

Summary
Electron crystallography determines the structure of membrane embedded proteins in the two-dimensionally crystallized state by cryo-transmission electron microscopy imaging and computer structure reconstruction. Milestones on the path to the structure are high-level expression, purification of functional protein, reconstitution into two-dimensional lipid membrane crystals, high-resolution imaging, and structure determination by computer image processing. Here we review the current state of these methods. We also created an Internet information exchange platform for electron crystallography, where guidelines for the imaging and data processing method are maintained. The server (http://2dx.org) provides the electron crystallography community with a central information exchange platform, which is structured in blog and Wiki form, allowing visitors to add comments or discussions. It currently offers a detailed step-by-step introduction to image processing with the MRC software program. The server is also a repository for the 2dx software package, a user-friendly image processing system for 2D membrane protein crystals.
Keywords: 2dx, electron crystallography, membrane proteins, image processing, 2D crystallization, electron microscopy
Introduction
Membrane proteins are of central importance for health and disease. They correspond to more than 25% of predicted proteins and act as transporters, channels, receptors, and scaffolding components. Because of their strategic location, membrane proteins play critical roles in many functions such as cell proliferation, nerve and muscle contraction, and regulation of blood pressure, salt and water balance. Consequently, loss of function typically results in critical physiological effects such as neurological disorders, cancer, digestive diseases, heart failure etc. Because of their biological importance, membrane proteins represent the majority of today’s drug targets in pharmaceutical research and are the major targets for current drugs ranging from Aspirin (cyclo-oxygenase) to Zoloft (serotonin transporter) [1]. Structural biology of membrane proteins is thus one of the most important fields of modern biology. Nevertheless, less than 300 structures of membrane proteins have been determined (see http://blanco.biomol.uci.edu/Membrane_Proteins_xtal.html), about one third of which are considered as unique structures. These structures were determined mostly by X-ray diffraction (XRD), electron crystallography and nuclear magnetic resonance (NMR), to a resolution sufficient to at least identify trans-membrane helices. Compared to more than 37,000 available structures of soluble proteins available in the Protein Data Bank (PDB, [2]), this low number of determined membrane protein structures is in stark contrast to their biological importance.
Structure determination of membrane proteins – eukaryotic membrane proteins in particular - faces several technical hurdles. Difficulties in heterologous over-expression, non-denaturing detergent solubilization and gentle purification limit the amount of functional membrane protein sample that are available for structural studies. Nevertheless, in cases where it was successful, structure determination by X-ray diffraction of three-dimensional (3D) crystals, nuclear magnetic resonance and cryo-electron microscopy (cryo-EM) of two-dimensional (2D) crystals revealed amazing structural concepts and mechanisms that nature employs to solve the challenging tasks posed to these proteins. Recent highlights include the 1.35Å structure by XRD of the ammonium transporter AmtB [3], the structure of the waterchannel Aqp0 by cryo-EM at 1.9Å [4] and XRD at 2.2Å resolution [5], and the structure of Mistic [6] by NMR [7], to give only a few examples.
Over the past decade, electron crystallography has established itself as a viable alternative to X-ray crystallographic structure determination, especially of membrane proteins. While the resolution often is lower than that which can be obtained by XRD, electron crystallography takes advantage of the fact that it analyses the structure of the protein embedded in a native membrane environment. Using this approach, atomic models for seven membrane proteins and tubulin were so far determined: BR [8], LHCII [9], AQP1 [10,11], nAChR [12], and AQP0 [4,13], AQP4 [14], MGST1 [15], and Tubulin [16]. Several other membrane proteins classified as transporters, ion pumps, receptors and membrane bound enzymes have been studied by electron crystallography at lower resolution allowing localization of secondary structure motifs such as trans-membrane helices, and are likely to produce atomic models in the near future (e.g. [17–20]).
Milestones
Structural investigation of membrane proteins by electron crystallography involves milestones as depicted in Figure 1. Before 2D crystals can be obtained identified targets have to be cloned, expressed and purified. Structural analysis by transmission electron microscopy firstly establishes a two-dimensional projection map of the membrane protein as seen from above the membrane. Combination of this map with data from tilted 2D crystal samples allows the reconstruction of the 3D map of the protein. At a resolution of 7 Å, alpha-helical transmembrane segments can be identified. At 4.5 Å the first densities for larger amino acid side-chains can be recognized, and an atomic model can be proposed at this or better resolution. Figure 1 also gives estimations for the required average time to reach the next milestone, as well as the expected success rates for those steps. These values are estimates based on the current state of the technology. We estimate an average time of 15 months for the production of highly pure, homogeneous and active protein, followed by one year to grow well-ordered 2D crystals. Structure determination by an experienced laboratory would then require ~3 years, during which time the quality of the 2D crystals would be further refined. Accumulative failure rates imply that from 20 targeted membrane proteins one atomic structure can be expected after ~ 5 years. In comparison with structure determination by XRD, obtaining well-ordered 2D crystals has a higher likelihood and lower estimated required time. However, the structure determination from such 2D crystals is still a labor- and time-intensive procedure, where only a handful of laboratories have so far demonstrated success. In the following we describe the different steps of electron crystallography and then describe how we suggest accelerating the process and increasing the success rate.
Figure 1.
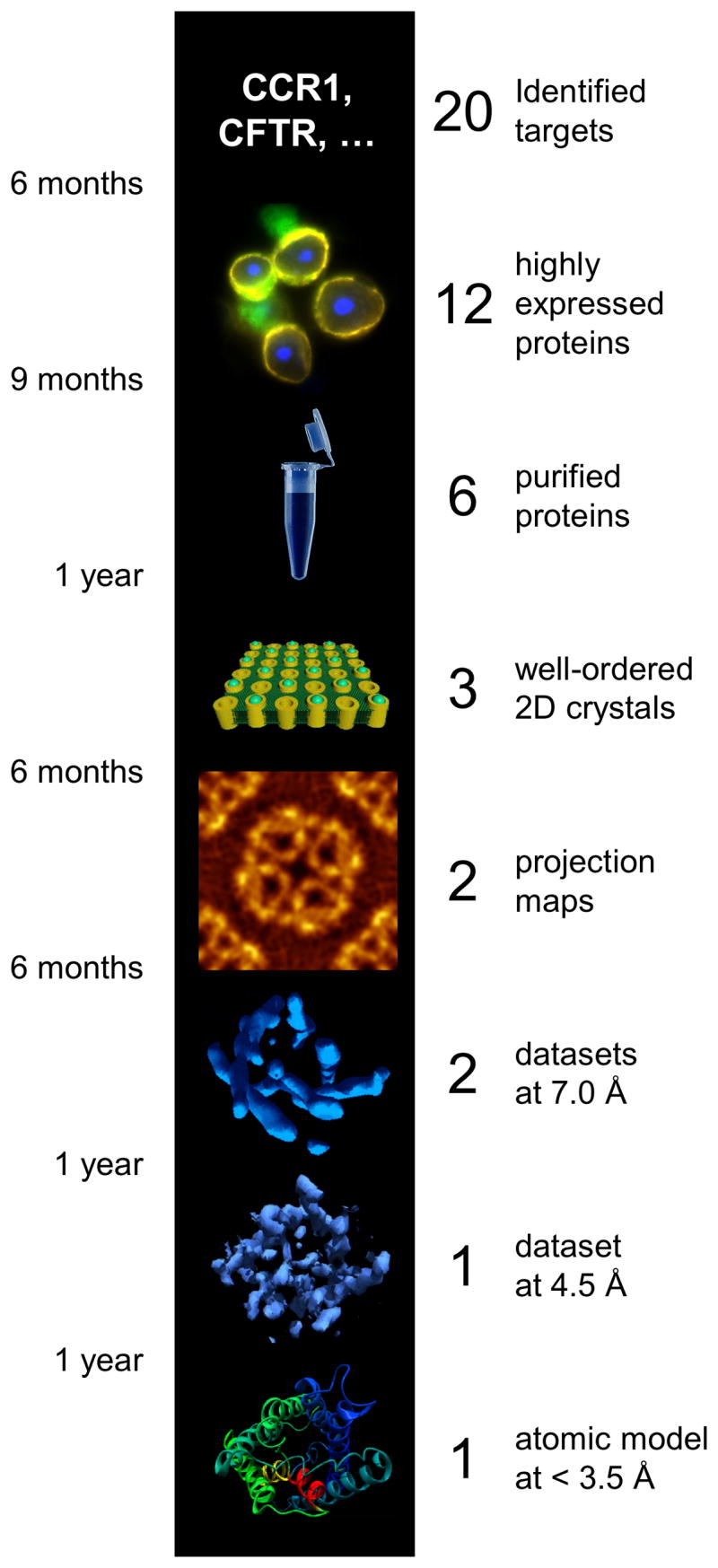
Milestones in the structure determination pipeline in electron crystallography (center). Our estimates for the required time to reach the next milestone for one experienced researcher are given on the left, and a crude estimate of the thinning of the pipeline is given on the right. These values are rough estimates based on the current state of the technology. Pooling resources, high-throughput and automation approaches, and open knowledge exchange is likely to significantly accelerate and broaden the pipeline in the future.
Membrane protein production and purification
Identified target sequences usually have to be cloned, inserted into a vector and over-expressed in a heterologous host. In the past the use of bacterial expression systems has been particularly useful. However, bacterial systems are frequently of little use for over-expression of mammalian trans-membrane proteins, which require many post-translational modifications and processing events for correct protein folding. The study of bacterial homologues as representatives of human membrane proteins will be of limited use, as no bacterial or archaeal homologues have been identified for about 50% of human membrane proteins [21].
Several membrane proteins affect the function of the host cell, e.g. insertion of the mammalian membrane proteins into bacterial cell membranes often results in cell death. Thus the search for a suitable expression system may be challenging [22]. Cell-free expression may be a valuable alternative route for highly cyto-toxic membrane proteins. Cell-free systems allow specific labeling of the sample, and the resulting expression product usually does not need further purification [23,24]. However, cell-free synthesis systems generally lack posttranslational modification machinery, which may be disadvantageous in the case of mammalian membrane proteins. Nevertheless, Klammt et al., (2004) could produce several mg of membrane protein per ml of reaction volume of apparently correctly folded protein after dissolving from a protein precipitate by the addition of detergents and lipids [25]. Recently the same authors reported that the addition of detergents and lipids to the translation mix stabilized some of the proteins tested [26]. In addition to these approaches, bacterial expression of eukaryotic membrane proteins can be facilitated by addition of the Mistic sequence to the target membrane protein sequence. Mistic is a small, highly charged protein from Bacillus subtilis, which facilitated bacterial expression and membrane insertion of several mammalian membrane proteins [6,27].
The need to reside in a membrane environment requires membrane proteins to be amphiphilic. These physicochemical properties of membrane proteins necessitate the use of detergents for solubilization from the membrane and for purification. Finding the best detergent for any given membrane protein is a matter of trial and error. Nevertheless, many membrane proteins can be solubilized and purified using a small collection of detergents [28]. The purification of solubilized membrane proteins uses similar techniques to those employed for soluble proteins. Chromatographic methodologies are widely used. In contrast to soluble proteins, membrane proteins may interact strongly with the column media, thus lowering purification efficiency. Purification steps that remove bound endogenous lipids from the solubilized membrane proteins may also induce conformational changes or loss of correct folding of the protein with subsequent loss of function and may also result in protein precipitation [29,30]. The use of poly-His or FLAG tags facilitates purification on specific affinity columns, yet the overall efficiency tends to be lower. Specific cleavage sites can be inserted to remove unwanted tags. Yet in this case, compatibility of the chosen protease with detergents should be tested in advance. Fortunately, well-ordered 2D crystals can also be grown in the presence of the purification tags (e.g. [31]) thus making the addition of protease cleavage sites mostly optional.
Solubilization, purification, and crystallization may have different detergent requirements. Therefore, it is necessary to perform a detergent-screen for all three steps. Moreover, addition of solubilized lipids to the purification buffers may help stabilize the membrane protein during the purification, as indicated by the beneficial effect of addition of phospholipids in the 3D crystallization of lactose permease (LacY) (Guan & Kaback, personal communication, see also [32]).
The brief synopsis given in the previous paragraph emphasizes the intricacies involved in membrane protein production for structural studies, regardless of the approach that is chosen for the actual data collection. With this in mind, centralization of membrane protein production is extremely beneficial to the membrane protein community by supplying pure protein for structural analysis This approach is implemented by several centers supported by the protein structure initiative (PSI) of the National Institute of General Medicine and Sciences (NIGMS, http://www.nigms.nih.gov/Initiatives/PSI/Centers). For example, the Membrane Protein Expression Center (MPEC, http://mpec.ucsf.edu) performs large-scale membrane protein production that benefits structural studies by several methods and laboratories. Similar approaches are underway in Canada (http://www.sgc.utoronto.ca/), Europe (e.g. E-MeP, http://www.e-mep.org/) and Japan (BIRC, http://unit.aist.go.jp/birc/index_e.html, [33]).
Two-dimensional membrane protein crystallization
The most widely used approach to achieve 2D crystallization of membrane proteins is the slow removal of the detergent from the protein-detergent micellar solution in the presence of added solubilized lipids at a low lipid-to-protein ratio, to allow reconstitution of the protein into a newly formed lipid bilayer (Figure 2). The goal is to reduce the detergent concentration to below its critical micellar concentration (cmc) for which precise knowledge of the starting detergent concentration is required. Kaufmann et al. (2005) have recently presented a device that allows exact determination of detergent concentrations during purification and crystallization trials using minimal sample quantities [34]. Here we briefly list and summarize the key aspect of the different approaches that have been successfully used in the past to generate 2D-crystals [28,35–38].
Figure 2.
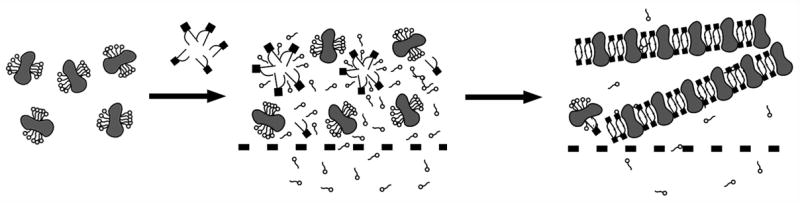
2D crystallization of membrane proteins by detergent removal via dialysis. Detergent-solubilized and purified membrane proteins (left) are mixed with solubilized lipids. The membrane proteins will then either precipitate or reconstitute into lipid membranes where they may form 2D crystals (right).
Dialysis
The dialysis method consists of dialyzing 20–100 μl samples against a large volume of buffer. This method allows slow and controlled removal of the detergent, but is difficult with low-cmc detergents, which require a longer dialysis time. As with 3D-crystallization, many factors affect the membrane reconstitution and two-dimensional crystallization, such as the choice of detergent, lipid, lipid-to-protein ratio, pH value, the kinds of salts, the salt concentrations, temperature, and other factors [36]. The use of a computer-controlled dialysis machine can reduce the dialysis time, and also allows a precise control of the temperature profile [35], which helps to make the entire process more reproducible. Still, fragile membrane proteins that do not withstand the presence of detergent for a long period of time are difficult to crystallize with this method.
Dilution
The principle of the dilution method is to dilute the protein-lipid-detergent solution to a detergent concentration below that of its cmc, thus allowing reconstitution of the protein into the lipid bilayer. It has the advantage that the detergent concentration can be very quickly brought to below its cmc. This method is also suited for low-cmc detergents [39,40].
Hydrophobic adsorption
The detergent in a protein-lipid-detergent mixture is removed by hydrophobic adsorption onto polystyrene beads, which usually can selectively bind the detergent but not protein or lipids. This procedure allows detergent removal in a short period of time, but the rate of removal cannot easily be controlled. This method can be used to remove both high- and low-cmc detergents [41,42].
Lipid monolayer crystallization
This method is based on the specific interaction between the solubilized membrane protein and a lipid monolayer that covers an air/water interface. The lipid monolayer is usually composed of two different kinds of lipids, a ligand lipid for the protein and a dilution lipid. Lipids need to be chosen that can maintain the monolayer even in the presence of the detergent. The lipid film is spread at an air/water interface of an aqueous solution containing the protein to be crystallized. The lipid-protein complex diffuses in the plane of the film, where the protein concentration is increased due to binding to the surface. Once bound, the membrane proteins can be reconstituted into a phospholipid membrane by adding lipids to the sub-phase and removing the detergent with the addition of Biobeads. This approach requires very small amounts of protein, less than 1 μg per incubation trial [43,44]. A similar approach was developed by Auer et al. (1999), who grew 2D crystals on an electron microscopy grid [45].
Crystal screening
As for three-dimensional membrane protein crystallization, 2D crystallization trials have to be screened for the presence of crystals. Due to the small size of the crystal this is so far done by transmission electron microscopy (TEM) imaging of negatively stained specimens. This step is labor intensive and monotonous, and represents a major bottleneck in the structure determination pipeline, given sufficient supply of purified protein. The use of an automated imaging system such as Leginon [46] or AutoEM [47] will greatly benefit crystal screening. The challenge for automatic systems lies in distinguishing 2D crystals from other precipitates, protein-free lipid membranes and stain and other artifacts. 2D crystals can have a multitude of shapes and appearances and are accompanied by a highly variable background. Nevertheless, the ability to calculate power spectra of recorded images is a powerful tool to assess crystallinity of a sample. Progress in automated crystal screening systems will greatly benefit systematic crystallization trials.
Electron microscopy data collection
Once suitable 2D crystals have been produced, a cryo-EM sample preparation method has to be established that maintains the high-resolution order of the 2D crystal and at the same time presents the 2D crystals with good contrast in the electron microscope. Several sample preparation methods are available and have to be adapted to the individual sample. 2D crystals usually are adsorbed to a thin carbon film, covering an electron microscopy grid. The grid with the crystals is then quick-frozen to liquid nitrogen temperature to vitrify the buffer and thereby preserve the membrane protein structure for cryo-EM imaging (e.g. [48]). Alternatively, the crystal solution can be quick-frozen on a fenestrated carbon film grid, and the crystals are then imaged in the vitrified ice over the holes of the carbon film [49]. However, the viscous (liquid) vitrified ice provides inferior stability, electrical conductivity and sample flatness than a supporting continuous carbon film can provide [50], which can only be partly compensated by surface coating [51]. Sugar embedding is another sample preparation method that involves partial drying of the carbon-film adsorbed membrane protein crystals in the presence of sugars. Tannic acid [9], trehalose [10,16,52] or glucose [53] can preserve the ultra-structure of most 2D crystals while facilitating the sample preparation and reducing the amount of ice contamination on the frozen samples. Nevertheless, as far as we know the latter approaches have not yet worked with any membrane protein that has large loops and/or termini residing outside the membrane.
The three-dimensional structure of the membrane protein is reconstructed by combining images from tilted 2D crystal samples. Depending on the actual structure and the goal of the analysis, this requires the recording of cryo-EM images of flat 2D crystal samples at angles up to 60° or 70° of sample tilt. Unfortunately, the electron-beam/sample interaction complicates the recording of images of tilted samples, which frequently show anisotropic diffraction that is poor in the direction perpendicular to the tilt axis. This beam-induced resolution loss was attributed to buildup of positive charge during the electron exposure, sample rearrangements under the electron beam, and/or a drum-head movement of the sample during image recording [50,54,55]. This phenomenon reduces the efficiency in data collection and can void high-resolution data in almost all of the recorded images of tilted samples. The data collection efficiency can be significantly enhanced by use of the SpotScanning data collection method [56–58] or by the sandwich sample preparation method [59]. The first consists of illuminating only a small area of the sample, while the concentrated electron beam “jumps” over the tilted sample and records a pattern of spots on one large image. This SpotScanning method reduces the effect of the beam-induced image drift with the dimensions of the illuminated sample area. The sandwich sample preparation method embeds the 2D crystals between two symmetrically arranged carbon films, thereby increasing the electrical conductivity and stability of the sample under the beam. Combination of both methods can further increase the efficiency of the data collection.
Recorded cryo-EM images of the 2D crystals are so-called real-space images, which provide amplitudes and phases for the reconstruction of the structure of the membrane protein. The resolution of such real-space images, however, is affected by sample vibration or movement, beam-induced sample- or image-drift as well as limited coherence of the electron microscope under high defocusing conditions. Similar to X-ray diffraction, the electron microscope can also be used to record electron diffraction patterns from the 2D crystals. Such electron diffraction patterns provide the precise amplitudes of the structure, but do not give immediate access to phase information. However, electron diffraction data collection is not sensitive to sample movement and does not significantly suffer from beam-induced effects onto the sample. Therefore, electron diffraction is an efficient way to collect high-resolution data as long as highly ordered and flat 2D crystals of sufficient size (generally diameter > 1μm) are available [60]. As in XRD studies, the phases of the structure then have to be obtained by other means, for example from homologous protein models [4] or by phase extension from a lower resolution 3D dataset [53]. A remarkable achievement from electron diffraction data collection is the recently solved 1.9 Å resolution 3D structure of Aquaporin-0 in the membrane-embedded state [4]. This map also allowed the building of an atomic model of the protein-surrounding lipid membrane and the protein-lipid interactions.
Computer image processing
Extensive computer image processing of the recorded electron crystallography data is needed to extract the structure factors (amplitudes and phases) for the membrane protein structure. Electron diffraction patterns are quantitatively evaluated and the diffraction intensities of the different diffraction spots are integrated to yield the intensities of the diffracted electron rays. The square root of these intensities then gives the amplitudes for the structure reconstruction. The real-space images of 2D crystals have to be Fourier transformed to produce a pattern similar to the diffraction pattern. The computed complex Fourier transformation, however, contains amplitude and phase information. While the high-resolution content of the real-space images is likely affected by the microscope and sample stability limitations, the real-space image has the advantage that any 2D crystal defects can be recognized and computationally corrected, before the Fourier transformation is calculated. This “unbending” of the 2D crystal images was first introduced by Henderson and Unwin [61], and results in significant sharpening of the diffraction peaks in the calculated Fourier transformation, after which the values for amplitudes and phases for the spots can be measured with much better precision.
Each real-space image or electron diffraction pattern contributes data along a plane in the three-dimensional Fourier space. Measurements from several images or diffraction pattern have to be combined, merged and interpolated to obtain a coherent 3D dataset in Fourier space, which can then be used to calculate a 3D map of the membrane protein density. If this map has sufficiently high resolution, an atomic model of the protein structure can be built. Due to the fact that images or diffraction patterns of 2D crystal samples can only be collected at sample tilts up to 60 or 70 degrees tilt, a 3D dataset from electron crystallography is devoid of information in the vertical direction perpendicular to the membrane plane and within a cone of at best 20–30° degrees opening angle to the vertical axis. This “missing cone” phenomenon is inherent to the method and is the reason why structures from electron crystallography have a lower resolution in the vertical direction: All details in the structure are smeared out vertically, and thinner horizontal elements, for example the surface loops of helical transmembrane proteins, may become undetectable (see for example the lower panels in Figure 1). In addition, regions of the membrane protein crystal that are less-well ordered will also appear at lower or even absent density in the 3D map. Less-well ordered regions are usually the surface loops of a membrane protein crystal due to deformations caused by adsorption of the crystal to the underlying carbon film. This is the reason why most electron crystallography structures do not show densities for the helix-connecting loops.
The computer image processing of the recorded images or diffraction pattern is usually done with the “MRC programs” for image processing [62]. Over many years a large set of programs has been written for processing images of two-dimensional crystals and electron diffraction patterns, usually in Fortran-77 [8–10,63]. While this software collection offers a vast and invaluable repertoire of tools for the processing of 2D crystal images, its usage involves a high amount of interactive time. An interesting new development is the IPLT software, the Image Processing Library and Toolbox [64]. Bsoft is another powerful software system that is partly applicable for 2D crystals [65].
Information exchange – 2dx.org
Electron crystallography so far lacks the community infrastructure that became available for X-ray diffraction through the CCP4 initiative [66]. While the MRC programs for electron crystallography are a powerful and well-maintained software collection, the MRC software and electron crystallography in general so far did not have a detailed manual, a tutorial, a school or a dedicated workshop, or conferences. This made it extremely difficult for newcomers to start using this method, since it usually required for a beginning student to be introduced to the method and software by one of the handful of experts in the field.
To address this need for information exchange, we have created a web server 2dx.org to provide the community of electron crystallography with a central information exchange platform for electron crystallography of membrane proteins. This server is maintained by the Stahlberg laboratory at UC Davis, and is based on a Zope platform (http://www.zope.org), running Plone (http://www.plone.org) and a ZWiki engine (http://www.zwiki.org). The 2dx.org server intends to collect information about electron crystallography sample preparation, imaging and computer image processing. It should give the newcomer to the method all the information needed to quickly familiarize him/herself with the method. It should also serve the advanced electron crystallography user as a central database for expert knowledge and information exchange. Currently, the 2dx.org server contains a detailed step-by-step introduction to the philosophy and usage of the MRC software programs, as well as documentation about the MRC software and the conventions for 2D crystal image processing, which were contributed by Vinzenz Unger (Yale University) and Anchi Cheng (Scripps Research Institute). The MRC documentation section includes the description of the functions and interfaces of the MRC programs, a collection of tips and guidelines for the usage of these programs, as well as definitions of the involved file formats, the conventions for the tilt geometry and other parameters. All manual or documentation pages of the 2dx.org server allow the visitors to add comments, corrections or questions in form of an online blog at the end of each page.
We also produce a software system 2dx for the user-friendly image processing of electron crystallography data [67]. 2dx provides a graphical user-interface (GUI) that guides the user through the required processing steps, displays processing parameters and processing results in a clearly structured way, and offers extensive help information and functions in all phases of the image processing, see
Figure 3. The 2dx software is partly based on the MRC programs, and assists in the usage of the MRC software and the interpretation of the processing results. 2dx in addition offers optionally a high level of automation, upto fully automatic processing of electron crystallography data. While the current implementation of 2dx is based on the MRC software, it can equally well be used as user-friendly GUI for other backend processing packages, for example for single particle processing tasks using the Spider software [68]. The 2dx software is available under the Gnu Public License (GPL), and is freely available as open source software on the 2dx.org web server. 2dx runs natively on Mac OSX and Linux/X11 (Linux, IRIX and other Unix variants). The 2dx GUI has an interactive help function (right-mouse click), which offers a context-sensitive direct link to the documentation on the 2dx.org web server. This allows the user throughout the different image processing steps a close interaction with the manual and documentation of the 2dx software, where the users can also make use of the online discussion blog for every section of the manual, to directly pose questions or add comments or suggestions or contribute their experiences or expert knowledge.
Figure 3.

The graphical user interface of the 2dx_image program for user-friendly image processing of 2D crystals. The “Standard Scripts” in the top left panel indicate the required workflow, which the user should accomplish in order to process one image. Each workflow step (script) requires parameters, which are displayed in the central pane. 2dx_image can optionally determine the required parameters and perform the entire processing fully automatically, which is a pre-requisite for high-throughput image processing in electron crystallography.
B. Hankamer from the University of Queensland, Australia, H. Stahlberg and R. Hill also organize a bi-annual international workshop on electron crystallography of membrane proteins, of which the first took place at UC Davis from August 6–11, 2006 (see: http://2dx.org/workshop). This first electron crystallography workshop featured 23 speakers, and was oversubscribed and limited to 20 students, showing the need in the community for information exchange in electron crystallography. Also for the workshops, the 2dx.org web server is the central information exchange platform.
Conclusions
Here we briefly summarized the different steps of a structural study of membrane proteins by electron crystallography. Each step presents difficulties, and improvements are required but within reach. For now, development of cell-free expression systems, dialysis machines, automated imaging systems, and a variety of different image processing software packages are underway, thus accelerating the path to the structure. Electron crystallography is an excellent alternative to X-ray crystallography. Obtaining the structure of the membrane protein embedded in a lipid bilayer instead of detergent molecules may better reflect the native structure of the membrane protein. Currently, atomic models for seven membrane proteins and twelve medium resolution models for which no X-ray structure is available have so far been determined by this method. Information included in these medium resolution maps can be combined with other techniques such as homology modeling to gain insights into the structure-function relationships of more membrane proteins, not amenable to 3D crystallization [21,69]. To provide the community of electron crystallography with a platform for information exchange, we have created a web server 2dx.org, where information about electron crystallography sample preparation, imaging and computer image processing is maintained. The server hosts user manuals for image processing software, the software system 2dx, and documentation about a bi-annual workshop on electron crystallography of membrane proteins.
Acknowledgments
This work was in part supported by the NSF, grant number MCB-0447860 and by the NIH, grant number U54-GM074929. V Unger is supported by NIH grants GM66145 and GM071590. We thank Y. Fujiyoshi and J. Fethière for providing material for Figure 1.
Abbreviations
- 2D
two-dimensional
- cmc
critical micellar concentration
- cryo-EM
cryo-electron microscopy
- NMR
nuclear magnetic resonance
- XRD
X-ray diffraction
References
- 1.Russell RB, Eggleston DS. Nat Struct Biol. 2000;7(Suppl):928–930. doi: 10.1038/80691. [DOI] [PubMed] [Google Scholar]
- 2.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nuc Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khademi S, O’Connell J, 3rd, Remis J, Robles-Colmenares Y, Miercke LJ, Stroud RM. Science. 2004;305:1587–1594. doi: 10.1126/science.1101952. [DOI] [PubMed] [Google Scholar]
- 4.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harries WE, Akhavan D, Miercke LJ, Khademi S, Stroud RM. Proc Natl Acad Sci U S A. 2004;101:14045–14050. doi: 10.1073/pnas.0405274101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 7.Wüthrich K. Nat Struct Biol. 1998;5 Suppl:492–495. doi: 10.1038/728. [DOI] [PubMed] [Google Scholar]
- 8.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. J Mol Biol. 1990;213:899– 929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 9.Kühlbrandt W, Wang DN, Fujiyoshi Y. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 10.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 11.Ren G, Reddy VS, Cheng A, Melnyk P, Mitra AK. Proc Natl Acad Sci U S A. 2001;98:1398–1403. doi: 10.1073/pnas.041489198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyazawa A, Fujiyoshi Y, Unwin N. Nature. 2003;424:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- 13.Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Nature. 2004;429:193–197. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- 14.Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, et al. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 15.Holm PJ, Bhakat P, Jegerschöld C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. J Mol Biol. 2006 doi: 10.1016/j.jmb.2006.05.056. in press. [DOI] [PubMed] [Google Scholar]
- 16.Nogales E, Wolf SG, Downing KH. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 17.Hirai T, Heymann JA, Shi D, Sarker R, Maloney PC, Subramaniam S. Nat Struct Biol. 2002;9:597–600. doi: 10.1038/nsb821. [DOI] [PubMed] [Google Scholar]
- 18.Tate CG, Ubarretxena-Belandia I, Baldwin JM. J Mol Biol. 2003;332:229–242. doi: 10.1016/s0022-2836(03)00895-7. [DOI] [PubMed] [Google Scholar]
- 19.Kukulski W, Schenk AD, Johanson U, Braun T, de Groot BL, Fotiadis D, Kjellbom P, Engel A. J Mol Biol. 2005;350:611–616. doi: 10.1016/j.jmb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Schenk AD, Werten PJ, Scheuring S, de Groot BL, Müller SA, Stahlberg H, Philippsen A, Engel A. J Mol Biol. 2005;350:278–289. doi: 10.1016/j.jmb.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 21.Fleishman SJ, Unger VM, Ben-Tal N. Trends Biochem Sci. 2006;31:106–113. doi: 10.1016/j.tibs.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 22.Grisshammer R. Biochim Biophys Acta. 2003;1610:1. [Google Scholar]
- 23.Baranov VI, Spirin AS. Meth Enzymol. 1993;217:123–142. doi: 10.1016/0076-6879(93)17059-e. [DOI] [PubMed] [Google Scholar]
- 24.Spirin AS, Baranov VI, Ryabova LA, Ovodov SY, Alakhov YB. Science. 1988;242:1162–1164. doi: 10.1126/science.3055301. [DOI] [PubMed] [Google Scholar]
- 25.Klammt C, Lohr F, Schafer B, Haase W, Dotsch V, Ruterjans H, Glaubitz C, Bernhard F. Eur J Biochem. 2004;271:568–580. doi: 10.1111/j.1432-1033.2003.03959.x. [DOI] [PubMed] [Google Scholar]
- 26.Klammt C, Schwarz D, Fendler K, Haase W, Dotsch V, Bernhard F. FEBS J. 2005;272:6024–6038. doi: 10.1111/j.1742-4658.2005.05002.x. [DOI] [PubMed] [Google Scholar]
- 27.Roosild TP, Vega M, Castronovo S, Choe S. BMC Struct Biol. 2006;6:10. doi: 10.1186/1472-6807-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rigaud J, Chami M, Lambert O, Levy D, Ranck J. Biochim Biophys Acta. 2000;1508:112–128. doi: 10.1016/s0005-2736(00)00307-2. [DOI] [PubMed] [Google Scholar]
- 29.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 30.Long SB, Campbell EB, Mackinnon R. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 31.Braun T, Philippsen A, Borgnia M, Agre P, Kühlbrandt W, Engel A, Stahlberg H. GLPF: A structural variant of the aquaporin tetramer. In: Hohmann S, Nielsen S, editors. Molecular biology and physiology of water and solute transport. Kluwer Academic; New York: 2000. pp. 13–22. [Google Scholar]
- 32.Guan L, Smirnova IN, Verner G, Nagamori S, Kaback HR. Proc Natl Acad Sci USA. 2006;103:1723–1726. doi: 10.1073/pnas.0510922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kyogoku Y, Fujiyoshi Y, Shimada I, Nakamura H, Tsukihara T, Akutsu H, Odahara T, Okada T, Nomura N. Acc Chem Res. 2003;36:199–206. doi: 10.1021/ar0101279. [DOI] [PubMed] [Google Scholar]
- 34.Kaufmann TC, Engel A, Remigy HW. Biophys J. 2006;90:310–317. doi: 10.1529/biophysj.105.070193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jap BK, Zulauf M, Scheybani T, Hefti A, Baumeister W, Aebi U, Engel A. Ultramic. 1992;46:45–84. doi: 10.1016/0304-3991(92)90007-7. [DOI] [PubMed] [Google Scholar]
- 36.Kühlbrandt W. Q Rev Biophys. 1992;25:1–49. doi: 10.1017/s0033583500004716. [DOI] [PubMed] [Google Scholar]
- 37.Hasler L, Heymann JB, Engel A, Kistler J, Walz T. J Struct Biol. 1998;121:162–171. doi: 10.1006/jsbi.1998.3960. [DOI] [PubMed] [Google Scholar]
- 38.Ringler P, Heymann BJ, Engel A. Two-dimensional crystallization of membrane proteins. In: Baldwin SA, editor. Membrane Transport. Oxford University Press; Oxford, UK: 2000. pp. 229–268. [Google Scholar]
- 39.Dolder M, Engel A, Zulauf M. FEBS Lett. 1996;382:203–208. doi: 10.1016/0014-5793(96)00180-9. [DOI] [PubMed] [Google Scholar]
- 40.Remigy HW, Caujolle-Bert D, Suda K, Schenk A, Chami M, Engel A. FEBS Lett. 2003;555:160–169. doi: 10.1016/s0014-5793(03)01105-0. [DOI] [PubMed] [Google Scholar]
- 41.Rigaud JL, Mosser G, Lacapere JJ, Olofsson A, Levy D, Ranck JL. J Struct Biol. 1997;118:226–235. doi: 10.1006/jsbi.1997.3848. [DOI] [PubMed] [Google Scholar]
- 42.Hankamer B, Morris EP, Barber J. Nat Struct Biol. 1999;6:560–564. doi: 10.1038/9341. [DOI] [PubMed] [Google Scholar]
- 43.Levy D, Mosser G, Lambert O, Moeck GS, Bald D, Rigaud JL. J Struct Biol. 1999;127:44–52. doi: 10.1006/jsbi.1999.4155. [DOI] [PubMed] [Google Scholar]
- 44.Levy D, Chami M, Rigaud JL. FEBS Lett. 2001;504:187–193. doi: 10.1016/s0014-5793(01)02748-x. [DOI] [PubMed] [Google Scholar]
- 45.Auer M, Scarborough GA, Kühlbrandt W. J Mol Biol. 1999;287:961–968. doi: 10.1006/jmbi.1999.2652. [DOI] [PubMed] [Google Scholar]
- 46.Carragher B, Kisseberth N, Kriegman D, Milligan RA, Potter CS, Pulokas J, Reilein A. J Struct Biol. 2000;132:33–45. doi: 10.1006/jsbi.2000.4314. [DOI] [PubMed] [Google Scholar]
- 47.Zhang P, Borgnia MJ, Mooney P, Shi D, Pan M, O’Herron P, Mao A, Brogan D, Milne JL, Subramaniam S. J Struct Biol. 2003;143:135–144. doi: 10.1016/s1047-8477(03)00124-2. [DOI] [PubMed] [Google Scholar]
- 48.Ren G, Cheng A, Reddy V, Melnyk P, Mitra AK. J Mol Biol. 2000;301:369–387. doi: 10.1006/jmbi.2000.3949. [DOI] [PubMed] [Google Scholar]
- 49.Cyrklaff M, Kühlbrandt W. Ultramic. 1994;55:141–153. doi: 10.1016/0304-3991(94)90165-1. [DOI] [PubMed] [Google Scholar]
- 50.Henderson R. Ultramic. 1992;46:1–18. doi: 10.1016/0304-3991(92)90003-3. [DOI] [PubMed] [Google Scholar]
- 51.Brink J, Gross H, Tittmann P, Sherman MB, Chiu W. J Microsc. 1998;191( Pt 1):67–73. doi: 10.1046/j.1365-2818.1998.00342.x. [DOI] [PubMed] [Google Scholar]
- 52.Kimura Y, Vassylyev DG, Miyazawa A, Kidera A, Matsushima M, Mitsuoka K, Murata K, Hirai T, Fujiyoshi Y. Nature. 1997;389:206–211. doi: 10.1038/38323. [DOI] [PubMed] [Google Scholar]
- 53.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 54.Glaeser RM, Downing KH. Microsc Microanal. 2004;10:790–796. doi: 10.1017/s1431927604040668. [DOI] [PubMed] [Google Scholar]
- 55.Typke D, Downing KH, Glaeser RM. Microsc Microanal. 2004;10:21–27. doi: 10.1017/S1431927604040164. [DOI] [PubMed] [Google Scholar]
- 56.Downing KH, Glaeser RM. Ultramicroscopy. 1986;20:269–278. doi: 10.1016/0304-3991(86)90191-9. [DOI] [PubMed] [Google Scholar]
- 57.Bullough P, Henderson R. Ultramic. 1987;21:223–230. [Google Scholar]
- 58.Downing KH. Science. 1991;251:53–59. doi: 10.1126/science.1846047. [DOI] [PubMed] [Google Scholar]
- 59.Gyobu N, Tani K, Hiroaki Y, Kamegawa A, Mitsuoka K, Fujiyoshi Y. J Struct Biol. 2004;146:325–333. doi: 10.1016/j.jsb.2004.01.012. [DOI] [PubMed] [Google Scholar]
- 60.Vonck J. Ultramic. 2000;85:123–129. doi: 10.1016/s0304-3991(00)00052-8. [DOI] [PubMed] [Google Scholar]
- 61.Henderson R, Unwin PN. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 62.Crowther RA, Henderson R, Smith JM. J Struct Biol. 1996;116:9–16. doi: 10.1006/jsbi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 63.Unwin PN, Henderson R. J Mol Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 64.Philippsen A, Schenk AD, Stahlberg H, Engel A. J Struct Biol. 2003;144:4–12. doi: 10.1016/j.jsb.2003.09.032. [DOI] [PubMed] [Google Scholar]
- 65.Heymann JB. J Struct Biol. 2001;133:156–169. doi: 10.1006/jsbi.2001.4339. [DOI] [PubMed] [Google Scholar]
- 66.Collaborative Computational Project, N. Acta Crystallog. 1994;50:760–763. [Google Scholar]
- 67.Gipson B, Zeng X, Zhang ZY, Stahlberg H. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.07.020. in press. [DOI] [PubMed] [Google Scholar]
- 68.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 69.Sali A, Glaeser R, Earnest T, Baumeister W. Nature. 2003;422:216–225. doi: 10.1038/nature01513. [DOI] [PubMed] [Google Scholar]