rucuca and Oregon R (WT). Partial sequences were obtained for brecP5 and its parental strain. In brec1 mutants, there is a 50 bp deletion in the fourth exon. In brec2 mutants, there is a 7 bp duplication in the 11th exon. brecP5 mutants containan I-element-like insert (P5 insert) ∼2 kb long between exons 6 and 7 and a predicted splice-site alteration at the end of exon 7 (asterisk).
Although none of the alleles isolated from the local hop P-element mutagenesis contained an intact lacW element as expected, significant genomic alterations were detected in all three of these alleles. brecP5 was analyzed in most detail; it contains an insertion of ∼2 kb between exons 6 and 7 that is present in neither wild-type nor the parental bon[S048706] stock used to generate the brec P-element mutants (Fig. 1). In addition, a predicted splice site is also altered at the end of exon 7 in brecP5 mutants (Fig. 1). Sequencing of the P5 insert showed that it matches (97-100% nucleotide identity) endogenous “LINE-like” transposons (I-elements) scattered throughout the genome. One or more of these was presumably inserted into CG18039 during the local hop mutagenesis. Dramatically reduced CG18039 protein is observed with immunocytochemistry and confocal imaging in brecP5 mutants, and brecP5 mutants are rescued to full viability using a transgene containing only the CG18039 genomic sequence. We therefore conclude that brec mutations are in CG18039, that brec1 and brec2 are protein null alleles, and that brecP1, brecP2, and brecP5 are hypomorphic alleles. The molecular genetic data for the alleles used in this study (brec1, brec2, and brecP5) are summarized in Figure 1.
The brec mutations disrupt the ionotropic glutamate receptor subunit GluRIID
A BLAST (basic local alignment search tool) search using the entire predicted 903 aa sequence of CG18039 suggests that CG18039 encodes a highly conserved ionotropic glutamate receptor subunit, with the closest sequence similarity to human, mouse, and rat kainate receptor subunit GluR6. Specifically, CG18039 displays ∼37% aa identity compared with human, mouse, and rat GluR6 (NP068775, P39087, and NP062182, respectively). Antibodies raised against the C terminus of the CG18039 GluR subunit (Qin et al., 2005) recognize the glutamatergic NMJ, suggesting that the protein encoded by CG18039 functions as part of an NMJ glutamate receptor [see companion paper by Qin et al. (2005) in this issue]. Specifically, CG18039 immunoreactivity in the embryonic NMJ forms discrete puncta similar to those described previously (Featherstone et al., 2002) (compare with Fig. 4) for the GluRIIA subunit (Fig. 2A, B). Furthermore, CG18039 immunoreactivity overlaps that of the postsynaptically abundant membrane-associated guanylate kinase (MAGUK) protein DLG (Fig. 2C). These results suggest that CG18039 encodes a glutamate receptor subunit in the NMJ postsynaptic density. Interestingly, only some CG18039 puncta are immunoreactive for GluRIIA. This is consistent with previous studies demonstrating that only a subset of Drosophila NMJ glutamate receptors contain the GluRIIA subunit (Marrus et al., 2004; Chen and Featherstone, 2005).
Figure 4.
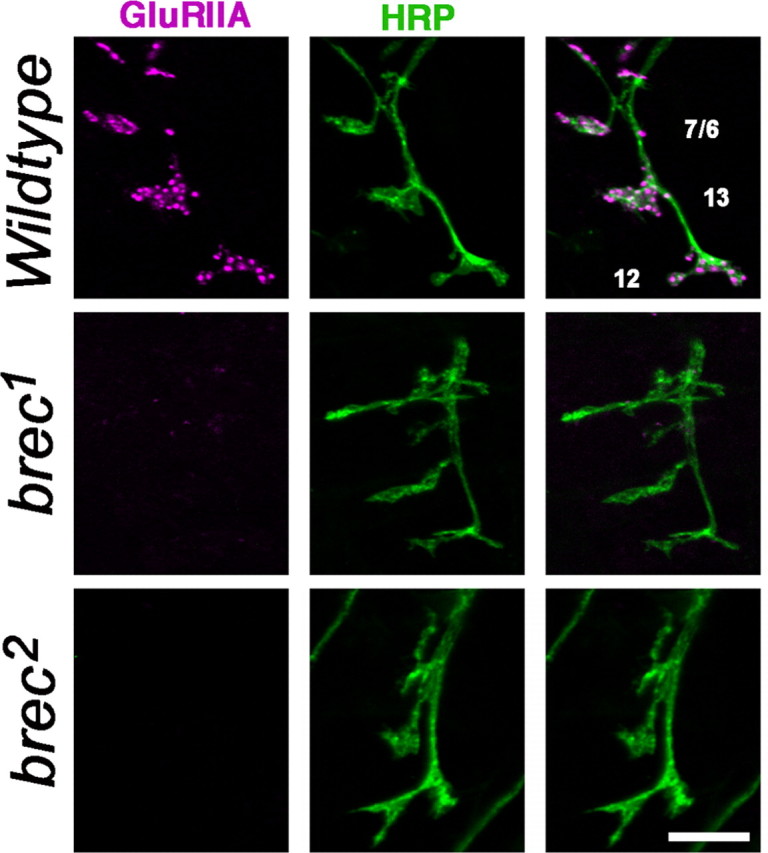
Homozygous brec mutant embryonic NMJs have no detectable glutamate receptors. Confocal images show intersegmental nerve branch b, visualized using anti-HRP antibody (neuronal membrane epitope; green), innervating several ventral longitudinal muscles. At WT NMJs (top row), glutamate receptors visualized using an anti-GluRIIA antibody (magenta) form distinct clusters opposite presynaptic terminals. In homozygous brec mutants (rows 2, 3), there are no detectable glutamate receptor clusters. Similar results were obtained using antibodies against the GluRIIB and GluRIIC subunits (see Fig. S1, available at www.jneurosci.org as supplemental material). Scale bar, 5 μm.
Figure 2.
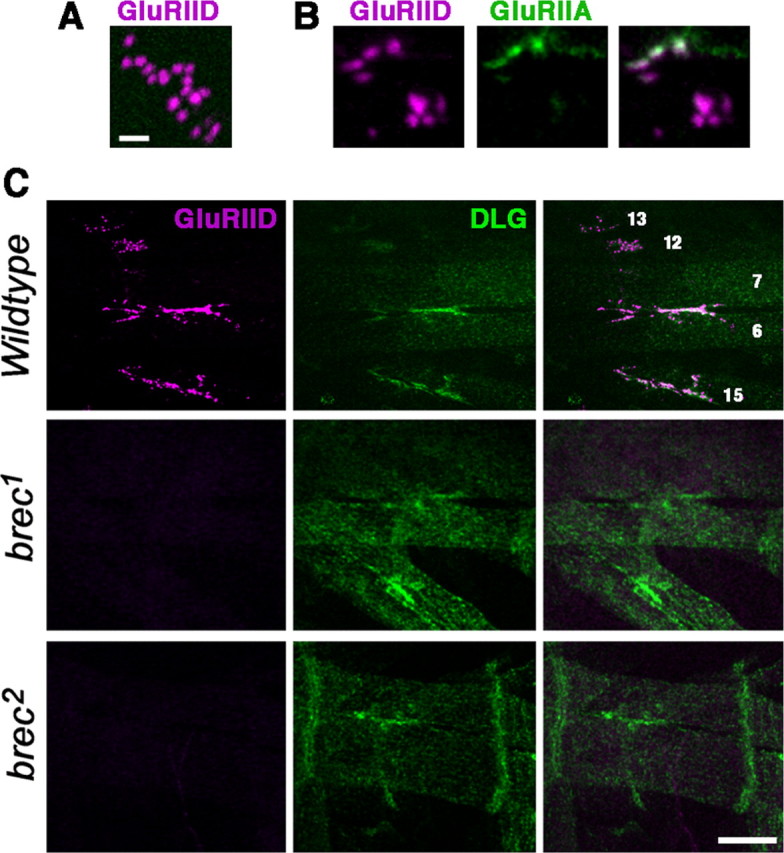
GluRIID is expressed in the embryonic NMJ. A, High-magnification confocal image of typical GluRIID immunoreactivity in a wild-type embryonic NMJ. Note that GluRIID immunoreactivity appears as small (∼300-nm-diameter) clusters. Scale bar, 1 μm. B, As in A but showing both GluRIID and GluRIIA immunoreactivity. Note that only some GluRIID clusters are colabeled with GluRIIA. C, Confocal images of glutamatergic NMJs on embryonic ventral muscles 7, 6, 13, 12, and 15, double stained using antibodies for GluRIID (magenta) and DLG (green). GluRIID immunoreactivity (but not DLG immunoreactivity) is eliminated in homozygous brec1 and brec2 mutants. Scale bar, 10 μm.
Based on the established naming convention for Drosophila NMJ ionotropic glutamate receptor subunits, we henceforth refer to the CG18039 protein as GluRIID. Of the previously identified Drosophila NMJ GluR subunits, GluRIID is most similar in sequence to GluRIIA, with 29% aa identity. GluRIID is 27% identical to GluRIIC and GluRIIB. However, several other predicted GluR subunits in the Drosophila genome are much more similar to GluRIID. CG31201 is 57% identical to GluRIID. CG31201 has been identified as an essential glutamate receptor subunit at the Drosophila NMJ (Qin et al., 2005) and is now referred to as “GluRIIE.” After GluRIIE, the genes most similar to GluRIID are CG5621, CG3822, CG11155, and clumsy, which are 49%, 39%, 37%, and 36% identical, respectively. There is currently no evidence that any of these four genes encodes an NMJ receptor subunit [see companion paper by Qin et al. (2005) in this issue].
GluRIID is required for NMJ transmission
To determine whether GluRIID plays a role in Drosophila synaptic transmission at the NMJ, we functionally characterized transmission in homozygous brec1 and brec2 mutant embryos, in which the GluRIID immunoreactivity is abolished (Fig. 2C). WT embryonic NMJs display robust sEJCs (Fig. 3A). In contrast, homozygous brec1 and brec2 mutants show no detectable endogenous NMJ transmission above baseline noise (WT sEJC frequency, 11.7 ± 1.6 Hz, n = 20; brec1, 0 ± 0 Hz, n = 6; brec2, 0.2 ± 0.5 Hz, n = 16; WT sEJC amplitude, 129.9 ± 14.9 pA, n = 20; brec1, 0 ± 0 pA, n = 6; brec2, 15 ± 22 pA, n = 16) (Fig. 3A, B). These results show that GluRIID is essential for endogenous glutamatergic neurotransmission at the NMJ.
Figure 3.

Homozygous null brec mutant embryonic NMJs have no functional glutamate receptors. A, B, brec mutants show no spontaneous synaptic currents (A) of any size (B). C, Evoked synaptic transmission is eliminated in brec mutant NMJs. D, Representative evoked synaptic currents; the robust EJCs observed at WT NMJs (top traces) are completely eliminated in the brec mutants (bottom traces). E, Pressure ejection of 1 mm glutamate directly onto the postsynaptic muscle elicits no response in the brec mutants. F, Representative glutamate-gated currents are large and reproducible in WT muscle (top traces) but completely absent in brec mutant muscles (bottom traces). Arrows mark the time of glutamate pressure ejection. Note also the spontaneous synaptic currents visible in the WT traces but absent in the recordings from the mutants.
Although endogenous NMJ activity in the brec mutants is abolished, mutant synapses could still be capable of transmission if strongly stimulated. To test evoked neurotransmission, the segmental nerve was stimulated directly with a suction electrode while monitoring postsynaptic EJCs in the voltage-clamped muscle. In wild-type mature embryonic NMJs, presynaptic stimulation triggers large EJCs mediated by the opening of ∼160 ionotropic glutamate receptors (mean EJC amplitude of 1964 pA; mean single GluR channel current of 12 pA). In homozygous brec1 and brec2 mutants, however, presynaptic stimulation fails to elicit any postsynaptic currents. In both mutants, evoked EJC amplitudes fall within the range of baseline noise (WT, 1964 ± 178 pA, n = 7; brec1, 8 ± 22 pA, n = 8; brec2, 2 ± 13 pA, n = 28).
To verify that the loss of synaptic transmission in brec mutants was attributable to a postsynaptic defect, we next functionally assayed postsynaptic glutamate receptor function directly by pressure ejecting 1 mm glutamate onto patch-clamped muscle (Fig. 3E, F). Both whole-cell capacitance (25-35 pA) and series resistance (15-40 MΩ) do not differ between wild-type and brec mutant muscle. In mature wild-type embryos, pressure ejection of glutamate onto the postsynaptic domain of muscle 6 opens ∼140 glutamate receptors (1726 pA divided by 12 pA). In brec1 and brec2 mutants the response to glutamate application is effectively eliminated; in both mutants, glutamate-gated current amplitudes fall within the range of baseline noise (WT, 1726 ± 134 pA, n = 21; brec1, 16 ± 5 pA, n = 5; brec2, 24 ± 9 pA, n = 13). Whole-cell patch-clamp recordings in Drosophila embryonic muscles are sensitive enough to detect single glutamate receptor channel openings (Nishikawa and Kidokoro, 1995; Featherstone et al., 2000, 2002). However, in homozygous brec1 and brec2 mutants, not even single channel openings are discernible. Together, these electrophysiology results strongly suggest that brec mutants completely lack functional postsynaptic glutamate receptors in the NMJ. Because pressure ejection of glutamate also activates unclustered receptors (Broadie and Bate, 1993; Featherstone et al., 2000; Chen and Featherstone, 2005), these results also suggest that there are no functional receptors anywhere in the membrane.
The GluRIID subunit is essential for glutamate receptor formation at the NMJ
Formation of the postsynaptic GluR field requires induction by the presynaptic motor neuron (Broadie and Bate, 1993). Thus, the lack of functional receptors could be caused by a lack of proper muscle innervation (Featherstone and Broadie, 2002). If muscles are innervated, the observed loss of functional GluRs could be caused by channel dysfunction, mistrafficking, or loss of the GluR subunit proteins. To determine whether muscles are correctly innervated and to determine whether glutamate receptor protein is absent or mislocalized, brec mutant NMJs were imaged with antibodies that recognize both the presynaptic terminal and postsynaptic glutamate receptors (Fig. 4).
In mature wild-type embryos, the intersegmental nerve shows a characteristic branching pattern as it forms NMJs on ventral longitudinal muscles 7, 6, 13, and 12 (Fig. 4, top row) (Johansen et al., 1989a,b; Bate and Broadie, 1995). This presynaptic morphology is normal in homozygous brec1 and brec2 mutants; the nerve forms normal branches to appropriate muscles, and axons terminate in anatomically normal presynaptic terminals (Fig. 4) (see also Fig. 7). In mature wild-type embryos, the GluRIIA subunit is localized into distinct punctate clusters in the postsynaptic membrane under the presynaptic boutons (Featherstone et al., 2002). In homozygous brec1 and brec2 mutants, however, anti-GluRIIA immunoreactivity is completely undetectable (Fig. 4, middle and bottom rows) (see also Fig. S1D, available at www.jneurosci.org as supplemental material). The GluRIIB and GluRIIC subunits are similarly undetectable in both brec1 and brec2 mutants (Fig. S1A, B, available at www.jneurosci.org as supplemental material). Thus, in the absence of GluRIID, all other known GluR subunits (GluRIIA, GluRIIB, and GluRIIC) are immunocytochemically undetectable [see companion paper by Qin et al. (2005) in this issue]. No detectable perinuclear immunoreactivity for any subunit is visible in brec mutants, suggesting that the lack of GluRIID does not result in accumulation of incompletely assembled receptors in endoplasmic reticulum or Golgi.
Figure 7.

NMJ presynaptic morphology is relatively normal in brec null mutant embryos but underdeveloped in hypomorphic brec mutant third-instar larvae. A, Confocal images of embryonic NMJ terminals visualized using antibodies against the synaptic vesicle marker CSP, from WT (top) and homozygous brec mutants (middle, bottom). B, C, Quantification of embryonic NMJ area (B) and number of presynaptic boutons (C). D, E, Quantification of larval NMJ area (D) and number of presynaptic boutons (E). P5, brecP5.
It is conceivable that loss of GluRIID could cause transcriptional downregulation of the other GluR subunits. To determine whether the loss of protein expression is secondary to changes in receptor gene transcription, real-time RT-PCR was performed on homozygous brec mutant embryos using glutamate receptor subunit-specific primers. The results show that brec mutants contain essentially normal levels of GluRII subunit mRNA [WT relative mRNA, 1.0; brec1/brec1 (n = 4), 0.89 ± 0.1 (GluRIIA), 0.82 ± 0.30 (GluRIIB); brec2/brec2, 0.94 ± 0.2 (GluRIIA), 1.1 ± 0.2 (GluRIIB) (n = 4); in separate experiments: WT (n = 3), 1.03 ± 0.04 (GluRIIA), 0.64 ± 0.02 (GluRIIB), 0.68 ± 0.02 (GluRIIC), 0.72 ± 0.03 (GluRIID), 0.71 ± 0.02 (GluRIIE); brecP5/TM3-GFP (n = 4), 0.92 ± 0.07 (GluRIIA), 0.66 ± 0.02 (GluRIIB), 0.67 ± 0.03 (GluRIIC), 0.68 ± 0.05 (GluRIID), 0.68 ± 0.03 (GluRIIE); brecP5/brecP5 (n = 5), 0.99 ± 0.04 (GluRIIA), 0.72 ± 0.07 (GluRIIB), 0.68 ± 0.05 (GluRIIC), 0.70 ± 0.06 (GluRIID), 0.72 ± 0.06 (GluRIIE)]. Other glutamate receptor subunit genes thus are normally transcribed in brec mutants, but receptor protein expression and function is not detectable at NMJs or elsewhere within the muscle cell (in brec1 or brec2), suggesting that receptors are not assembled and rapidly degraded in the absence of the GluRIID. If GluRIID forms functional receptors in combination with GluRIIA and/or GluRIIB, we would also predict that GluRIID should be lost in the absence of these subunits, because formation of the heteromeric receptor assembly would be similarly prevented. To test this prediction, GluRIID immunoreactivity was imaged in homozygous Df[SP22] mutants, a deficiency that eliminates both GluRIIA and GluRIIB genes (Petersen et al., 1997; DiAntonio et al., 1999). As predicted, GluRIID immunoreactivity at the NMJ is completely eliminated in Df[SP22] mutant embryos (Fig. S1C, available at www.jneurosci.org as supplemental material) [see companion paper by Qin et al. (2005) in this issue].
GluRIIA is the only Drosophila NMJ glutamate receptor subunit for which heterologous expression has been described (Schuster et al., 1991). In Xenopus oocytes, GluRIIA is able to form functional homomeric receptor channels (Schuster et al., 1991). We therefore next asked whether the apparent requirement for GluRIID could be bypassed by strong overexpression of GluRIIA in vivo. A myc-tagged GluRIIA transgene was overexpressed in body-wall muscle using the muscle-specific MHC promoter (DiAntonio et al., 1999), and transgenic receptor expression was visualized using anti-myc antibodies. As shown in Figure 5, overexpression of GluRIIA in a control (brec2/+) background results in a large increase of GluRIIA-containing receptors in both the NMJ and nonsynaptic muscle membrane (Fig. 5, top). When myc-tagged GluRIIA is similarly overexpressed in either brec1 or brec2 mutant background, however, anti-myc immunoreactivity at the NMJ is only very weakly detectable (Fig. 5, bottom). Furthermore, GluRIIA overexpression does not generate any detectable rescue of either brec1 or brec2 mutants, which still die as fully paralyzed, unhatched embryos. Thus, in the absence of GluRIID in vivo, GluRIIA appears unable to replace GluRIID function or form functional GluRIIA homomers. These results support the conclusion that GluRIID is absolutely required for the formation and stability of glutamate receptors in vivo and suggest that important differences must exist between receptor assembly in vivo and in heterologous expression systems. To test whether the abundance and/or localization of other postsynaptic proteins is altered in brec mutant NMJ terminals lacking glutamate receptors, we used antibodies raised against DLG, a homolog of the MAGUKs PSD-95 (postsynaptic density-95) and SAP97 (synapse-associated protein 97), which is also abundant postsynaptically in the fly NMJ (Lahey et al., 1994; Parnas et al., 2001). Anti-DLG immunoreactivity is not detectably altered in homozygous brec mutants (compare with Figs. 2, 5) [WT DLG immunofluorescence (arbitrary units), 69 ± 6.4, n = 12; brec1, 67 ± 8.6, n = 10; brec2, 66 ± 12.9, n = 6; Df(2)SP22, 77 ± 8.3, n = 9]. These results suggest that the absence of GluRIID (and glutamate receptors) does not result in a general mistrafficking of postsynaptically localized proteins or other obvious disruption of postsynaptic development.
Figure 5.
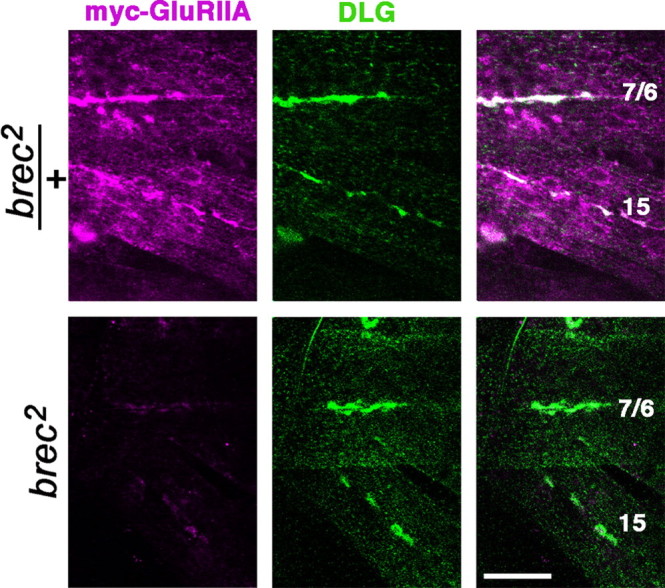
GluRIID is necessary for efficient expression of transgenic GluRIIA. In heterozygous brec mutants (top row), postsynaptically expressed transgenic myc-tagged GluRIIA (visualized using an anti-myc antibody; magenta) clusters predominantly at postsynaptic sites, as shown by the colocalization (white) with the postsynaptic marker DLG (green). In homozygous brec mutants (bottom row), overexpression of myc-tagged glutamate receptor protein does not rescue mutant lethality, and synaptic GluRIIA localization remains greatly reduced compared with controls.
NMJ differentiation is impaired in brec mutants
The complete absence of postsynaptic glutamate receptors in brec null mutants makes it difficult to determine whether presynaptic neurotransmitter release is changed in brec mutants. To answer this question, we studied brecP5 hypomorphs, which survive to become third-instar larvae. As shown in Figure 6A, brecP5 larval NMJs display a strong reduction in GluRIIA immunoreactivity, similar to, but not as severe as, the reduction observed in the brec null alleles. The loss of functional receptors was confirmed electrophysiologically (Fig. 6B, C); two-electrode voltage-clamp recordings from brecP5 larval muscle 6 NMJs show a strong reduction in sEJC amplitude (WT, 0.636 ± 0.002 nA, n = 8; brecP5, 0.484 ± 0.004 nA, n = 7). Note that the mean sEJC amplitude measured in brecP5 mutants underestimates the magnitude of the change (compare representative traces in Fig. 6B), probably because many events in brecP5 mutants are lost within baseline noise and thus are not measured. Consistent with this, brecP5 mutants also showed a large reduction in sEJC frequency (WT, 2.32 ± 0.17 Hz, n = 8; brecP5, 0.24 ± 0.03 Hz, n = 7). The magnitude of the frequency change was, however, surprisingly large. Because changes in sEJC frequency are typically indicative of presynaptic alterations, we carefully assayed presynaptic morphology in the brec mutants (Fig. 7). In homozygous brec null mutant embryos, immunoreactivity for CSP, a synaptic vesicle protein (Zinsmaier et al., 1994; Braun and Scheller, 1995), is normal (Fig. 7A). Quantitation of total NMJ area and bouton number (Fig. 7B, C) shows that homozygous brec1 or brec2 mutant embryos display relatively normal synaptic morphology [WT, 47 ± 5 μm2, n = 13; brec1, 39 ± 4, n = 16; brec2, 40 ± 5, n = 14 (Fig. 3B); and WT, 9 ± 3 boutons, n = 13; brec1, 11 ± 3, n = 16; brec2, 12 ± 2, n = 14 (Fig. 3C)]. In homozygous brecP5 mutants, however, presynaptic morphology is severely disrupted by the end of larval development (Fig. 7D, E). The mutant total NMJ area was approximately one-half of controls, and the number of boutons is reduced to approximately one-third of controls (area: brecP5/TM6, 554 ± 56 μm2, n = 9; brecP5/brecP5, 291 ± 55, n = 8; and bouton number: brecP5/TM6, 76 ± 5 boutons, n = 9; brecP5/brecP5, 26 ± 4, n = 8).
Figure 6.

Hypomorphic brec mutants have greatly reduced NMJ glutamate receptor expression and synaptic activity. A, Confocal images of third-instar larval NMJs on adjacent muscles 6 and 7, from heterozygous brecP5 mutant larvae (brecP5/TM6; controls) and homozygous brecP5 larvae. The presynaptic terminal is visualized using the neuronal membrane marker anti-HRP (green). Glutamate receptors, visualized using anti-GluRIIA antibodies (magenta), are reduced to barely detectable levels at the NMJ in homozygous brecP5 mutants. B, Sample voltage-clamp recordings from muscle 6, showing frequent spontaneous EJCs (downward deflections) in wild-type larvae (top trace), which are essentially abolished in homozygous brecP5 larvae (bottom trace). C, Cumulative frequency histogram of sEJC amplitudes from WT and homozygous brecP5 mutant larvae, quantifying a significant decrease in sEJC amplitudes measured in mutants. D, Quantification of sEJC frequency in WT and homozygous brecP5 mutant larvae, showing greatly decreased frequency in mutants. P5, brecP5; K-S test, Kolmogorov-Smirnov test.
The dramatic changes in larval NMJ morphology observed with reduction in postsynaptic glutamate receptors could be attributable to a requirement for glutamate receptors in an activity-dependent retrograde developmental signaling pathway that controls presynaptic morphology. However, studies suggest that NMJ morphology does not change when postsynaptic CaMKII (calcium/calmodulin-dependent kinase II) is altered (Beumer et al., 2002; Haghighi et al., 2003) (but see Koh et al., 1999) or when the ability of muscles to depolarize is genetically inhibited (Paradis et al., 2001; White et al., 2001). Overexpression of GluRIIA causes increased growth of larval NMJs, but knock-out of GluRIIB also causes NMJ overgrowth (Sigrist et al., 2002). Overexpression of GluRIIB has no effect (Sigrist et al., 2002). Smaller NMJs in the absence of GluRs have never been reported. Thus, there is no evidence for a mechanism by which loss of postsynaptic receptors could alter presynaptic morphology, although we cannot rule out the possibility. Conversely, it is well established that presynaptic NMJ morphology depends on neural electrical activity (for review, see Budnik, 1996; Hannan and Zhong, 1999; Koh et al., 2000). To test whether GluRIID might be required for normal presynaptic electrical activity, we recorded from presynaptic neurons within the ventral ganglion.
GluRIID is required in CNS neuropil for motor activity output
Unexpectedly, strong GluRIID immunoreactivity is present in the synaptic neuropil of the embryonic ventral ganglion (Fig. 8A). This CNS immunoreactivity is absent in brec1 or brec2 mutants (data not shown). This observation suggests that GluRIID must play a role in the CNS in additional to its function in GluRs at the NMJ. To assay this predicted CNS function, we recorded from dorsal neurons in the ventral ganglion of brec null mutants. These neurons, which include both identified motor and interneurons (Baines et al., 1999, 2001), have been shown to receive predominantly excitatory cholinergic input from upstream interneurons and therefore express ionotropic acetylcholine receptors (Baines and Bate, 1998; Rohrbough and Broadie, 2002; Rohrbough et al., 2003). Neuronal current responses elicited by acetylcholine application are normal in both amplitude and kinetics in brec null mutants (WT, 137 ± 25 pA, n = 9; brec2, 128 ± 17 pA, n = 7) (Fig. 8B, D). This shows that cholinergic receptor development and function is normal is the absence of GluRIID, consistent with expectations.
Figure 8.
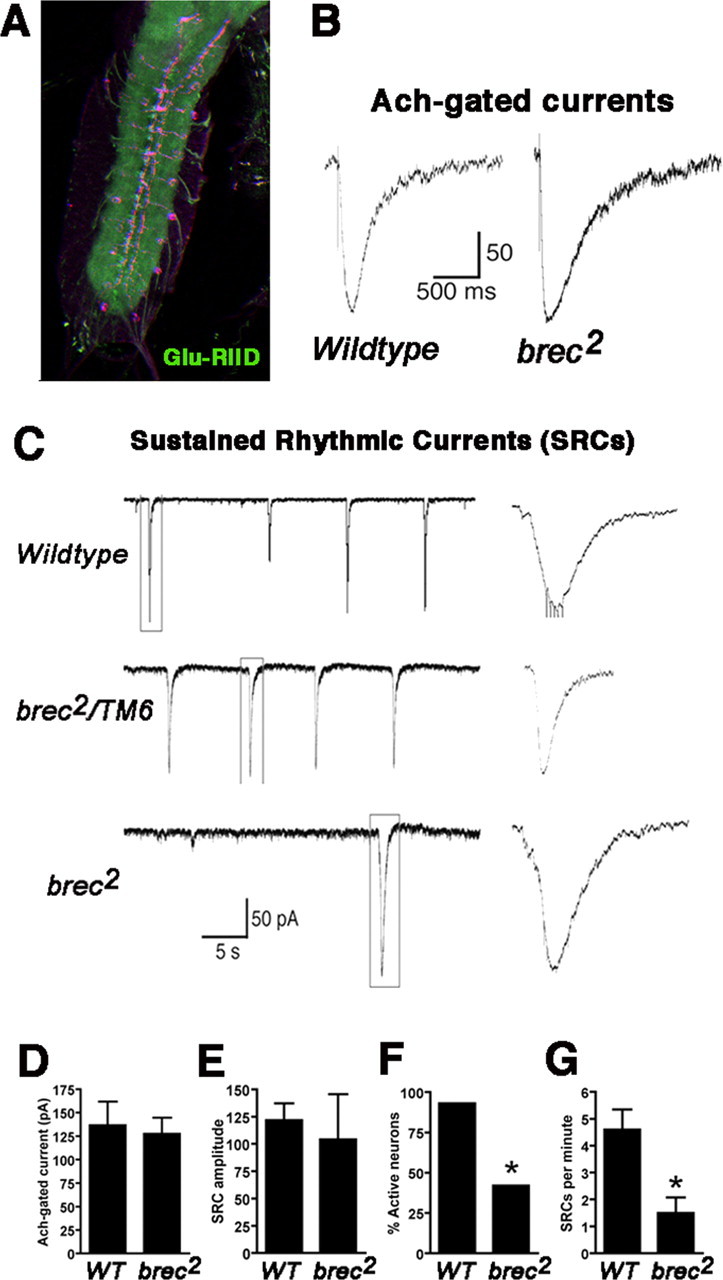
GluRIID is localized to the CNS neuropil and modulates excitatory neuronal activity. A, Confocal image showing an embryonic ventral ganglion stained with GluRIID antibodies (green) and anti-DLG antibodies (magenta). Note the GluRIID immunoreactivity throughout the synaptic neuropil. B, Representative acetylcholine-gated currents recorded from WT and homozygous brec2 mutant neurons. C, Voltage-clamp recordings from WT, heterozygous brec2/TM6 controls, and homozygous brec2 mutant motor neurons, showing endogenous SRCs. Events outlined by boxes are shown at an expanded time scale to the right. D-G, Quantification of ACh-gated current amplitudes (D), SRC amplitudes (E), percentage of neurons displaying SRCs (F), and SRC frequency in the neurons that display SRCs (G) in control (WT and heterozygous brec2/TM6 mutants) and homozygous brec2 mutant embryos.
Neurons in both wild-type and brec2/TM6 control preparations display large spontaneous endogenous excitatory currents, or “sustained rhythmic currents” (SRCs), on average four to five times per minute (Fig. 8C) (Rohrbough and Broadie, 2002; Rohrbough et al., 2003). These events require cholinergic presynaptic input and underlie the endogenous patterned motor activity driving locomotion (Rohrbough and Broadie, 2002; Rohrbough et al., 2003). In homozygous brec2 mutants, the amplitude of SRCs is unchanged in neurons exhibiting endogenous activity (WT, 122 ± 16 pA, n = 13; brec2, 104 ± 42 pA, n = 3) (Fig. 8E), but the frequency of SRCs is reduced more than threefold (WT, 4.6 ± 0.75 pA, n = 13; brec2, 1.5 ± 0.5 pA, n = 13) (Fig. 8F, G). Additionally, the incidence of neurons with SRC activity was reduced from >90% in controls to ∼40% in brec mutants. Taking into account both the reduced frequency of excitatory activity and fewer active cells, SRCs are thus reduced to ∼15% of normal. Thus, GluRIID is not only required for GluR assembly and synaptic transmission at the NMJ, but it is also vital for maintained patterned activity within the CNS. We therefore conclude that the motor output of the CNS relies on GluRIID-dependent glutamatergic modulation. Together, these studies identify GluRIID as the first Drosophila glutamate receptor subunit that functions in both the peripheral NMJ and the CNS.
Discussion
Because it is increasingly apparent that many aspects of glutamate receptor assembly and trafficking depend on the presence of specific receptor subunits (for review, see Barry and Ziff, 2002; Molnar and Isaac, 2002; Sheng and Hyoung Lee, 2003; van Zundert et al., 2004), a complete grasp of glutamate receptor composition is critical for studies of postsynaptic development and receptor regulation. The Drosophila NMJ is a genetically tractable glutamatergic synapse that is widely exploited for molecular studies of synaptic development, function, and plasticity. This synapse has been shown previously to contain three ionotropic glutamate receptor subunits: GluRIIA, GluRIIB, and GluRIIC (also known as GluRIII) (Schuster et al., 1991; Petersen et al., 1997; DiAntonio et al., 1999; Marrus et al., 2004). This study reveals a fourth NMJ subunit, GluRIID. The companion paper by Qin et al. (2005) supports the findings reported here and also introduces a fifth NMJ subunit, GluRIIE. Thus, the Drosophila NMJ contains, at least, five different ionotropic glutamate receptor subunits, each encoded by a different gene: GluRIIA, GluRIIB, GluRIIC, GluRIID, and GluRIIE. Null mutations in GluRIIC, GluRIID, and GluRIIE each cause embryonic lethality, loss of functional NMJ glutamate receptors, and decreased immunoreactivity for other subunits (Marrus et al., 2004; Qin et al., 2005; this paper). This suggests that GluRIIC, GluRIID, and GluRIIE are essential subunits contained by each glutamate receptor at the NMJ. In contrast, GluRIIA and GluRIIB are each individually dispensable, although at least one of these subunits is required for a functional receptor because deletion of both GluRIIA and GluRIIB is lethal (Petersen et al., 1997; DiAntonio et al., 1999). The subunit stoichiometry of mammalian non-NMDA glutamate receptors has never been definitively solved, but recent evidence from partial crystal structures strongly suggests that each ionotropic glutamate receptor is a “dimer of dimers,” e.g., composed of four subunits (Sun et al., 2002; Gouaux, 2004; Mayer and Armstrong, 2004). If Drosophila NMJ glutamate receptors are similarly tetrameric, then all existing data suggest that they are heterotetramers composed of one GluRIIC subunit, one GluRIID subunit, and one GluRIIE subunit, plus either one subunit of GluRIIA or one subunit of GluRIIB (but not both GluRIIA and GluRIIB). In other words, the Drosophila NMJ contains two subclasses of ionotropic glutamate receptor: (1) GluRIIA-containing receptors and (2) GluRIIB-containing receptors. This model is consistent with immunocytochemical and genetic results: (1) immunoreactivity for GluRIIA and GluRIIB is segregated such that clusters appear to contain either GluRIIA or GluRIIB but not both (Marrus et al., 2004; Chen and Featherstone, 2005); (2) only some GluRIID clusters are immunoreactive for GluRIIA (compare with Fig. 2) (Chen and Featherstone, 2005); and (3) GluRIIA and GluRIIB are differentially trafficked and stabilized (Chen and Featherstone, 2005) (K. Chen, F. Liebl, and D. E. Featherstone unpublished data). If GluRIIA and GluRIIB can be found in the same receptor (which presumably also contains the required subunits GluRIIC, GluRIID, and GluRIIE), then we must conclude that Drosophila NMJ glutamate receptors are likely pentameric.
Because at least four different GluR subunits are required in the Drosophila NMJ in vivo, it suggests that there are at least four distinct subunit-dependent requirements for receptor assembly, trafficking, and/or stabilization. It is not clear how the different subunits play these roles. Amino acid sequence alignment shows that GluRIIA, GluRIIB, GluRIIC, GluRIID, and GluRIIE subunits differ most from each other near their N termini, a region that is known to be involved in ligand binding and possibly receptor assembly (Gouaux, 2004; Mayer and Armstrong, 2004). However, GluRIIA and GluRIIB show no obvious similarity in this region to explain why they might to be able to substitute for each other. GluRIIC has a class II C-terminal consensus PDZ (PSD-95/DLG/zona occludens-1)-binding domain (Marrus et al., 2004), suggesting that GluRIIC might have a unique anchoring role. Neither GluRIIA nor GluRIIB contains recognizable PDZ-binding motifs, although stabilization of GluRIIB-containing receptors requires (apparently indirectly) the presence of the PDZ domain protein DLG (Chen and Featherstone, 2005). Thus, it remains unclear how individual Drosophila GluR subunits contribute to receptor assembly and function. For mammalian receptors, answers to this question have typically been sought using heterologously expressed receptor subunits. However, our study suggests that there may be important differences in the mechanisms of receptor assembly and function in vivo. GluRIIA forms functional homomeric receptors when expressed in Xenopus oocytes (Schuster et al., 1991), but in vivo overexpression of GluRIIA in muscle is essentially unable to over-come the requirement for GluRIID or form functional GluRs (compare with Fig. 5). It is conceivable that Xenopus oocytes contain endogenous proteins similar to kainite receptor subunits, and these proteins are sufficient for GluRIIA assembly and/or stabilization. Indeed, Xenopus oocytes are known to contain an endogenous protein (XenU1) that can substitute for the mammalian NMDA receptor subunit NR2 (Soloviev and Barnard, 1997). Alternatively, homomeric GluRIIA receptors may be only very inefficiently formed in oocytes, similar to the in vivo situation, but this inefficiency is not apparent outside of a synaptic context. Heterologous expression of other Drosophila NMJ GluR subunits has not been reported. Thus, our results suggest caution when interpreting some results using expressed subunits and highlight the importance of in vivo studies.
Our study also suggests, for the first time, a functional role for glutamate receptors in the Drosophila CNS. Uniquely among known fly GluR subunits, GluRIID is expressed both in the NMJ and at central synapses. Excitatory transmission in the Drosophila CNS is thought to be predominantly cholinergic, although in situ data for several putative ionotropic glutamate receptor subunits shows that many subunits are expressed in the CNS (Schuster et al., 1993; Tomancak et al., 2002). We show that GluRIID is expressed at high levels throughout the synaptic neuropil of the ventral nerve cord, indicating that glutamatergic synapses in Drosophila might be much more widespread and pervasive than has been speculated previously. Consistent with this, we show that, in the absence of GluRIID, there is severe disruption of endogenous central motor pattern output. Interestingly, the only glutamategated responses that have been demonstrated in Drosophila neurons are inhibitory; glutamate-gated currents in voltage-clamped larval CNS neurons are prolonged (2-5 s), reverse at -55mV, and are blocked by picrotoxin (Rohrbough and Broadie, 2002). Nevertheless, GluRIID in the CNS could be part of an excitatory receptor that remains functionally unidentified. More intriguing is the possibility that GluRIID is an essential component of a kainate receptor-like glutamate-gated cation channel in Drosophila muscle but part of a glutamate-gated anion channel in the CNS. Glutamate-gated currents in embryos are, unfortunately, so far undetectable in embryos; thus, we have been unable to determine whether GluRIID is required for CNS glutamate-gated anion currents. Although the nature of the GluRIID-containing receptor is unknown, loss of its function clearly causes dramatic changes in endogenous patterned activity within motor neurons. In mutants, many motor neurons lack detectable patterned motor output, and all cells show a striking reduction in the frequency of patterned motor output. This result minimally demonstrates that GluRIID-dependent glutamatergic transmission plays a vital modulatory role in controlling motor output from the CNS.
Although embryonic synaptogenesis appears normal in the absence of GluRIID, partial loss of GluRIID in viable brec mutants dramatically reduces postembryonic synaptic growth and differentiation. The loss of glutamate receptors in either the CNS or NMJ could cause NMJ morphology defects in two ways: (1) loss of muscle depolarization could disrupt a retrograde signal that induces presynaptic growth, or (2) disruption of endogenous central motor pattern activity could alter electrical activity-dependent presynaptic growth. In Drosophila, there is not good support for the former mechanism, because inhibition of muscle depolarization does not detectably alter NMJ arborization (Paradis et al., 2001; White et al., 2001). In contrast, it is well established that neuronal electrical activity is positively correlated with the growth of the Drosophila presynaptic motor terminal (for review, see Budnik, 1996; Hannan and Zhong, 1999; Koh et al., 2000). Thus, the second model is the most parsimonious explanation. This conclusion raises the exciting prospect that the endogenous pattern of central electrical activity plays a critical role in sculpting postembryonic NMJ development.
Footnotes
This work was supported by National Institutes of Health Grants GM54544 (K.B.) and NS045628 (D.E.F.). We thank Stephan Sigrist and his laboratory staff for GluRIID transgene, antibodies, independent mutant alleles for complementation tests, sharing data before publication, and many useful discussions. We also thank Aaron DiAntonio and Konrad Zinsmaier for antibodies vital to this study and Jay Newby for fly work.
Correspondence should be addressed to either of the following: David Featherstone, Department of Biological Sciences, University of Illinois at Chicago, 840 West Taylor Street (M/C 067), Chicago, IL 60607, E-mail: def@uic.edu; or Kendal Broadie, Department of Biological Sciences, Vanderbilt University, VU Station B, Box 351634, Nashville, TN 37235-1634, E-mail: kendal.broadie@vanderbilt.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253199-10$15.00/0
References
- Baines RA, Bate M (1998) Electrophysiological development of central neurons in the Drosophila embryo. J Neurosci 18: 4673-4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Robinson SG, Fujioka M, Jaynes JB, Bate M (1999) Postsynaptic expression of tetanus toxin light chain blocks synaptogenesis in Drosophila Curr Biol 9: 1267-1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines RA, Uhler JP, Thompson A, Sweeney ST, Bate M (2001) Altered electrical properties in Drosophila neurons developing without synaptic transmission. J Neurosci 21: 1523-1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry MF, Ziff EB (2002) Receptor trafficking and the plasticity of excitatory synapses. Curr Opin Neurobiol 12: 279-286. [DOI] [PubMed] [Google Scholar]
- Bate M, Broadie K (1995) Wiring by fly: the neuromuscular system of the Drosophila embryo. Neuron 15: 513-525. [DOI] [PubMed] [Google Scholar]
- Beckstead R, Ortiz JA, Sanchez C, Prokopenko SN, Chambon P, Losson R, Bellen HJ (2001) Bonus, a Drosophila homolog of TIF1 proteins, interacts with nuclear receptors and can inhibit betaFTZ-F1-dependent transcription. Mol Cell 7: 753-765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer K, Matthies HJ, Bradshaw A, Broadie K (2002) Integrins regulate DLG/FAS2 via a CaM kinase II-dependent pathway to mediate synapse elaboration and stabilization during postembryonic development. Development 129: 3381-3391. [DOI] [PubMed] [Google Scholar]
- Braun JE, Scheller RH (1995) Cysteine string protein, a DnaJ family member, is present on diverse secretory vesicles. Neuropharmacology 34: 1361-1369. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bate M (1993) Innervation directs receptor synthesis and localization in Drosophila embryo synaptogenesis. Nature 361: 350-353. [DOI] [PubMed] [Google Scholar]
- Budnik V (1996) Synapse maturation and structural plasticity at Drosophila neuromuscular junctions. Curr Opin Neurobiol 6: 858-867. [DOI] [PubMed] [Google Scholar]
- Chen K, Featherstone DE (2005) Discs-large (DLG) is clustered by presynaptic innervation and regulates postsynaptic glutamate receptor subunit composition in Drosophila BMC Biol 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS (1999) Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci 19: 3023-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Featherstone D, Broadie K (2002) Response: meaningless minis? Trends Neurosci 25: 386-387. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Rushton EM, Hilderbrand-Chae M, Phillips AM, Jackson FR, Broadie K (2000) Presynaptic glutamic acid decarboxylase is required for induction of the postsynaptic receptor field at a glutamatergic synapse. Neuron 27: 71-84. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Rushton E, Broadie K (2002) Developmental regulation of glutamate receptor field size by nonvesicular glutamate release. Nat Neurosci 5: 141-146. [DOI] [PubMed] [Google Scholar]
- Gouaux E (2004) Structure and function of AMPA receptors. J Physiol (Lond) 554: 249-253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS (2003) Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron 39: 255-267. [DOI] [PubMed] [Google Scholar]
- Hannan F, Zhong Y (1999) Second messenger systems underlying plasticity at the neuromuscular junction. Int Rev Neurobiol 43: 119-138. [DOI] [PubMed] [Google Scholar]
- Johansen J, Halpern ME, Johansen KM, Keshishian H (1989a) Stereotypic morphology of glutamatergic synapses on identified muscle cells of Drosophila larvae. J Neurosci 9: 710-725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen J, Halpern ME, Keshishian H (1989b) Axonal guidance and the development of muscle fiber-specific innervation in Drosophila embryos. J Neurosci 9: 4318-4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Popova E, Thomas U, Griffith LC, Budnik V (1999) Regulation of DLG localization at synapses by CaMKII-dependent phosphorylation. Cell 98: 353-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Gramates LS, Budnik V (2000) Drosophila larval neuromuscular junction: molecular components and mechanisms underlying synaptic plasticity. Microsc Res Tech 49: 14-25. [DOI] [PubMed] [Google Scholar]
- Kubo M, Ito E (2004) Structural dynamics of an ionotropic glutamate receptor. Proteins 56: 411-419. [DOI] [PubMed] [Google Scholar]
- Lahey T, Gorczyca M, Jia XX, Budnik V (1994) The Drosophila tumor suppressor gene dlg is required for normal synaptic bouton structure. Neuron 13: 823-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Ganetzky B (2000) Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron 26: 35-43. [DOI] [PubMed] [Google Scholar]
- Marrus SB, DiAntonio A (2004) Preferential localization of glutamate receptors opposite sites of high presynaptic release. Curr Biol 14: 924-931. [DOI] [PubMed] [Google Scholar]
- Marrus SB, Portman SL, Allen MJ, Moffat KG, DiAntonio A (2004) Differential localization of glutamate receptor subunits at the Drosophila neuromuscular junction. J Neurosci 24: 1406-1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Armstrong N (2004) Structure and function of glutamate receptor ion channels. Annu Rev Physiol 66: 161-181. [DOI] [PubMed] [Google Scholar]
- McFeeters RL, Oswald RE (2004) Emerging structural explanations of ionotropic glutamate receptor function. Faseb J 18: 428-438. [DOI] [PubMed] [Google Scholar]
- Molnar E, Isaac JT (2002) Developmental and activity dependent regulation of ionotropic glutamate receptors at synapses. ScientificWorldJournal 2: 27-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan DT, Wenthold RJ, eds (1997) The ionotropic glutamate receptors. Totowa, NJ: Humana.
- Nishikawa K, Kidokoro Y (1995) Junctional and extrajunctional glutamate receptor channels in Drosophila embryos and larvae. J Neurosci 15: 7905-7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737-749. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS (2001) Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron 32: 415-424. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19: 1237-1248. [DOI] [PubMed] [Google Scholar]
- Qin G, Schwarz T, Kittel RJ, Schmid A, Rasse TM, Kappei D, Ponimaskin E, Heckmann M, Sigrist SJ (2005) Four different subunits are essential for expressing the synaptic glutamate receptor at neuromuscular junctions of Drosophila J Neurosci 25: 3209-3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J, Broadie K (2002) Electrophysiological analysis of synaptic transmission in central neurons of Drosophila larvae. J Neurophysiol 88: 847-860. [DOI] [PubMed] [Google Scholar]
- Rohrbough J, O'Dowd DK, Baines RA, Broadie K (2003) Cellular bases of behavioral plasticity: establishing and modifying synaptic circuits in the Drosophila genetic system. J Neurobiol 54: 254-271. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schloss P, Cox JA, Schmitt B, Betz H (1991) Molecular cloning of an invertebrate glutamate receptor subunit expressed in Drosophila muscle. Science 254: 112-114. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Ultsch A, Schmitt B, Betz H (1993) Molecular analysis of Drosophila glutamate receptors. EXS 63: 234-240. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hyoung Lee S (2003) AMPA receptor trafficking and synaptic plasticity: major unanswered questions. Neurosci Res 46: 127-134. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Schuster CM (2002) The postsynaptic glutamate receptor subunit DGluR-IIA mediates long-term plasticity in Drosophila J Neurosci 22: 7362-7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simeone TA, Sanchez RM, Rho JM (2004) Molecular biology and ontogeny of glutamate receptors in the mammalian central nervous system. J Child Neurol 19: 343-360. [DOI] [PubMed] [Google Scholar]
- Soloviev MM, Barnard EA (1997) Xenopus oocytes express a unitary glutamate receptor endogenously. J Mol Biol 273: 14-18. [DOI] [PubMed] [Google Scholar]
- Sprengel R, Aronoff R, Volkner M, Schmitt B, Mosbach R, Kuner T (2001) Glutamate receptor channel signatures. Trends Pharmacol Sci 22: 7-10. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E (2002) Mechanism of glutamate receptor desensitization. Nature 417: 245-253. [DOI] [PubMed] [Google Scholar]
- Tomancak P, Beaton A, Weiszmann R, Kwan E, Shu S, Lewis SE, Richards S, Ashburner M, Hartenstein V, Celniker SE, Rubin GM (2002) Systematic determination of patterns of gene expression during Drosophila embryogenesis. Genome Biol 3: RESEARCH0088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zundert B, Yoshii A, Constantine-Paton M (2004) Receptor compartmentalization and trafficking at glutamate synapses: a developmental proposal. Trends Neurosci 27: 428-437. [DOI] [PubMed] [Google Scholar]
- White BH, Osterwalder TP, Yoon KS, Joiner WJ, Whim MD, Kaczmarek LK, Keshishian H (2001) Targeted attenuation of electrical activity in Drosophila using a genetically modified K+ channel. Neuron 31: 699-711. [DOI] [PubMed] [Google Scholar]
- Wollmuth LP, Sobolevsky AI (2004) Structure and gating of the glutamate receptor ion channel. Trends Neurosci 27: 321-328. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, Greenbaum MP, Bellen HJ (2003) Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci USA 100: 10860-10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S (1994) Paralysis and early death in cysteine string protein mutants of Drosophila Science 263: 977-980. [DOI] [PubMed] [Google Scholar]