

Abstract
Infection with the intracellular protozoan parasite Toxoplasma gondii causes serious public health problems and is of great economic importance worldwide. The micronemal protein MIC3, which is a potent adhesin of T. gondii, could be a significant candidate vaccine against toxoplasmosis. In this study, all CBA/J mice intramuscularly vaccinated with a plasmid encoding the immature form of the MIC3 protein (pMIC3i) produced specific anti-MIC3 immunoglobulin G (IgG) antibodies, and their sera displayed high antibody titers. This response was increased by the coadministration of a plasmid encoding the granulocyte-macrophage colony-stimulating factor (pGM-CSF). Similarly, a specific and significant cellular immune response was obtained in mice immunized with pMIC3i, and this response was markedly enhanced by pGM-CSF coadministration. The cellular immune response was associated with the production of gamma interferon IFN-γ and interleukin-2 (IL-2), indicating that this was a Th1-type response. This was confirmed by the production of large amounts of IgG2a. Mice immunized with pMIC3i displayed significant protection against an oral challenge with T. gondii 76K cysts, exhibiting fewer brain cysts than did the control mice. Coadministration of pGM-CSF enhanced this protection. In conclusion, this study describes the design of a potent DNA vaccine encoding the novel T. gondii target antigen, MIC3 protein, that elicits a strong specific immune response as well as providing effective protection against T. gondii infection. In the attempt to achieve complete protection against toxoplasmosis, MIC3 is a good candidate vaccine which could be combined with other relevant and previously described candidates, such as SAG1 and GRA4.
Toxoplasma gondii is an obligate intracellular protozoan parasite that infects all warm-blooded animals, including humans, and causes toxoplasmosis. This broad host range makes it one of the most successful protozoan parasites. In primary human infections, various mild symptoms may be observed, such as lymphadenopathy, low-grade fever, mild malaise, sore throat, and lethargy. Immunosuppressed patients may exhibit severe symptoms, including encephalitis, myocarditis, pneumonitis, hepatitis, splenomegaly, polymyositis, dermatomyositis, chorioretinitis, and multisystem organ failure. In pregnant women, congenital infection can lead to miscarriage, neonatal malformations, or other defects occurring during the development of the fetus, such as blindness or severe cognitive impairment (22, 37). In animals, toxoplasmosis is of great economic importance worldwide because it causes abortions, stillbirth, and neonatal loss in all types of livestock, especially in sheep and goats (10). In addition, the tissue cysts of T. gondii in meat of infected livestock are an important source of infection for humans (21).
This great worldwide importance for public health and economics of T. gondii infection makes the development of an effective vaccine for controlling this infection an important goal. So far, the only developed vaccine is the live, attenuated tachyzoite S48 (11). However, this vaccine is not widely accepted because of its side effects, short shelf life, and high cost. Live vaccines also carry a risk of accidental infection of humans and unexpected harmful reverse mutations. In an attempt to overcome these problems, current research is investigating subunit, recombinant and DNA vaccines, but they do not provide complete protection against T. gondii infection (7).
We have focused on the development of a DNA-based vaccine because such vaccines have been shown to elicit potent, long-lasting humoral and cell-mediated immunity, as well as providing protection against viral, bacterial, and parasitic infections (4).
The most common method used to deliver DNA vaccines is the intramuscular injection, which is known to induce a Th1-type response (31), which is generally thought to protect the host against T. gondii infection (32).
Several trials of DNA-based vaccines against toxoplasmosis have been conducted, mainly with mice and various T. gondii antigens, such as membrane-associated surface antigen SAG1 (5, 32), excreted-secreted dense-granule proteins GRA1 (33, 38), GRA7 (38), and GRA4 (18), and rhoptry proteins ROP2 (29, 38) and ROP1 (14). These trials have been encouraging, in that they have demonstrated the development of different levels of protection in mice. Among the putative vaccine candidates, the micronemal protein MIC3 (90 kDa) looks particularly promising because it is a potent adhesin of T. gondii (12, 23), that is expressed in all three infectious stages of T. gondii (tachyzoites, bradyzoites, and sporozoites) and that elicits early and powerful immune responses in mice and humans (M. Lebrun, personal communication).
A number of approaches are being explored that could enhance the efficacy of DNA vaccines, such as the coadministration of cytokine-encoding plasmids (28). Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a potent cytokine, and its role as potential vaccine adjuvant has already been investigated (25, 39). Coadministration of plasmid GM-CSF enhances the DNA vaccine-elicited humoral and cellular immune responses, as well as protection, in several models (26, 40, 41). All these properties support the use of plasmid encoding GM-CSF as an adjuvant vaccine in this study. The mechanism underlying the adjuvant properties of plasmid encoding GM-CSF may involve increased recruitment of macrophages and dendritic cells at the site of injection (8, 24, 27).
We describe here the development and evaluation of a DNA vaccine based on a plasmid encoding the immature form of the MIC3 protein, either alone or combined with another plasmid encoding GM-CSF.
In this study, the 76K strain has been used for challenge infection. This strain is a type II isolate, and type II isolates are the predominant isolates in human congenital toxoplasmosis (2, 3). Protection was evaluated in CBA/J mice, which are markedly resistant to acute toxoplasmosis infection but susceptible to cyst formation and development of toxoplasmosis encephalitis in chronic infection. As a protective criterion, we chose to evaluate the decrease in brain cyst load, since the number of brain cysts is one of the most important factors that determine the development of toxoplasmic encephalitis (9, 16, 36).
MATERIALS AND METHODS
Animals.
Female CBA/J mice (H-2k), 6 to 8 weeks old, were obtained from Janvier (Le Genest St. Isle, France) and maintained under pathogen-free conditions in our animal house for use throughout these experiments.
Parasites.
Two strains of T. gondii (RH and 76K) were used in this study. The RH strain tachyzoites were harvested from the peritoneal fluids of Swiss OF1 mice that had been intraperitoneally infected 3 to 4 days earlier. The 76K strain cysts were obtained from the brains of orally infected CBA/J mice and maintained by monthly passage.
Preparation of TAg.
T. gondii antigen (TAg) was prepared from RH strain tachyzoites as previously described (34). Briefly, the obtained tachyzoites were washed and sonicated for three 10-min periods at 60 W/s. The toxoplasma sonicate was centrifuged at 2,000 × g for 30 min. The protein concentration was determined in the supernatant, which was later used as the source of antigen, by the Micro BCA protein assay reagent kit using bovine serum albumin (BSA) as the standard (Pierce, Rockford, Ill.). The TAg was stored at −80°C until use.
Plasmid construction.
The complete MIC3 open reading frame was obtained by PCR amplification of the MIC3 gene from pBluescript II SK-MIC3 (23), using primers 5′-GTGTAAGCTTCTTGTCCAACACTGGGTA-3′ (forward) and 5′-CACGGATATCTGCGAATGGGCG-3′ (reverse), which introduce the HindIII and EcoRV restriction sites, respectively. The amplified DNA fragment was cloned in the HindIII and EcoRV sites of the eukaryotic expression vector pcDNA3 (Invitrogen). The resulting plasmid was named immature pcDNA3-MIC3 (pMIC3i). The plasmid encoding murine GM-CSF (pGM-CSF) was kindly provided by H. C. J. Ertl (Wistar Institute, Philadelphia, Pa.).
Plasmid purification.
All the plasmids were purified from transformed Escherichia coli DH5α by anion-exchange chromatography (EndoFree plasmid Giga or Mega kit; Qiagen GmBH, Hilden, Germany) as specified by the manufacturer. The purified plasmids were dissolved in sterile endotoxin-free phosphate-buffered saline (PBS; Sigma) and stored at −20°C. The integrity of the DNA plasmids was checked by agarose gel electrophoresis after digestion with appropriate restriction enzymes. The DNA concentration was determined by measuring the optical density at 260 nm (OD260). The OD 260/280 ratios for the purified DNA were 1.80 to 1.95, indicating that preparations were free of any major protein contamination.
Expression of pMIC3i in vitro.
Baby hamster kidney 21 (BHK-21) cells were transfected with either pMIC3i or a control plasmid, pcDNA3, using a polycationic liposome reagent (LipofectAmine; GIBCO BRL) as instructed by the manufacturer. Six-well tissue culture plates were seeded with BHK-21 cells (3 × 105 cells/well) in BHK-21 medium (GIBCO BRL) supplemented with 5% fetal calf serum, 2% tryptose, 100 U of penicillin per ml, and 100 μg of streptomycin per ml, and the cells were grown until they were about 70 to 80% confluent (18 to 24 h). For each transfection, 1 μg of DNA and 10 μl of LipofectAmine were used.
Western blot analysis.
The pellets and supernatant of transfected cells (24 h after transfection) and purified T. gondii tachyzoites (RH strain) were suspended in sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer (with or without reducing agent), sonicated, heated at 100°C for 3 min, and separated on a 10% polyacrylamide gel. After electrophoresis, proteins were transferred onto a nitrocellulose membrane, which was probed with anti-MIC3 monoclonal antibody (MAb) T8 2C10 or T4 2F3 (23) or serum obtained from a T. gondii-infected mouse and diluted 1:200 in 5% nonfat dried milk-TNT (15 mM Tris-HCl, 140 mM NaCl, 0.05% Tween 20) as previously described (13). Bound antibodies were detected using anti-mouse immunoglobulin G (IgG)-alkaline phosphatase conjugate (Sigma) diluted 1:1000 in 5% nonfat dried milk-TNT. Alkaline phosphatase activity was detected using the 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium (BCIP/NBT) liquid substrate system (Sigma). Molecular masses standards (prestained SDS-PAGE standards, low range; Bio-Rad) were used.
DNA immunization.
The mice (12 per group) were injected intramuscularly (i.m.), using syringes with 30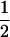 -gauge needles (Microlance; Becton Dickinson), with 100 μl of 10 μM cardiotoxin (Latoxan, Rosans, France) into each tibialis anterior muscle 5 days before DNA immunization, to enhance the uptake of the plasmid DNA (15). On days 0 and 14, the regenerating muscles of these mice were injected with 50 μg of pMIC3i, either alone or combined with 50 μg of pGM-CSF, in 100 μl of sterile endotoxin-free PBS (50 μl in each muscle) and then boosted on day 28 with 50 μg of pMIC3i alone. The control groups were composed of mice injected i.m. with 50 μg of empty plasmid, pcDNA3, or 50 μg of pGM-CSF three times at intervals of 2 weeks.
-gauge needles (Microlance; Becton Dickinson), with 100 μl of 10 μM cardiotoxin (Latoxan, Rosans, France) into each tibialis anterior muscle 5 days before DNA immunization, to enhance the uptake of the plasmid DNA (15). On days 0 and 14, the regenerating muscles of these mice were injected with 50 μg of pMIC3i, either alone or combined with 50 μg of pGM-CSF, in 100 μl of sterile endotoxin-free PBS (50 μl in each muscle) and then boosted on day 28 with 50 μg of pMIC3i alone. The control groups were composed of mice injected i.m. with 50 μg of empty plasmid, pcDNA3, or 50 μg of pGM-CSF three times at intervals of 2 weeks.
Measurement of humoral antibody responses.
Serum IgG antibody responses to MIC3 were measured by Western blotting and enzyme-linked immunosorbent assay (ELISA) as follows.
(i) Western blotting.
Western blotting was performed as described above. The TAg (60 μg/1-cm-wide slot) was separated on a 12% polyacrylamide gel under nonreducing conditions.
(ii) ELISA.
Levels of antigen-specific IgG antibodies in serum samples were determined by standards procedures. Briefly, the 96 flat-bottom wells of microtiter plates (Maxisorp; Nunc, Roskilde, Danemark) were coated overnight at 4°C with TAg at 10 μg/ml in 50 mM sodium carbonate buffer (pH 9.6). The plates were washed with PBS containing 0.05% Tween 20 (PBS-T20), pH 7.4; nonspecific binding sites were blocked with PBS containing 4% bovine serum albumin (PBS-4% BSA) for 1 h at 37°C. Twofold serial dilutions of serum samples in PBS were added to the wells and incubated for 1 h at 37°C. After the plates were washed, bound antibodies were detected by incubation for 2 h at 37°C with a goat anti-mouse IgG-alkaline phosphatase conjugate (Sigma) diluted 1:2000 in PBS-4% BSA. After the plates were washed in PBS-T20, the bound phosphatase activity was measured with p-nitrophenylphosphate at 1 mg/ml in 1 M diethanolamine buffer (pH 9.8). The reaction was stopped after incubating for 15 min at 37°C by addition of 3 N NaOH; the OD405 of each sample was then read in a Titertek Multiskan ELISA reader (Flow Laboratories, McLean, Va.). The antigen-specific antibody titer is given as the reciprocal of the highest dilution producing an OD that was 2.5-fold greater than that of the serum of mice injected with the empty plasmid, pcDNA3, at the same dilution. Results are expressed as the means of log2 titers ± standard deviations (SD).
Determination of the anti-MIC3 IgG subclass.
The anti-MIC3 IgG subclass was determined by ELISA as described above, except that sera were added to the plates at a single dilution (1:400 for sera of mice immunized with pMIC3i and 1:800 for sera of mice immunized with pMIC3i combined with pGM-CSF). The alkaline phosphatase-conjugated anti-mouse IgG1, IgG2a, IgG2b, and IgG3 (Cappel, West Chester, Pa.) were used at 1:100 in PBS-4% BSA. The results are expressed as mean OD405 ± SD.
In vitro lymphocyte proliferation studies.
The spleens were aseptically removed from mice (three per group) 2 weeks after the third immunization and pressed through stainless steel mesh. Single-cell suspensions were obtained by filtration through nylon mesh to remove any tissue debris. After erythrocytes were eliminated by hypotonic shock, the spleen cells were resuspended in RPMI 1640 medium (GIBCO) supplemented with 5% fetal calf serum, 10 mM HEPES, 2 mM l-glutamine, 1 mM sodium pyruvate, 50 μM β-mercaptoethanol, 100 U of penicillin per ml, and 100 μg of streptomycin per ml. They were seeded in triplicate in flat-bottom 96-well microtiter plates (Costar) at 5 × 105 cells per well in 200 μl of culture medium either alone or with various concentrations of TAg or 10 μg of concanavalin A (Con A) per ml. The plates were incubated for 3 days under 5% CO2 at 37°C and pulsed with 37 kBq of [3H]thymidine per well for an additional 18 h. The cells were ruptured by freezing and thawing, and the cell contents were collected on fiberglass filters. Finally, the incorporated radioactivity (counts per minute [cpm]) was measured by liquid scintillation counting. The results are expressed as Δcpm, calculated as the cpm obtained in the presence of the stimulating antigen minus the cpm in the medium alone.
Cytokine assays.
Spleen cell proliferation was assayed as described above, but the cells were stimulated by being incubated with 15 μg of TAg per ml or with the medium alone. Cell-free supernatants were harvested and assayed for interleukin-2 (IL-2) and IL-4 activities at 24 h, IL-10 activity at 72 h, and gamma interferon (IFN-γ) activity at 96 h. The concentrations of IL-2, IL-4, IL-10, and IFN-γ were determined with an ELISA kit (OptEIA set; Pharmingen, San Diego, Calif.) as specified by the manufacturer. The sensitivity limits for the assays were 10 pg/ml for IL-2 and IL-4, 20 pg/ml for IFN-γ, and 50 pg/ml for IL-10.
Challenge infection.
CBA/J mice were infected orally with 70 cysts of the 76K strain 2 weeks after the last immunization. One month after the challenge, mice were sacrificed and their brains were removed. Each brain was homogenized in 5 ml of RPMI medium with a pestle and mortar. The mean number of cysts per brain was determined microscopically by counting eight samples (10 μl each) of each homogenate. The results are expressed as means ± SD for each group.
Statistical analysis.
Levels of significance of the differences between groups of mice were determined by an analysis of variance test.
RESULTS
Synthesis of MIC3 in vitro.
The entire MIC3 nucleotide-coding sequence (1,155 bp) was inserted into the HindIII and EcoRV sites of the eukaryotic expression vector pcDNA3, under the transcriptional control of the cytomegalovirus early promoter. The resulting plasmid was named pMIC3i. BHK-21 cells were transfected with pMIC3i or empty plasmid, pcDNA3, and the supernatant and the lysates of transfected cells were analyzed on immunoblots (Fig. 1).
FIG. 1.

Immunoblot analysis of the MIC3 protein (immature form) expressed by mammalian cells. Supernatant (lanes 3) and lysate (lanes 2) of BHK-21 cells transfected with pMIC3i compared with tachyzoite lysate (lanes 1, positive control) and supernatant (lanes 5) or lysate (lanes 4) of BHK-21 transfected with pcDNA3 (negative control) were electrophoresed in an SDS-10% polyacrylamide gel under nonreducing (A) or reducing (B) conditions and then transferred onto a nitrocellulose membrane that was probed with anti-MIC3 MAb T8 2C10 (a) or serum from a T. gondii-infected mouse (b). Bounds antibodies were detected using anti-mouse-alkaline phosphatase conjugate. The positions of molecular masses standards are shown on the left.
The supernatant (panel a, lanes 3) and lysate (panel a, lanes 2) from BHK-21 cells transfected with pMIC3i showed specific expression of a MIC3 protein recognized by anti-MIC3 MAb T8 2C10 (Fig. 1). Anti-MIC3 MAb T8 2C10 reacted with protein bands migrating with the molecular weights expected for MIC3 dimers under nonreducing conditions and for monomers under reducing conditions, indicating that dimerization of recombinant MIC3 was obtained in mammalian cells. However, the recombinant MIC3 protein migrated at a higher molecular mass (105 kDa) than the native MIC3 (90 kDa) found in tachyzoites lysate (Fig. 1A, panel a, lanes 1 and 3). This finding is consistent with the data reported by Cérède et al. (12), who reported that the recombinant MIC3 produced in BHK-21 cells migrated at a higher molecular mass than native MIC3 did. This difference was attributable to a failure to cleave the MIC3 propeptide in BHK-21 cells. Antiserum to native MIC3 from a T. gondii-infected mouse recognized the recombinant MIC3 dimer (Fig. 1A, panel b, lanes 2 and 3) but did not efficiently recognize reduced recombinant MIC3 (Fig. 1B, panel b, lane 3). These results suggested that the B-cell epitopes are conformational and that the recombinant protein is correctly folded and targeted in mammalian cells. In contrast, no immunoreactive material was found in supernatant or lysate of BHK-21 cells transfected with empty plasmid, pcDNA3 (Fig. 1, panels a and b, lanes 4 and 5). Therefore, pMIC3i appeared to direct the synthesis of an antigenic MIC3 protein by mammalian cells in vitro.
Humoral immune response induced by DNA vaccination.
A specific IgG antibody response was generated 2 weeks after the first immunization. In the immunoblot analysis, all tested sera from mice immunized with pMIC3i alone reacted strongly with a single protein band at the expected molecular mass (90 kDa) of the corresponding antigen (Fig. 2). In contrast, antibodies recognizing the 90-kDa antigen were absent from the sera of mice injected with control plasmid pcDNA3. These findings show that DNA immunization induces antibodies that are able to recognize the native (natural) MIC3 protein found in T. gondii.
FIG. 2.

Detection of specific anti-MIC3 IgG antibodies in sera of CBA/J mice immunized with pMIC3i alone, by Western blotting using T. gondii antigen. Sera from individual mice were collected on day 14 and diluted 1:100. The reactivities of sera from immunized mice and from mice receiving empty plasmid pcDNA3 are shown. The band corresponding to the native MIC3 at 90 kDa is indicated by an arrow. The molecular masses are shown on the left; results from one of three similar experiments are shown. T+, positive control (anti-MIC3 MAb T4 2F3); T−, negative control (serum from a naive mouse).
To determine the levels of antibody titers, all sera were tested by ELISA using TAg (Fig. 3). A very high level of specific anti-MIC3 IgG antibody titers was found in the sera of mice immunized with pMIC3i alone or combined with pGM-CSF, especially after the third immunization (the level of antibody titers increased with successive immunizations). The IgG antibody titer was greater in the sera of mice coimmunized with pGM-CSF (17.2 ± 1) than in the sera of mice immunized with pMIC3i alone (14.6 ± 1). In contrast, mice injected with control plasmids pcDNA3 or pGM-CSF did not generate antibodies against MIC3 (Fig. 3).
FIG. 3.

Determination of specific anti-MIC3 antibody titers in the sera of CBA/J mice immunized with 50 μg of pMIC3i alone or combined with 50 μg of pGM-CSF on days 0 and 14 and then boosted with 50 μg of pMIC3i on day 28. Sera were collected on days 14, 21, and 35 and tested by ELISA using TAg. The titer is given as the reciprocal of the highest dilution with an OD405 that was 2.5-fold greater than the OD of untreated mouse sera at the same dilution. Results are expressed as the mean log2 titers and SD and represent one of three similar experiments.
Specific anti-MIC3 IgG subclass induced by DNA vaccination.
We were also interested in finding whether a Th1 and/or Th2 response to MIC3 was generated by MIC3 DNA immunization. We therefore analyzed the distribution of IgG subtypes (IgG1, IgG2a, IgG2b, and IgG3) specific for MIC3 (Fig. 4). Both IgG2a and IgG2b were found in the sera of mice immunized with pMIC3i alone or combined with pGM-CSF, and there was no statistically significant difference between the two groups (P > 0.05). In contrast, neither IgG1 nor IgG3 was detected in the sera of immunized mice. These results indicate a shift toward the Th1 type of response.
FIG. 4.

Determination of the specific anti-MIC3 IgG subclass profile in the sera of CBA/J mice immunized with 50 μg of pMIC3i alone or pMIC3i plus 50 μg of pGM-CSF on days 0 and 14 and boosted with 50 μg of pMIC3i on day 28. Sera were collected on day 35 and analyzed by ELISA using TAg. Results are expressed as the mean of the OD405 and SD and represent one of three similar experiments.
Cellular proliferative response induced by DNA vaccination.
The splenocytes from mice immunized with pMIC3i alone or combined with pGM-CSF were prepared 2 weeks after the third immunization to assess the proliferative immune responses to MIC3 (Fig. 5). Marked specific lymphoproliferation was observed in splenocyte cultures from mice immunized with pMIC3i alone when stimulated with TAg (Δcpm, 25,379 at 30 μg of TAg per ml). This proliferation was further increased when pMIC3i was combined with pGM-CSF (Δcpm, 50,457 at 30 μg of TAg per ml). In contrast, splenocytes from mice immunized with control plasmid pcDNA3 did not proliferate when stimulated with TAg.
FIG. 5.

In vitro proliferation of splenocytes from CBA/J mice immunized with 50 μg of pMIC3i alone or combined with 50 μg of pGM-CSF on days 0 and 14 and then boosted with 50 μg of pMIC3i on day 28, in response to TAg. Control splenocytes were from mice receiving empty plasmid pcDNA3. The results are expressed as Δcpm, calculated by subtracting the mean cpm of unstimulated cells from the mean cpm of stimulated cells. These results represent one of three similar experiments.
Cytokine production.
The supernatants of splenocytes cultured from mice immunized with pMIC3i alone or combined with pGM-CSF were harvested at different times after the restimulation with TAg and assessed for the production of IFN-γ, IL-2, IL-4, and IL-10 (Table 1). Very large specific amounts of IFN-γ were produced in the supernatants of restimulated splenocyte cultures from mice immunized with pMIC3i combined with pGM-CSF. The splenocytes from mice immunized with pMIC3i alone displayed stong specific proliferation when stimulated with TAg, but nonspecific production of IFN-γ was observed. Specific amounts of IL-2 were also synthesized by the restimulated splenocytes from the mice immunized with pMIC3i alone or combined with pGM-CSF. In contrast, no specific release of IL-4 and IL-10 from any culture supernatant was demonstrable. These findings confirm the results obtained with the anti-MIC3 IgG subclass, indicating that a Th1 immune response was induced by MIC3 DNA immunization.
TABLE 1.
Cytokine production by splenocytes of immunized CBA/J mice after stimulation by TAg
| Group | Cytokine production (mean ± SD)a
|
|||
|---|---|---|---|---|
| IL-2 (pg/ml) | IFN-γ (ng/ml) | IL-4 (pg/ml) | IL-10 (pg/ml) | |
| pcDNA3 | 27 ± 9 | 10 ± 1.3 | 29 ± 10 | 281 ± 167 |
| pMIC3i | 210 ± 45 | 11.5 ± 0.3 | 32 ± 14 | 332 ± 228 |
| pMIC3i plus pGM-CSF | 242 ± 57 | 111.9 ± 11 | 33 ± 10 | 343 ± 226 |
Cell-free supernatants were harvested and assyed for IL-2 and IL-4 activities at 24 h, IL-10 activity at 72 h, and IFN-γ activity at 96 h.
Effective protection of DNA vaccination in mice.
To test whether DNA vaccination induced effective protection against T. gondii infection, the immunized mice were orally challenged with 70 cysts of T. gondii 76K 2 weeks after the last immunization. One month after the oral challenge, the cysts in the brains of the mice were counted (Table 2). Effective and highly significant protection was obtained in the mice immunized with pMIC3i alone, which had 58, 45, and 57.5% fewer brain cysts than did the mice immunized with pcDNA3 (negative control) in experiments 1, 2, and 3 respectively (Table 2). Coadministration of pGM-CSF potentiated the protection. The mice immunized with pMIC3i plus pGM-CSF had 67, 73, and 74% fewer brain cysts than did those immunized with pcDNA3 in experiments 2, 3, and 4 respectively (Table 2). These results demonstrated the protective effect of MIC3 DNA against T. gondii infection.
TABLE 2.
Protection of CBA/J mice against T. gondii oral infectiona
| Immunization | Dose (no. of cysts) | Mean no. of brain cysts ± SD | % Reduction |
|---|---|---|---|
| Expt 1 | 70 | ||
| pcDNA3 | 7,059 ± 2,047 | ||
| pMIC3i | 2,966 ± 1,756b | 58 | |
| Expt 2 | 70 | ||
| pcDNA3 | 5,468 ± 1,048 | ||
| pMIC3i | 3,048 ± 911b | 45 | |
| pMIC3i + pGM-CSF | 1,839 ± 781b | 67 | |
| Expt 3 | 70 | ||
| pcDNA3 | 6,109 ± 829 | ||
| pMIC3i | 2,600 ± 936b | 57.5 | |
| pMIC3i + pGM-CSF | 1,652 ± 494b | 73 | |
| Expt 4 | 70 | ||
| pcDNA3 | 5,088 ± 1,064 | ||
| pMIC3i + pGM-CSF | 1,349 ± 362b | 74 |
The mice were immunized with 50 μg of pMIC3i alone or combined with 50 μg of pGM-CSF on days 0 and 14 and then boosted with 50 μg of pMIC3i on day 28. The vaccinated mice (nine per group) were orally challenged with 70 cysts of T. gondii strain 76K 2 weeks after the last immunization. The brain cyst load was determined 1 month after the challenge. Mice inoculated with empty plasmid pcDNA3 were used as controls.
P < 0.001 with respect to control.
DISCUSSION
In this study, we have demonstrated that a DNA vaccine encoding the novel target antigen, MIC3 protein, of T. gondii is a simple and potent vaccine that elicits a strong specific immune response, as well as providing significant protection against T. gondii infection. We also demonstrated that combination of a plasmid encoding GM-CSF enhances both the immune response and the protection provided.
The first characteristic of DNA vaccination is that the antigen is expressed directly by the cells of the host to be vaccinated, i.e., by a eucaryotic system, thus avoiding the possible problems of incorrect folding or lack of some posttranslational modifications due to procaryotic expression of recombinant proteins (4). This property is of great interest for MIC3, which is a dimeric 90-kDa, cysteine-rich, soluble micronemal protein, synthesized as 40-kDa precursors that are proteolytically processed to 38 kDa (1). MIC3 contains five partially overlapping epidermal growth factor-like domains and a lectin-like domain. It has a putative hydrophobic N-terminal signal sequence, a potential site of N glycosylation, 34 cysteine residues, and 12 putative phosphorylation sites (23). The lectin-like domain is responsible for the adhesion. The N-terminal propeptide cleavage and the C-terminal dimer formation are needed for the cell binding function of the MIC3 protein (12).
The BHK-21 cells transfected with pMIC3i expressed a dimeric recombinant MIC3 protein that was secreted into the supernatant as well as being present in the cell pellets. It had an apparent molecular mass (∼105 kDa) that was ∼15 kDa greater than that of the native MIC3 protein found in the tachyzoite lysate. These results are consistent with those of Cérède et al. (12), who demonstrated that the increase in size of the recombinant protein was due to a failure to cleave the MIC3 propeptide in BHK-21 cells; this was confirmed by probing the immunoblot with a specific anti-propeptide serum (12). These authors also showed that the presence of the propeptide inhibited the adhesive function of the MIC3 protein but that it remained antigenic, because it was well recognized by serum from a T. gondii-infected mouse.
Furthermore, we have shown that the recombinant MIC3 protein produced in vivo by DNA vaccination was immunogenic, because all the CBA/J mice immunized i.m. with pMIC3i either alone or combined with pGM-CSF produced specific anti-MIC3 IgG antibodies that were able to recognize the native MIC3 protein found in T. gondii as detected by Western blotting. Very high specific anti-MIC3 IgG antibody titers developed in mice immunized with pMIC3i alone as determined by ELISA, especially after the third immunization. This could be attributable to the secretion of the MIC3 protein by the cells (20). Association with pGM-CSF increased the antibody titers, in agreement with findings reported by a number of investigations (35, 41).
A specific cellular immune response, characterized by significant splenocyte proliferation, was observed in response to TAg in mice immunized with pMIC3i alone. This proliferation was greater in mice immunized with pMIC3i plus pGM-CSF, as had already been reported (26, 35).
One of the goals of a vaccination protocol is to be able to direct the T-helper response in the appropriate direction. For T. gondii, it has been demonstrated that a naturally occurring infection gives rise to a Th1-biased response (17). This suggests that a good vaccination protocol will direct T-helper cells toward a Th1 rather than a Th2 response. We evaluated the T-helper response of vaccinated mice in two ways. The first was to determine the isotype nature of the IgG response achieved during DNA vaccination. The immunized mice exhibited preferential production of IgG2a and IgG2b, without any statistically significant difference between the mice immunized with pMIC3i alone and those which also received pGM-CSF. This indicates that the response was oriented toward a Th1-type response, which was confirmed by analysis of the cytokine production. As a result, large amounts of IFN-γ were produced in the supernatants of restimulated splenocyte cultures from mice immunized with pMIC3i plus pGM-CSF. The splenocytes from mice immunized with pMIC3i alone exhibited strong specific proliferation when stimulated with TAg, but nonspecific production of IFN-γ was observed. Some studies indicate that the IFN-γ production by antigen-specific T cells may have been obscured by the high level of nonspecific production induced by total T. gondii lysate (38). Specific amounts of IL-2 were also synthesized by the restimulated splenocytes from the mice immunized with either pMIC3i alone or pMIC3i plus pGM-CSF, whereas there was no detectable production of IL-4 and IL-10, which is consistent with a Th1-type response. GM-CSF is generally thought to enhance antibody production and T-cell proliferation without changing the orientation towards a Th1- or Th2-type immune response (18).
We evaluated the protection induced by MIC3 DNA vaccination by orally infecting vaccinated mice with 70 cysts of the T. gondii 76K strain. An effective and highly significant degree of protection was obtained in mice immunized with pMIC3i either alone or combined with pGM-CSF, in comparison with the protection in control pcDNA3 mice. Coadministration of a plasmid encoding GM-CSF enhanced the protection against T. gondii infection, which was in agreement with previous data (18) showing that immunization of C57BL/6 mice with a DNA plasmid encoding the GRA4 protein of T. gondii plus plasmid encoding GM-CSF enhanced protection against the acute and chronic phases of T. gondii infection. The plasmid encoding GM-CSF was also used with a DNA plasmid encoding the SAG1 protein of T. gondii (5).
In conclusion, this study shows that a plasmid DNA encoding the MIC3 protein of T. gondii induced strong specific humoral and cellular immune responses, as well as effective, highly significant protection in immunized CBA/J mice. Moreover, this is the first demonstration that the MIC3 gene of T. gondii is a potent and effective vaccine candidate against toxoplasmosis. Coadministration of the plasmid encoding GM-CSF considerably enhanced both the immune response and the protection. The effects of GM-CSF could be increased by adopting a bicistronic DNA vaccine strategy (6). Because the main objective of vaccination against T. gondii is to prevent infection of the fetus, the efficacy of the pMIC3i vaccine against congenital toxoplasmosis in animal models and in relevant animals, such as sheep, needs to be evaluated. Finally, the coadministration of plasmids encoding potential candidates for vaccination can probably enhance the protection (19, 30). It may be possible to provide complete protection against T. gondii infection by such a strategy, if pMIC3i is combined with other relevant and previously described candidates, such as SAG1 and GRA4.
Acknowledgments
This work was supported by grants from the Conseil Général d'Indre et Loire and from the Région Centre.
We are indebted to J. F. Dubremetz for allowing us to work with the pBluescript II SK-MIC3 plasmid. We appreciate the gift of GM-CSF plasmid from H. C. J. Ertl. We thank R. Magné, J. Pierre, J. M. Rith, and T. Papin for skillful technical assistance. We also thank D. Tabareau for secretarial assistance.
Editor: J. M. Mansfield
REFERENCES
- 1.Achbarou, A., O. Mercereau-Puijalon, J. M. Authemen, B. Fortier, D. Camus, and J. F. Dubremetz. 1991. Characterization of microneme proteins of Toxoplasma gondii. Mol. Biochem. Parasitol. 47:223-233. [DOI] [PubMed] [Google Scholar]
- 2.Ajzenberg, D., A. L. Banuls, M. Tibayrenc, and M. L. Darde. 2002. Microsatellite analysis of Toxoplasma gondii shows considerable polymorphism structured into two main clonal groups. Int. J. Parasitol. 32:27-38. [DOI] [PubMed] [Google Scholar]
- 3.Ajzenberg, D., N. Conge, L. Paris, M. H. Bessiers, P. Thulliez, D. Filisetti, H. Pelloux, P. Marty, and M. L. Darde. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J. Infect. Dis. 186:684-689. [DOI] [PubMed] [Google Scholar]
- 4.Alarcon, J. B., G. W. Waine, and D. P. McManus. 1999. DNA vaccines: technology and application as anti-parasite and anti-microbial agents. Adv. Parasitol. 42:343-410. [DOI] [PubMed] [Google Scholar]
- 5.Angus, C. W., D. Klivington-Evans, J. P. Dubey, and J. A. Kovacs. 2000. Immunization with a DNA plasmid encoding the SAG1 (P30) protein of Toxoplasma gondii is immunogenic and protective in rodents. J. Infect. Dis. 181:317-324. [DOI] [PubMed] [Google Scholar]
- 6.Barouch, D. H., S. Santra, K. Tenner-Racz, P. Racz, M. J. Kuroda, J. E. Schmitz, S. S. Jackson, M. A. Lifton, D. C. Freed, H. C. Perry, M.-E. Davies, J. W. Shiver, and N. L. Letvin. 2002. Potent CD4+ T cell responses elicited by a bicistronic HIV-1 DNA vaccine expressing gp 120 and GM-CSF. J. Immunol. 168:562-568. [DOI] [PubMed] [Google Scholar]
- 7.Bout, D. T., M.-N. Mévélec, F. Velge-Roussel, I. Dimier-Poisson, and M. Lebrun. 2002. Prospects for a human Toxoplasma vaccine. Curr. Drug Targets—Immune Endocr. Metab. Dis. 2:227-234. [DOI] [PubMed] [Google Scholar]
- 8.Bowne, W. B., J. D. Wolchok, W. G. Hawkins, R. Srinivasan, P. Gregor, N. E. Blachere, Y. Moroi, M. E. Engelhorn, A. N. Houghton, and J. J. Lewis. 1999. Injection of DNA encoding granulocyte-macrophage colony-stimulating factor recruits dendritic cells for immune adjuvant effects. Cytokines Cell Mol. Ther. 5:217-225. [PubMed] [Google Scholar]
- 9.Brown, C. R., A. Hunter, R. G. Estes, E. Beckmann, J. Formann, C. David, J. S. Remington, and R. McLeod. 1995. Definitive identification of a gene that confers resistance against Toxoplasma gondii cyst burden and encephalitis. Immunology 85:419-428. [PMC free article] [PubMed] [Google Scholar]
- 10.Buxton, D. 1998. Protozoan infections (Toxoplasma gondii, Neospora caninum and Sarcocystis spp.) in sheep and goats: recent advances. Vet. Res. 29:289-310. [PubMed] [Google Scholar]
- 11.Buxton, D., K. Thomson, S. Maley, S. Wright, and H. J. Bos. 1991. Vaccination of sheep with a live incomplete strain (S48) of Toxoplasma gondii and their immunity to challenge when pregnant. Vet. Rec. 129:89-93. [DOI] [PubMed] [Google Scholar]
- 12.Cérède, O., J. F. Dubremetz, D. Bout, and M. Lebrun. 2002. The Toxoplasma gondii protein MIC3 requires pro-peptide cleavage and dimerization to function as adhesin. EMBO. J. 2:1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chardès, T., I. Bourguin, M.-N. Mévélec, J. F. Dubremetz, and D. Bout. 1990. Antibody responses to Toxoplasma gondii in sera, intestinal secretions and milk from orally infected mice and characterization of target antigens. Infect. Immun. 58:1240-1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen, G., H. Guo, F. Lu, and H. Zheng. 2001. Construction of a recombinant plasmid harbouring the rhoptry protein 1 gene of Toxoplasma gondii and preliminary observation on DNA immunity. Chin. Med. J. 114:837-840. [PubMed] [Google Scholar]
- 15.Davis, H. L., M.-L. Michel, M. Mancini, M. Schleef, and R. G. Whalen. 1994. Direct gene transfer in skeletal muscule: plasmide DNA based immunization against the hepatitis B virus surface antigen. Vaccine 12:1503-1509. [DOI] [PubMed] [Google Scholar]
- 16.Debard, N., D. Buzoni-Gatel, and D. Bout. 1996. Intranasal immunization with SAG1 protein of Toxoplasma gondii in association with cholera toxin dramatically reduces development of cerebral cysts after oral infection. Infect. Immun. 64:2158-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denkers, E. Y., and R. T. Gazzinelli. 1998. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 11:569-588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desolme, B., M.-N. Mévélec, D. Buzoni-Gatel, and D. Bout. 2000. Induction of protective immunity against toxoplasmosis in mice by DNA immunization with a plasmid encoding Toxoplasma gondii GRA4 gene. Vaccine 18:2512-2521. [DOI] [PubMed] [Google Scholar]
- 19.Doolan, D. L., and S. L. Hoffman. 2001. DNA-based vaccines against malaria: status and promise of the Multi-Stage Malaria DNA Vaccine Operation. Int. J. Parasitol. 31:753-762. [DOI] [PubMed] [Google Scholar]
- 20.Drew, D. R., M. Lightowlers, and R. A. Strugnell. 2000. Humoral immune responses to DNA vaccines expressing secreted membrane bound and non-secreted forms of the Tania ovis 45W antigen. Vaccine 18:2522-2532. [DOI] [PubMed] [Google Scholar]
- 21.Dubey, J. P. 2000. The scientific basis for prevention of Toxoplasma gondii infection: studies on tissue cyst survival, risk factors and hygiene measures, p. 271-275 In P. Ambroise-Thomas and E. Petersen (ed.), Congenital toxoplasmosis: scientific background, clinical management and control. Springer-Verlag, Paris, France.
- 22.Gagne, S. S. 2001. Toxoplasmosis. Prim. Care Update Ob/Gyn. 8:122-126. [DOI] [PubMed] [Google Scholar]
- 23.Garcia-Réguet, N., M. Lebrun, M. N. Fourmaux, O. Mercereau-Puijalon, T. Mann, C. J. Beckers, B. Samyn, J. Van Beeumen, D. Bout, and J. F. Dubremetz. 2000. The microneme protein MIC3 of Toxoplasma gondii is a secretory adhesin that binds to both the surface of the host cells and the surface of the parasite. Cell Microbiol. 2:353-364. [DOI] [PubMed] [Google Scholar]
- 24.Haddad, D., J. Ramprakash, M. Sedegah, Y. Charoenvit, R. Baumgartner, S. Kumar, S. L. Hoffman, and W. R. Weiss. 2000. Plasmid vaccine expressing granulocyte-macrophage colony-stimulating factor attracts infiltrates including immature dendritic cells into injected muscles. J. Immunol. 165:3772-3781. [DOI] [PubMed] [Google Scholar]
- 25.Jones, T., A. Stern, and R. Lin. 1994. Potential role of granulocyte-macrophage colony-stimulating factor as vaccine adjuvant. Eur. J. Clin. Microbiol. Infect. Dis. 2:47-53. [DOI] [PubMed] [Google Scholar]
- 26.Kamath, A. T., T. Hanke, H. Briscoe, and W. J. Britton. 1999. Co-immunization with DNA vaccines expressing granulocyte-macrophage colony-stimulating factor and mycobacterial secreted proteins enhances T-cell immunity but not protective efficacy against Mycobacterium tuberculosis. Immunology 96:511-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim, J. J., J. S. Yang, D. J. Lee, D. M. Wilson, L. K. Nottingham, L. Morrison, A. Tsai, J. Oh, K. Dang, T. Dentchev, M. G. Agadjanyan, J. I. Sin, A. A. Chalian, and D. B. Weiner. 2000. Macrophage colony-stimulating factor can modulate immune responses and attract dendritic cells in vivo. Hum. Gene Ther. 11:305-321. [DOI] [PubMed] [Google Scholar]
- 28.Kowalczyk, D. W., and H. C. Ertl. 1999. Immune responses to DNA vaccines. Cell Mol. Life Sci. 55:751-770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leyva, R., P. Herion, and R. Saavedra. 2001. Genetic immunization with plasmide DNA coding for the ROP2 protein of Toxoplasma gondii. Parasitol. Res. 87:70-79. [DOI] [PubMed] [Google Scholar]
- 30.Mendez, S., Y. Belkaid, R. A. Seder, and D. Sacks. 2002. Optimization of DNA vaccination against cutaneous leishmaniasis. Vaccine 20:3702-3708. [DOI] [PubMed] [Google Scholar]
- 31.Montgomery, D. L., J. B. Ulmer, J. J. Donnelly, and M. A. Liu. 1997. DNA vaccines. Pharmacol. Ther. 74:195-207. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen, H.V., S. L. Lauemøller, L. Christiansen, S. Buus, A. Fomsgaard, and E. Petersen. 1999. Complete protection against lethal Toxoplasma gondii infection in mice immunized with a plasmid encoding the SAG1 gene. Infect. Immun. 67:6358-6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scorza, T., S. D'Souza, M. Laloup, J. Dewit, J. De Braekeleer, H. Verschueren, M. Vercammen, K. Huygen, and E. Jongert. 2003. A GRA1 DNA vaccine primes cytolytic CD8+ T cells to control acute Toxoplasma gondii infection. Infect. Immun. 71:309-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma, S. D., J. Mullenack, F. G. Araujo, H. A. Erlich, and J. S. Remington. 1983. Western blot analysis of the antigens of toxoplasma gondii recognized by human IgM and IgG antibodies. J. Immunol. 131:977-983. [PubMed] [Google Scholar]
- 35.Sin, J. I., J. J. Kim, K. E. Ugen, R. B. Ciccarelli, T. J. Higgins, and D. B. Weiner. 1998. Enhancement of protective humoral (Th2) and cell-mediated (Th1) immune responses against herpes simplex virus-2 through co-delivery of granulocyte-macrophage colony-stimulating factor expression casettes. Eur. J. Immunol. 28:3530-3540. [DOI] [PubMed] [Google Scholar]
- 36.Suzuki, Y., M. A. Orellana, S. Y. Wong, F. K. Conley, and J. S. Remington. 1993. Susceptibility to chronic infection with Toxoplasma gondii does not correlate with susceptibility to acute infection in mice. Infect. Immun. 61:2284-2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tenter, A. M., A. R. Heckeroth, and L. M. Weiss. 2000. Toxoplasma gondii: from animals to humans. Int. J. Parasitol. 30:1217-1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vercammen, M., T. Scorza, K. Huygen, J. De Braekeleer, R. Diet, D. Jacobs, E. Saman, and H. Verschueren. 2000. DNA vaccination with genes encoding Toxoplasma gondii antigens GRA1, GRA7 and ROP2 induces partially protective immunity against lethal challenge in mice. Infect. Immun. 68:38-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warren, T. L., and G. J. Weiner. 2000. Uses of granulocyte-macrophage colony-stimulating factor in vaccine development. Curr. Opin. Hematol. 7:168-173. [DOI] [PubMed] [Google Scholar]
- 40.Weiss, W. R., R. J. Ishii, R. C. Hedstrom, M. Sedegah, M. Ichino, K. Barnhart, D. M. Klinman, and S. L. Hoffman. 1998. A plasmid encoding murine granulocyte-macrophage colony-stimulating factor increases protection conferred by a malaria DNA vaccine. J. Immunol. 161:2325-2332. [PubMed] [Google Scholar]
- 41.Xiang, Z. Q., and H. C. J. Ertl. 1995. Manipulation of the immune response to a plasmid-encoded viral antigen by coinoculation with plasmids expressing cytokines. Immunity 2:129-135. [DOI] [PubMed] [Google Scholar]