


Abstract
The c-FMS gene encodes the macrophage colony-stimulating factor receptor (M-CSFR or CSF1-R), which is a tyrosine kinase growth factor receptor essential for macrophage development. We have previously characterized the chromatin features of the mouse gene; however, very little is known about chromatin structure and function of the human c-FMS locus. Here we present a side-by-side comparison of the chromatin structure, histone modification, transcription factor occupancy and cofactor recruitment of the human and the mouse c-FMS loci. We show that, similar to the mouse gene, the human c-FMS gene possesses a promoter and an intronic enhancer element (c-fms intronic regulatory element or FIRE). Both elements are evolutionarily conserved and specifically active in macrophages. However, we demonstrate by in vivo footprinting that both murine and human c-FMS cis-regulatory elements are recognised by an overlapping, but non-identical, set of transcription factors. Despite these differences, chromatin immunoprecipitation experiments show highly similar patterns of histone H3 modification and a similar distribution of chromatin modifying and remodelling activities at individual cis-regulatory elements and across the c-FMS locus. Our experiments support the hypothesis that the same regulatory principles operate at both genes via conserved cores of transcription factor binding sites.
INTRODUCTION
The c-FMS gene encodes the tyrosine kinase receptor for macrophage colony-stimulating factor (M-CSFR) that plays a critical role in the differentiation and maintenance of macrophages (1). The same gene is also expressed in trophoblasts and low level mRNA expression has been reported in haematopoietic stem cells (2). However, the receptor protein is only detected on the surface of committed myeloid precursor cells and only from a distinct point in development onwards do these cells become responsive to macrophage colony-stimulating factor (M-CSF) (3,4). The regulation of the c-FMS gene has been the focus of considerable attention, as mutation of the gene can generate proteins with oncogenic potential (5). In addition, the regulatory regions of the human c-FMS gene have been shown to be targets of nuclear oncoproteins involved in myeloid leukaemia, such as AML1-ETO (6,7).
Transcription of the human c-FMS gene in trophoblasts and macrophages starts from two different promoters. Exon 1 is transcribed only in trophoblasts, whereas exon 2 is located 25 kb downstream and is the first exon found in macrophage-specific transcripts (8,9). In mice, macrophage-specific transcription starts at the same sequences as in the human gene; however, trophoblast transcription initiation occurs at several sites within 1 kb upstream of the macrophage initiation site (10). A number of experiments looking at the mouse gene have addressed the question of how the lineage-specific and stage-restricted expression of c-fms is regulated. The proximal promoter contains multiple binding sites for the transcription factor PU.1 and other members of the Ets transcription factor family (11,12). To identify other cis-regulatory elements, we have mapped macrophage-specific DNase I hypersensitive sites (DHSs) in the promoter region and in the second intron (13). One of the intronic DHSs contains an important tissue-specific enhancer element (c-fms intron regulatory element or FIRE) that is absolutely required for c-fms expression in macrophage cell lines and transgenic mice (10,13). Lipopolysaccharide (LPS) treatment has been shown to down-regulate c-fms expression (14). This has an effect not only on c-fms transcription, but also affects mRNA elongation downstream of the ATG. It was shown that intronic sequences including FIRE are required for this block in mRNA elongation (15).
We have previously investigated the developmental regulation of the mouse c-fms gene at the level of chromatin structure. To this end, we purified early, multipotent myeloid precursor cells from mouse bone marrow and differentiated these cells to macrophages in vitro. In these cells we investigated c-fms chromatin fine structure and transcription factor occupancy within the promoter and FIRE and correlated this with M-CSFR mRNA and protein expression (4). We showed that the c-fms gene is already transcribed in early myeloid precursors and that low level transcription is associated with complete transcription factor complex assembly on the c-fms promoter. Subsequently, alterations in transcription factor occupancy at FIRE accompany terminal differentiation and activation. Until now it has not been known which chromatin remodelling and modification activities are recruited to the different c-fms regulatory elements of the murine gene.
The regulation of the human c-FMS gene is less well understood. Sequence comparisons between the human and mouse c-FMS genes revealed that the regulatory elements required for correct expression of the murine gene, such as FIRE, are highly conserved between mouse and man (13). It was therefore likely that these elements would fulfil a similar regulatory role in human cells. We could indeed show that they form a DHS in macrophage cells and bind macrophage-specific transcription factors (7). Interestingly, in spite of their high sequence conservation, we found a significant deviation in factor occupancy at cores of specific factor binding sites between the human and the mouse genes.
To this end, we have carried out a comparison of chromatin structure and transcription cofactor recruitment as well as transcription factor occupancy at the human and the mouse c-FMS loci. We show that although the transcription factor composition at the cis-elements of each respective gene locus is different, the distribution of chromatin modifying activities and the pattern of chromatin remodelling across both loci is highly similar. Our experiments suggest that conserved cores of transcription factor binding sites are responsible for the conservation of regulatory features of the human and mouse c-FMS genes.
MATERIALS AND METHODS
Cell culture
HL-60 cells (ATCC CCL-240) were grown in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin and 100 µg/ml streptomycin (P/S). When indicated, HL-60 cells were cultured with 50 ng/ml phorbol-12-myristate acetate (PMA) (Sigma) for 72 h prior to harvesting. HeLa, Hep3B, 3T3 and RAW 264 cells and human fibroblasts (ATCC CCL-110) were grown in Dulbecco’s modified Eagle’s medium with 10% FCS and P/S. Normal human monocytes/macrophages were obtained from peripheral blood by culturing mononuclear cells in IMDM containing 10% FCS, P/S and 50 ng/ml M-CSF (R+D Systems). Adherent monocytes/macrophages were harvested after 24 h in culture. Mouse macrophages were obtained from bone marrow by culturing in IMDM containing 10% FCS, P/S and 10% L cell conditioned medium containing M-CSF (4) for 7 days.
RT–PCR
Total RNA was extracted and reverse transcribed using standard techniques. PCR were performed using real-time quantitative PCR (ABI Prism 7700 sequence detection system; Perkin Elmer) with SYBR Green. Relative expression was calculated as the c-FMS/GAPDH ratio expressed relative to human and mouse cell line cDNA. Primers were designed using Primer Express™ 1.5 software. Mouse and human c-FMS- and GAPDH-specific primer sequences were as published before (4,7).
Reporter constructs and expression plasmids
Figure 1B shows a map of constructs 1–6. The c-FMS promoter sequence from –540 to –20 was PCR amplified and subcloned into a pGL2-basic vector (Promega) (construct 4). The FIRE sequence from +4143 to +4785 was PCR amplified and subcloned into pGL2 basic with an inserted c-FMS promoter sequence (constructs 5 and 6) or subcloned into a pGL2 SV40 promoter vector (constructs 2 and 3) in both directions at the BamHI site downstream of the luciferase gene. Construct 1 was the standard pGL2 SV40 promoter vector.
Figure 1.

FIRE acts as an orientation-independent enhancer with maximal activity in RAW 264 macrophage cells. (A) A schematic map of the 8 kb of upstream regulatory sequences of the human c-FMS gene and 6 kb upstream regulatory sequences of the murine c-fms gene showing boxed non-coding regions with significant sequence homology. The difference in size between human and murine intronic sequences is indicated by a triangle in the map of the mouse gene (P, promoter; F-1kb, FIRE-1 kb; F, FIRE). Numbers indicate base pairs relative to ATG and the dotted line on the mouse sequence indicates sequence inserted in the human gene. The transcription start site in macrophages is indicated by a horizontal arrow. Black boxes indicate the primer sites for ChIP experiments presented in Figures 5–7 and black arrows represent macrophage-specific DNase I hypersensitive sites. (B) Schematic representation of luciferase reporter genes used for transient transfection assays represented in (C)–(E). (C) FIRE enhances transcriptional activity from the SV40 promoter in the sense and antisense orientations in RAW 264 cells, which represent a model for macrophages. (D) The c-FMS promoter construct has markedly increased activity in RAW 264 cells compared with c-FMS non-expressing HeLa and Hep3B cells. (E) FIRE enhances transcriptional activity from the c-FMS promoter in the sense and antisense orientations in RAW 264 cells. Data presented are average values ± SD from at least two independent transfection experiments.
Transient transfection assays
Transient transfections of RAW 264, HeLa and Hep3B cells were carried out in triplicate experiments in 6-well plates using lipofectamine (Invitrogen) and standard techniques. A sample of 1 µg of each plasmid was used per transfection and firefly luciferase activities from constructs 1–6 were determined 24 h post-transfection using a luciferase reporter assay (Promega). To control for transfection efficiency, luciferase activities were normalised to LacZ activities from a co-transfected RSVlacZ construct, which were determined using a luminescent β-galactosidase assay kit (BD Bioscience). Results presented are means ± SEM of between two and four independent experiments
In vivo footprinting analysis
In vivo DMS footprinting was performed as described previously (4) with 1 µg of purified and piperidine-cleaved genomic DNA (16) from DMS-treated cells used as starting material. DMS-treated and piperidine-cleaved naked genomic DNA was used as a control. Alterations in G(N7) DMS reactivity at c-FMS cis-regulatory elements were visualised by LM-PCR. In vivo UV footprinting was performed as described (17) with 1 µg of purified genomic DNA extracted from UV-treated cells used as starting material. Alterations in UV-induced pyrimidine dimer formation were visualised using terminal transferase-dependent PCR (TD–PCR) amplification. UV-treated genomic DNA was used as a naked DNA control. Primer sequences used for DMS and UV footprinting were as published before (4,7).
Ligation-mediated PCR (LM–PCR) and TD–PCR generated bands were visualised and quantified with a phosphorimager screen on a Molecular Imager™ FX (Bio-Rad), using Quantity One software. The mouse experiment in Figure 4D was run on a LiCor sequencing system as detailed previously (17).
Figure 4.

DMS and UV in vivo footprinting of human and mouse FIRE reveals similarities and differences in transcription factor occupancy and associated alterations in chromatin fine structure in c-FMS expressing cells. (A) DMS in vivo footprinting of the human FIRE on the upper strand shows protections of GC-rich regions flanking the AML1/Sp1/Ets site, which displays enhanced DMS reactivity at G+4434 in PMA-treated HL-60 and primary macrophages. The GC–2 footprint is also found in murine FIRE (4). (B) DMS in vivo footprinting of the human FIRE on the lower strand shows extensive protection, particularly over the AML1/SP-1/Ets binding sites which are present in all c-FMS expressing cells. These DMS footprints are conserved between mouse and human (4). There is further protection in c-FMS expressing cells over a GC-rich region (GC–3) and enhancement of G+4516 at a putative Ets site. (C and D) In vivo UV footprints of the lower DNA strand of human and mouse FIRE, respectively. Four regions of altered dimer formation are identified (W, X, Y and Z) in the human sequence, with X and Z showing preserved differences between mouse and human. X and Z overlap the GC–2 and the putative Ets binding sites, respectively, which are conserved with similar DMS footprinting between the species. (E) The FIRE sequence with protections and enhancements indicated as white and black circles, respectively. Numbers represent base pairs relative to human ATG. Sequences conserved between mouse and human are indicated by a line, sequence deviations are displayed as *. Hatched areas (X, W, Y and Z) represent areas of altered pyrimidine dimer formation, with conserved regions in bold. Transcription factor binding sites are labelled, with sites conserved between mouse and human labelled in bold and sites only found in mouse labelled in italic. Lane labelling for (A)–(D): lane G, G reaction; lane N, naked DNA; lane 1, HeLa; lane 2, HL-60; lane 3, HL-60 + PMA; lane 4, human fibroblasts; lane 5, human macrophages; lane 6, 3T3; lane 7, RAW 264.
Chromatin immunoprecipitation (ChIP) assay and real-time PCR analysis
ChIP assays were performed and normalized as detailed previously (7,18). Sonicated chromatin from 107 cells was used for each immunoprecipitation. The following antibodies were used for the respective immunoprecipitations: 5 µl of normal rabbit IgG (12-370; Upstate Biotechnology), 25 µl of anti-CBP, 50 µl of anti-Brg-1 (Santa-Cruz) or 5 µl of anti-dimethyl-histone H3 (Lys9), anti-acetyl-histone H3 (Lys9), anti-TATA box binding protein (TBP) or anti-HDAC1 (Upstate Biotechnology). Precipitated DNA was quantified using real-time quantitative PCR (ABI Prism 7700 sequence detection system; Perkin Elmer) with SYBR Green. The location of the human and mouse c-FMS-specific primers is shown in Figure 1. The human primer sequences were as published (7). The primer sequences specific for the mouse c-fms and α-fetoprotein genes are as follows: Prom-1.5kb: forward, CAGGCCGGCTGAGTGTCT; reverse, TCCACGTAGATGGTGTCAGCAT. Prom: forward, CTGC TGCTGGCCACAGTTT; reverse, CAGCGATGCCCTCTT TGC. FIRE-1.0kb: forward, GTGTCCTGTGACCTTTAG GGTTAAG; reverse, TTCTCAGGCCGTACTGCTAATG. FIRE: forward, CCTCAGAGGCTGTGAATCAGTTC; reverse, TGCCTGTGTGGTGTCAGCAT. FIRE+1.5: forward, CCATGGCGAGGGTTCATTAT; reverse, AGGTA TTGCTGTCAAGGACTTTAGC. α-fetoprotein promoter: forward, TGCCTAACTTCAACGTAAGGAAATAG; reverse, TCCTGTTTAAGGGATGCCTGTT.
Relative PCR signals for all primers were first calculated as a signal ratio obtained with the specific antibody versus signals observed with an IgG control (non-specific background). To correct for the efficiency of precipitation in different experiments with different human cell types, signals were then normalised to those obtained with the GAPDH exon 6 primers, except for H3 methylation mapping experiments, where signals were normalised to α-fetoprotein exon 3. For the analysis of the murine c-fms locus, signals were normalised to the mouse α-fetoprotein promoter. Data presented in Figures 5–7 represent the means ± SD of quantifications from at least two separate immunoprecipitations on at least two separate chromatin preparations for each cell type.
Figure 5.

Chromatin immunoprecipitation of (A) acetylated lysine 9 (K9) of histone H3 and (B) methylated K9 of histone H3 in HeLa, HL-60, HL-60 + PMA, 3T3 and RAW 264 cells with quantification of recovered DNA at 6 and 5 points along the human and murine c-FMS locus, respectively. The location of the promoter, FIRE-1 kb and FIRE regions are indicated (P, F-1kb and F) and DNA was quantified as described in Materials and Methods. Differentiation of HL-60 cells with PMA induces a marked increase in H3 K9 acetylation upstream and downstream of FIRE while FIRE remains under-acetylated. A similar pattern of H3 K9 acetylation is seen in RAW 264 cells. Both HL-60 and HeLa H3 are methylated at K9 across the locus while PMA differentiation of HL-60 cells induces H3 K9 demethylation.
Figure 7.
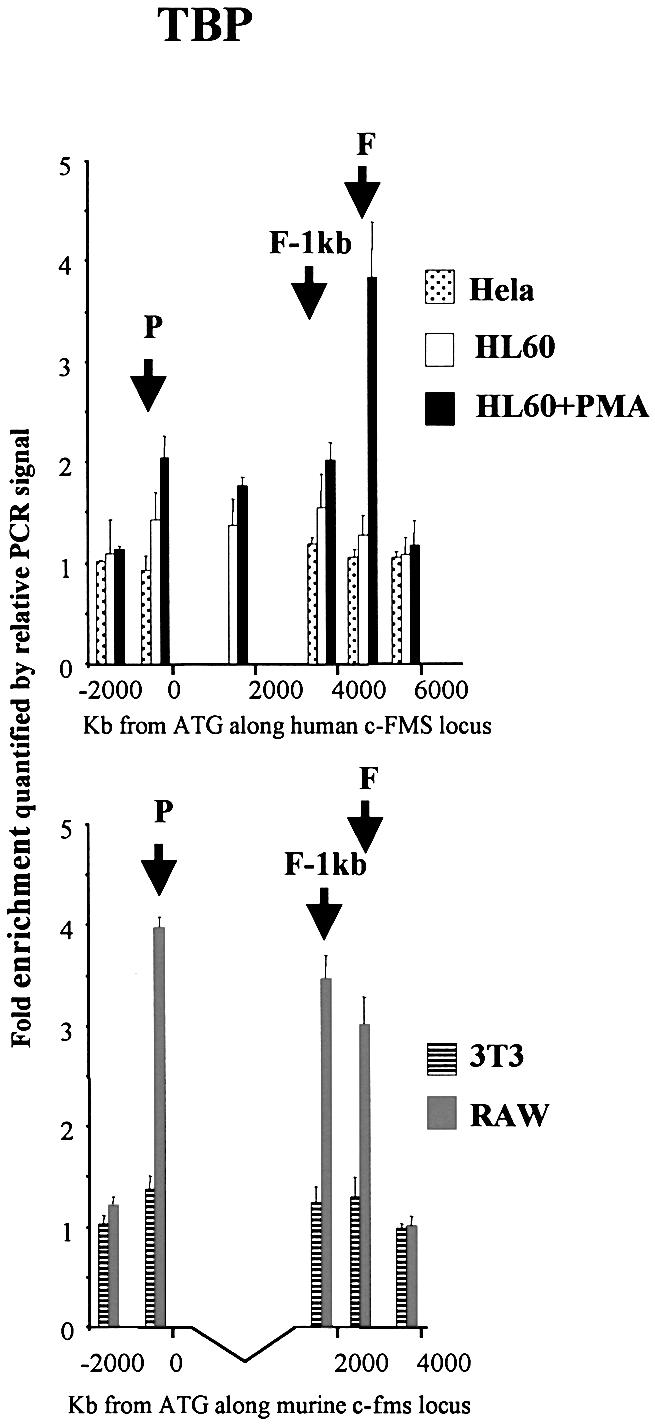
Chromatin immunoprecipitation of TBP in HeLa, HL-60, HL-60 + PMA, 3T3 and RAW 264 cells with quantification of recovered DNA at 6 and 5 points along the human and murine c-FMS locus, respectively. The location of the promoter, FIRE-1 kb and FIRE regions are indicated (P, F-1kb and F) and DNA was quantified as in Materials and Methods. TBP binding at both the promoter and FIRE was increased with PMA-induced differentiation of HL-60 cells. In RAW 264 cells increased amounts of TBP were precipitated at all three elements relative to 3T3 cells.
RESULTS
Human FIRE has macrophage-specific enhancer activity
Murine FIRE has been shown to possess enhancer activity in transfected cells (13) and is essential for high level expression of a c-fms construct in transgenic mice (10). The corresponding element of the human gene had not yet been characterised. Figure 1A shows a map comparing the position of conserved regulatory elements and DHSs in the human and the mouse gene (7,13). Three major DHSs are shown corresponding to the promoter and intronic elements, one of which is FIRE. Although the mouse intronic elements are closer to the promoter, the position and spacing between the intronic DHSs are highly conserved between human and mouse. We performed transient transfection experiments in macrophage and non-macrophage cell lines and measured the activity of luciferase constructs carrying the SV40 basic promoter and the c-FMS promoter alone or together with FIRE inserted in its downstream position (Fig. 1B). The results are shown in Figure 1C–E. In both orientations, FIRE was able to stimulate the SV40 basic promoter as well as the c-FMS promoter. Our experiments clearly show that both the c-FMS promoter and FIRE are preferentially active in macrophage cell lines.
Chromatin fine structure alterations at c-FMS regulatory elements
For our chromatin fine structure analyses we used a number of human and mouse cell lines. As control cell lines that did not express the c-FMS gene we employed human fibroblasts and HeLa cells as well as murine 3T3 fibroblast cells. As myeloid c-FMS expressing cells we used the promyelocytic leukaemia cell line HL-60, which could be further differentiated into monocytes/macrophages by culturing them with PMA as described in Materials and Methods. RAW 264 cells served as a model for murine macrophages. Figure 2 displays the relative c-FMS mRNA expression level in the different cell lines as measured by RT–PCR. The differentiation of HL-60 cells led to a pronounced up-regulation of c-FMS mRNA, although levels were still lower than levels measured in primary monocytes/macrophages purified from peripheral blood. The level of c-fms mRNA in RAW 264 cells was also lower than in primary monocytes/macrophages from mouse bone marrow.
Figure 2.

Levels of c-FMS-specific mRNA increase significantly with monocyte/macrophage differentiation. Real-time PCR quantification of c-FMS mRNA levels relative to GAPDH in different human and mouse cell lines and cells from human donors and normal mice. The numbers indicate average relative mRNA levels from at least two independent RNA preparations. Fibroblasts, HeLa and 3T3 cells are c-FMS non-expressing cells. HL-60 cells express very low but detectable levels of c-FMS mRNA but PMA-induced monocyte/macrophage differentiation increases expression over 300-fold. Highest expression is observed in both normal human and mouse monocyte/macrophage cells and RAW 264 cells, a mouse macrophage cell line. ND, not done.
To obtain information about the chromatin fine structure of the c-FMS locus, we used two different in vivo footprint methods described in Kontaraki et al. (17), which depend on the fact that bound transcription factors and chromatin structure affect the reactivity of DNA with dimethyl sulphate (DMS) or UV, both of which can be applied to intact cells. After in vivo formation of alkylated or dimerised bases, respectively, the position of these lesions is determined at nucleotide resolution by use of LM–PCR or a related technique (TD–PCR) (see 19). DMS footprinting indicates the position of transcription factor binding sites occupied by high affinity DNA binding proteins, but does not reveal information about general chromatin components, such as histones. UV footprinting highlights UV-induced dimer formation between neighbouring bases (mostly pyrimidines) and generates information regarding DNA topology that can be used to indirectly visualise the action of general chromatin components as well.
Figure 3 shows the application of these methods to the human c-FMS promoter. The DMS footprinting experiment displayed in Figure 3A confirmed the DNA–protein interactions seen in previous experiments (7). The PU.1 binding site at –173 bp and the AML binding site at –196 bp were occupied in both undifferentiated and differentiated HL-60 cells as well as primary macrophages. Reduction and enhancement of bands as a result of differential alkylation was confirmed by quantification of the bands on a phosphorimager (Fig. 3C). The PU.1 site is located at a similar position in the mouse gene and was fully occupied in macrophage precursors and differentiated macrophages (4). However, the AML1 binding sequence located at –196 bp of the human gene is absent in the mouse gene. The human c-FMS promoter contains a conserved C/EBP site that has been shown to be functional in transient transfection assays (20). We were unable to see occupancy of this site by DMS in vivo footprinting on this DNA strand and footprinting experiments looking at the other strand were inconclusive. We therefore examined the same region by UV in vivo footprinting (Fig. 3B). This experiment clearly demonstrated significant alterations in chromatin fine structure over the C/EBP and AML1 sites in c-FMS expressing cells compared to c-FMS non-expressing cells. These latter cells showed a DNA topology similar to that of naked DNA. Interestingly, DNA topology at a highly conserved pyrimidine region upstream of the C/EBP binding site was also strongly altered. As indicated in Figure 3D, both alterations (alterations A and B) were also found in the mouse gene (4).
Figure 3.

DMS and UV in vivo footprinting of the human c-FMS promoter reveals transcription factor occupancy with associated alterations in chromatin fine structure in c-FMS expressing cells. (A) In vivo DMS footprinting reveals significant protections over the PU.1 and AML-1 sites previously identified with in vitro assays (20) in c-FMS expressing cells. (B) In vivo UV footprinting reveals significant alterations in DNA topology over three sites (A, B and C) over and upstream from the AML1 and C/EBP sites in c-FMS expressing cells. Both undifferentiated and PMA-differentiated HL-60 cells show similar occupancy and chromatin fine structure to primary macrophages. (C) Example of the quantification of bands generated by in vivo DMS footprinting depicting the region in the c-FMS promoter around the protected guanine at –194 bp at the AML1 site and the region of altered DMS reactivity around the PU.1 site at –173 bp. (D) The promoter sequence with protections indicated as white circles. Sequences conserved between mouse and human are indicated by a line (-); sequence deviations are displayed as *. The region shown as /// indicates a 20 bp poly(A) insert in the mouse sequence. Transcription factor binding sites in bold are conserved between mouse and human. Regions with altered UV pyrimidine dimer formation are indicated (A–C), with conserved dimer changes indicated in bold. Lane labelling for (A) and (B): lane G, G reaction; lane N, naked DNA; lane 1, HeLa; lane 2, HL-60; lane 3, HL-60 + PMA; lane 4, human fibroblasts; lane 5, human macrophages.
Figure 4 shows the same analysis for FIRE. Here we have shown that the human c-FMS element is bound by an overlapping, but non-identical, set of transcription factors compared to the mouse gene (4). The conserved occupied core consists of a central Ets/SP1/AML1 core element that is flanked by GC-rich sequences that may be binding sites for the transcription factor AP2 or MZF-1 (21). In addition, a conserved Ets binding site that is located downstream of the core elements was footprinted in c-FMS expressing cells. Two sites in murine FIRE known to bind PU.1 in vitro (4) differ from the human gene and did not appear to be occupied by any other transcription factor.
Transcription factor interactions were accompanied by conserved changes in DNA topology as revealed by UV footprinting (Fig. 4C and D, alterations X and Z). Changes in DNA topology at the human gene occurring in the vicinity of non-conserved sequences were not seen in the mouse (Fig. 4C and D, alterations Y and W). This included a marked increase in UV-induced dimer formation at T+4221. At this position the mouse gene has a different sequence. This increase was apparent in c-FMS non-expressing cells only and was not present with naked DNA. We conclude from these experiments that human and mouse c-FMS gene cis-regulatory elements show significant differences in their transcription factor occupancy, although they demonstrate a high degree of sequence similarity. However, a core of interacting factors with associated alterations in chromatin fine structure is conserved.
Human and mouse c-FMS regulatory elements display the same histone H3 K9 acetylation and methylation pattern
We have previously studied levels of histone acetylation and methylation across the human c-FMS locus during macrophage differentiation (7). We have repeated these experiments across the mouse c-fms locus and compared c-FMS gene chromatin in human and mouse cell lines by ChIP with antibodies against acetylated and methylated K9 of histone H3 (Fig. 5A and B, respectively). These modifications are mutually exclusive, whereby K9 methylation is found in inactive chromatin and acetylation is found at active loci (22,23). Histone H3 acetylation levels in both human HeLa cells and mouse 3T3 cells were uniformly low across the c-FMS gene regulatory region. Interestingly, although already bound by transcription factors, this also held true for uninduced HL-60 cells. In contrast, PMA-treated HL-60 cells displayed a significant increase in histone H3 K9 acetylation at the promoter and the intronic regulatory region. The highest level of histone H3 acetylation was observed at the promoter-proximal conserved region (FIRE-1 kb) (Fig. 5A, upper panel), however, not over FIRE itself. The same pattern was found in the mouse macrophage cell line (Fig. 5A, lower panel), where acetylation levels at FIRE were also low. We observed similar results with antibodies against diacetylated forms of histone H3 (K9 and K14) (data not shown).
Figure 5B shows the results of ChIP assays examining H3 K9 methylation. The c-FMS locus in HeLa cells and 3T3 cells displayed the same degree of histone H3 (K9) methylation as the respective control genes. Interestingly, this was also true for uninduced HL-60 cells, indicating that the binding of transcription factors was not generally inhibited by histone H3 K9 methylation. In both human and mouse macrophage-like cells, we observed a significant demethylation of histone H3 tails at K9 across the c-FMS gene regulatory region.
Human and mouse FIRE are dynamic elements which recruit both activating and repressing chromatin modifying activities
Transcription factors gain access to chromatin and initiate chromatin remodelling events by recruiting chromatin remodelling and chromatin modifying activities. As the transcription factor composition between human and mouse FIRE is different, we wanted to study whether the same type of activities are recruited to the human and mouse elements. Human and murine c-FMS gene expression is up-regulated during macrophage differentiation, but is down-regulated by signals such as bacterial LPS and M-CSF itself (14,15). With the murine gene, we have previously demonstrated that this is paralleled by a dynamic up- and down-regulation of transcription factor assembly at FIRE (4), suggesting interplay between activating and repressing factors. We therefore examined whether activating and repressing chromatin modifying complexes were recruited to FIRE in both species.
We performed ChIP assays with antibodies against the histone acetyltransferase (HAT) CBP, the histone deacetylase HDAC1 and Brg-1, which is the central part of the SWI/SNF chromatin remodelling complex (24,25). The results are depicted in Figure 6A–C, respectively. No CBP association was seen in HeLa and 3T3 cells. In c-FMS expressing cells, all cis-elements in both the human and mouse genes recruited CBP. In both human and mouse macrophage-like cells, the highest level of CBP association was found at FIRE. Despite the high level of histone H3 K9 methylation, a significant amount of CBP bound to all elements in uninduced HL-60 cells (Fig. 6A). In PMA-induced HL-60 cells, CBP recruitment at FIRE, but not at the promoter, was increased. The same pattern of Brg-1 recruitment was observed in human and mouse c-FMS expressing cells (Fig. 6B). We again saw that Brg-1 association was increased over FIRE, but not the promoter, after HL-60 differentiation (Fig. 6B, upper panel). Again, the high level of histone H3 K9 methylation appeared not to interfere with the binding of SWI/SNF-type complexes. We observed the same results with an antibody against another SWI/SNF complex component, Baf170 (data not shown).
Figure 6.

Chromatin immunoprecipitation of (A) CBP (B) Brg-1 and (C) HDAC-1 in HeLa, HL-60, HL-60 + PMA, 3T3 and RAW 264 cells with quantification of recovered DNA at 6 and 5 points along the human and murine c-FMS locus, respectively. The location of the promoter, FIRE-1 kb and FIRE regions are indicated (P, F-1kb and F) and DNA quantified as described in Materials and Methods. CBP clearly binds to FIRE in both HL-60 and PMA-treated HL-60 cells, with reduced binding at other points across the locus and no binding in HeLa cells. A similar pattern is seen in RAW 264 cells. A similar pattern of Brg-1 recruitment is observed in PMA-treated HL-60 cells and RAW 264 cells, with an increase in recruitment to FIRE with PMA differentiation of HL-60 cells.
The analysis of HDAC1 recruitment to c-FMS regulatory elements showed that although strongly bound by CBP and SWI/SNF complexes, all c-FMS cis-elements in c-FMS expressing cells of both species recruited significant amounts of HDAC1 (Fig. 6C). Differentiation of HL-60 cells led to a reduction in HDAC1 recruitment at the promoter and the downstream DHS, but not at FIRE itself (Fig. 6C, upper panel). From these experiments we consider it likely that c-FMS mRNA levels in human and mouse cells are regulated by the recruitment of similar chromatin remodelling activities.
Interaction of TATA box binding protein (TBP) with intronic regulatory elements
c-FMS mRNA levels are up-regulated by a combination of increased transcription frequency, as measured by nuclear run-on assays, and a relief of a block in mRNA elongation (14,15). Sequences within the second intron harbouring the cis-regulatory elements studied here are absolutely required for this block in mRNA elongation (15). However, up to now it was not clear how enhanced transcription frequency correlated with the recruitment of components of the basal transcription machinery. We therefore measured by ChIP the recruitment of a component of the TFIID complex, TBP, to the c-FMS promoter and, as a control, across the rest of the locus (Fig. 7). TBP was recruited to the c-FMS promoter, confirming the well-known fact that TATA-less promoters also bind TBP (26). TBP recruitment increased in induced HL-60 cells compared to uninduced cells. Interestingly, we observed an above background signal over all cis-regulatory elements and not just the promoter. No TBP recruitment could be seen in upstream and downstream flanking regions. This was particularly pronounced in mouse macrophage-like cells (Fig. 7, lower panel). Very little TBP, if any, is associated at FIRE in uninduced HL-60 cells. However, TBP recruitment increases strongly after PMA stimulation, where signals are higher than those of the promoter. No TBP binding was observed with control cells and no TBP recruitment could be observed using control primers far downstream of the c-FMS promoter and in the coding region of control genes (data not shown).
DISCUSSION
Mouse and human c-FMS cis-regulatory elements bind an overlapping, but non-identical set of transcription factors
With genes whose function is conserved throughout evolution, it has been observed in a number of instances that the relative position and the nature of cis-regulatory elements are highly conserved (for a review see 27). From these examples it is apparent that what is actually conserved are tissue-specific and ubiquitous transcription factor binding sites. However, very few studies have compared the actual occupancy of these transcription factors in vivo. The study presented here describes such a comparison and extends these studies to a macrophage-specific gene. The binding sites for PU.1 are conserved between the human and mouse c-FMS gene promoters and are occupied in both species (4,28), whereas the AML1 site found in the human gene is not conserved and the corresponding sequence is not occupied in the mouse. Both, human and mouse FIRE contain core elements that are present and occupied in macrophages. This includes a conserved Ets/Sp1/AML1 core, a juxtaposed GC-rich sequence and a downstream site shown to bind Ets factors (4). As indicated in Figure 4, a number of other factor binding sites are exclusively occupied in either the human or the mouse c-FMS gene.
These data suggest that these conserved and occupied transcription factor cores are important for the conservation of the regulatory features of this conserved gene. The data presented here and recent ChIP experiments (7) show that the AML1 site in the conserved Ets/Sp1/AML1 core of FIRE is occupied in both species. We therefore speculated that this AML1 site had a more critical impact on c-FMS gene regulation than the AML site in the promoter, although both are functional. This notion was confirmed by experiments with leukaemic cells carrying the t(8;21) translocation that encodes for a dominant negative form of AML1 (AML1-ETO), which has been shown to be involved in the repression of c-FMS expression (29). We could demonstrate the presence of an extensive repressing complex containing AML1-ETO over FIRE, but not the promoter (7). Similarly, the mouse c-fms FIRE element contains binding sites for PU.1, which are occupied in the mouse gene, but are not conserved in the human gene and are not occupied by any other factors. The presence of PU.1 is important for c-FMS gene expression (30). A combination of binding sites for PU.1 and other Ets factors is sufficient for macrophage-specific transcription initiation in transfection assays (11) and is the main determinant for the high activity of the c-FMS promoter in macrophage cells. Analogous to what is outlined above, it is therefore possible that the promoter sites are the most important targets for this transcription factor.
Human and mouse c-FMS genes show conserved regulatory features and a conserved pattern of chromatin rearrangements
Our data suggest highly conserved functions of c-FMS cis-regulatory elements in both species. In non-induced HL-60 cells c-FMS mRNA expression levels are low and chromatin is not fully DNase I hypersensitive (7) yet appears to be primed by the binding of transcription factors to the promoter. This confirms data obtained with the mouse gene, where we see complete transcription factor occupancy in precursor cells (4). The central ETS/SP1/AML1 core at FIRE and other FIRE elements are increasingly occupied in differentiated HL-60 cells and primary monocytes/macrophages. This is similar to what is observed in the mouse gene, where we see an up-regulation of FIRE occupancy during macrophage maturation and a down-regulation after LPS treatment (4).
The up-regulation of mRNA expression in PMA-treated HL-60 cells and the high level of expression in RAW 264 cells parallels a strong increase in histone H3 K9 acetylation at the promoter and the DHS at FIRE-1 kb. Furthermore, this is accompanied by demethylation of histone H3 K9, as well as respective increases and decreases in Brg-1 and HDAC1 recruitment to all cis-elements. In this context it is noteworthy that although histone H3 K9 in the promoter and the FIRE-1 kb DHS are hyperacetylated in macrophage-like cells, FIRE is not. Both human and mouse FIRE display low levels of histone H3 K9 acetylation and both recruit CBP and Brg-1 as well as HDAC1. Based on these observations we suggest that in both species FIRE is a dynamic cis-regulatory element that responds to developmental cues and other signals. FIRE may bind stimulating and repressing chromatin modifying and remodelling activities, probably in a mutually exclusive fashion. The molecular details of this dynamic regulation are currently under investigation.
A novel finding that merits further discussion is the discovery that a component of the basal transcription machinery, TBP, associates not only with the promoter, but also with the c-FMS intronic regulatory region. This could be interpreted as a direct interaction between the c-FMS intronic regulatory elements and the promoter, similar to what has been observed with the HNF4-α gene (31). However, it is also possible that c-FMS gene intronic elements harbour promoters and generate intragenic transcripts, similar to what has been found in the β-globin locus (32,33). Transient transfection experiments have indeed revealed that FIRE can have promoter activity (13) and may be responsible for the block in RNA elongation observed in LPS-treated cells (15). Further experiments beyond the scope of this study are required to clarify this issue.
We conclude from our data that although a significant difference exists in the composition of human and mouse transcription factor complexes binding to the different c-FMS gene cis-elements, the same chromatin modifying and remodelling activities are recruited. In both species, FIRE recruits TBP and chromatin remodelling as well as HAT and HDAC activities. In both species histone acetylation levels at FIRE are low compared to upstream regions. In addition, our UV photofootprinting experiments, which analysed chromatin fine structure alterations at the promoter and FIRE, showed that many, but not all, of these alterations occur near conserved core binding sites. It is possible that we missed some of them as UV photofootprinting relies on formation of UV-induced dimers between neighbouring bases (mostly pyrimidines), not all of which are conserved. However, our experiments indicate that conserved transcription factor complexes induce similar chromatin fine structure changes in both species.
Taken together, our data support the hypothesis that cores of conserved transcription factor elements are responsible for the conservation of regulatory features of genes whose function is conserved in evolution. This does not mean that binding sites in flanking regions are irrelevant. They may have evolved to support the expression of specific genes in different developmental contexts or may be involved in facilitating the interaction between different cis-regulatory elements, as suggested by experiments performed with elements of the β-globin locus (34). Whatever their role, the study presented here indicates that the experimental determination of transcription factor occupancy in cells of different species has to accompany sequence comparisons in order to obtain a true picture of the role of transcription factor complexes governing the regulation of conserved genes.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Charlotte Stephenson for technical support. This work was supported by the Medical Research Council, the Leukaemia Research Fund and the Wellcome Trust. G.A.F. is the recipient of a MRC Clinical Training Fellowship.
REFERENCES
- 1.Dai X.M., Ryan,G., Hapel,A.J., Dominguez,M.G., Russell,R.G., Kapp,S., Sylvestre,V. and Stanley,E.R. (2002) Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies and reproductive defects. Blood, 99, 111–120. [DOI] [PubMed] [Google Scholar]
- 2.Miyamoto T., Iwasaki,H., Reizis,B., Ye,M., Graf,T., Weissman,I.L. and Akashi,K. (2002) Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev. Cell, 3, 137–147. [DOI] [PubMed] [Google Scholar]
- 3.Bartelmez S.H., Bradley,T.R., Bertoncello,I., Mochizukim,D.Y., Tushinskim,R.J., Stanley,E.R., Hapel,A.J., Young,I.G., Kriegler,A.B. and Hodgson,G.S. (1989) Interleukin 1 plus interleukin 3 plus colony-stimulating factor 1 are essential for clonal proliferation of primitive myeloid bone marrow cells. Exp. Hematol., 17, 240–245. [PubMed] [Google Scholar]
- 4.Tagoh H., Himes,R., Clarke,D., Leenen,P.J., Riggs,A.D., Hume,D. and Bonifer,C. (2002) Transcription factor complex formation and chromatin fine structure alterations at the murine c-fms (CSF-1 receptor) locus during maturation of myeloid precursor cells. Genes Dev., 16, 1721–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sherr C.J., Rettenmeier,C.W., Sacca,R., Roussel,M.F., Look,A.T. and Stanley,E.R. (1985) The c-fms proto-oncogene product is related to the receptor for the mononuclear phagocyte growth factor, CSF-1. Cell, 41, 665–676. [DOI] [PubMed] [Google Scholar]
- 6.Rhoades K.L., Hetherington,C.J., Rowley,J.D., Hiebert,S.W., Nucifora,G., Tenen,D.G. and Zhang,D.E. (1996) Synergistic up-regulation of the myeloid-specific promoter for the macrophage colony-stimulating factor receptor by AML1 and the t(8;21) fusion protein may contribute to leukemogenesis. Proc. Natl Acad. Sci. USA, 93, 11895–11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Follows G., Tagoh,H., Lefevre,P., Hodge,D., Morgan,G. and Bonifer,C. (2003) Epigenetic consequences of AML1-ETO action at the c-FMS locus. EMBO J., 22, 2798–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Visvader J. and Verma,I. (1989) Differential transcription of exon 1 of the human c-fms gene in placental trophoblasts and monocytes. Mol. Cell. Biol., 9, 1336–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts W.M., Shapiro,L.H., Ashmun,R.A. and Look,A.T. (1992) Transcription of the human colony-stimulating factor-1 receptor gene is regulated by separate tissue-specific promoters. Blood, 79, 586–593. [PubMed] [Google Scholar]
- 10.Sasmono R.T., Oceandy,D., Pollard,J.W., Tong,W., Pavli,P., Wainwright,B.J., Ostrowski,M.C., Himes,S.R. and Hume,D.A. (2003) A macrophage colony-stimulating factor receptor-green fluorescent protein transgene is expressed throughout the mononuclear phagocyte system of the mouse. Blood, 101, 1155–1163. [DOI] [PubMed] [Google Scholar]
- 11.Ross I.L., Yue,X., Ostrowski,M.C. and Hume,D.A. (1998) Interaction between PU.1 and another Ets family transcription factor promotes macrophage-specific basal transcription initiation. J. Biol. Chem., 273, 6662–6669. [DOI] [PubMed] [Google Scholar]
- 12.Rehli M., Lichanska,A., Cassady,A.I., Ostrowski,M.C. and Hume,D.A. (1999) TFEC is a macrophage-restricted member of the microphthalmia-TFE subfamily of basic helix-loop-helix leucine zipper transcription factors. J. Immunol., 162, 1559–1565. [PubMed] [Google Scholar]
- 13.Himes S.R., Tagoh,H., Goonetilleke,N., Sasmono,T., Oceandy,D., Clark,R., Bonifer,C. and Hume,D.A. (2001) A highly conserved c-fms gene intronic element controls macrophage-specific and regulated expression. J. Leukoc. Biol., 70, 812–820. [PubMed] [Google Scholar]
- 14.Gusella G.L., Ayroldi,E., Espinoza-Delgado,I. and Varesio,L. (1990) Lipopolysaccharide, but not IFN-gamma, down-regulates c-fms mRNA proto-oncogene expression in murine macrophages. J. Immunol., 144, 3574–3580. [PubMed] [Google Scholar]
- 15.Yue X., Favot,P., Dunn,T.L., Cassady,A.I. and Hume,D.A. (1993) Expression of mRNA encoding the macrophage colony-stimulating factor receptor (c-fms) is controlled by a constitutive promoter and tissue-specific transcription elongation. Mol. Cell. Biol., 13, 3191–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maxam A.M. and Gilbert,W. (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol., 65, 499–560. [DOI] [PubMed] [Google Scholar]
- 17.Kontaraki J., Chen,H.H., Riggs,A. and Bonifer C. (2000) Chromatin fine structure profiles for a developmentally regulated gene: reorganization of the lysozyme locus before trans-activator binding and gene expression. Genes Dev., 14, 2106–2122. [PMC free article] [PubMed] [Google Scholar]
- 18.Lefevre P., Melnik,S., Riggs,A.D. and Bonifer,C. (2003) Developmentally regulated recruitment of transcription factors and chromatin modification activities to chicken lysozyme cis-regulatory elements in vivo. Mol. Cell. Biol., 23, 4386– 4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H.H., Kontaraki,J., Bonifer,C. and Riggs,A.D. (2001) Terminal transferase-dependent PCR (TDPCR) for in vivo UV photofootprinting of vertebrate cells. Sci. STKE, 2001, PL1. [DOI] [PubMed] [Google Scholar]
- 20.Zhang D.E., Hetherington,C., Meyers,S., Rhoades,K.L., Larson,C.J., Chen,H.M., Hiebert,S.W. and Tenen,D.G. (1996) CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF alpha2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol., 16, 1231–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris J.F., Hromas,R. and Rauscher,F.J.,III (1994) Characterization of the DNA-binding properties of the myeloid zinc finger protein MZF1: two independent DNA-binding domains recognize two DNA consensus sequences with a common G-rich core. Mol. Cell. Biol., 14, 1786–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eberharter A. and Becker,P. (2002) Histone acetylation: a switch between repressive and permissive chromatin. Second in review series on chromatin dynamics. EMBO Rep., 3, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kouzarides T. (2002) Histone methylation in transcriptional control. Curr. Opin. Genet. Dev., 12, 198–209. [DOI] [PubMed] [Google Scholar]
- 24.Khavari P.A., Peterson,C.L., Tamkun,J.W., Mendel,D.B. and Crabtree,G.R. (1993) BRG1 contains a conserved domain of the SWI2/SNF2 family necessary for normal mitotic growth and transcription. Nature, 366, 170–174. [DOI] [PubMed] [Google Scholar]
- 25.Richmond E. and Peterson,C.L. (1996) Functional analysis of the DNA-stimulated ATPase domain of yeast SWI2/SNF2. Nucleic Acids Res., 24, 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zenzie-Gregory B., Khachi,A., Garraway,I.P. and Smale,S.T. (1993) Mechanism of initiator-mediated transcription: evidence for a functional interaction between the TATA-binding protein and DNA in the absence of a specific recognition sequence. Mol. Cell. Biol., 13, 3841–3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hardison R.C. (2000) Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet., 16, 369–372. [DOI] [PubMed] [Google Scholar]
- 28.DeKoter R.P., Lee,H.J. and Singh,H. (2002) PU.1 regulates expression of the interleukin-7 receptor in lymphoid progenitors. Immunity, 16, 297–309. [DOI] [PubMed] [Google Scholar]
- 29.Heidenreich O., Krauter,J., Riehle,H., Hadwiger,P., John,M., Heil,G., Vornlocher,H.P. and Nordheim,A. (2003) AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood, 15, 3157–3163. [DOI] [PubMed] [Google Scholar]
- 30.DeKoter R.P., Walsh,J.C. and Singh,H. (1998) PU.1 regulates both cytokine-dependent proliferation and differentiation of granulocyte/macrophage progenitors. EMBO J., 17, 4456–4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hatzis P. and Talianidis,I. (2002) Dynamics of enhancer-promoter communication during differentiation-induced gene activation. Mol. Cell, 10, 1467–1477. [DOI] [PubMed] [Google Scholar]
- 32.Ashe H.L., Monks,J., Wijgerde,M., Fraser,P. and Proudfoot,N.J. (1997) Intergenic transcription and transinduction of the human beta-globin locus. Genes Dev., 11, 2494–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Routledge S.J. and Proudfoot,N.J. (2002) Definition of transcriptional promoters in the human beta globin locus control region. J. Mol. Biol., 323, 601–611. [DOI] [PubMed] [Google Scholar]
- 34.Molete J.M., Petrykowska,H., Bouhassira,E.E., Feng,Y.Q., Miller,W. and Hardison,R.C. (2001) Sequences flanking hypersensitive sites of the beta-globin locus control region are required for synergistic enhancement. Mol. Cell. Biol., 21, 2969–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]