

Abstract
The role of peroxynitrite in hypoxia–reoxygenation-induced coronary vasospasm was investigated in isolated bovine coronary arteries. Hypoxia–reoxygenation selectively blunted prostacyclin (PGI2)-dependent vasorelaxation and elicited a sustained vasoconstriction that was blocked by a cyclooxygenase inhibitor, indomethacin, and SQ29548, a thromboxane (Tx)A2/prostaglandin H2 receptor antagonist, but not by CGS13080, a TxA2 synthase blocker. The inactivation of PGI2 synthase, as evidenced by suppressed 6-keto-PGF1α release and a decreased conversion of 14C-prostaglandin H2 into 6-keto-PGF1α, was paralleled by an increased nitration in both vascular endothelium and smooth muscle of hypoxia–reoxygenation-exposed vessels. The administration of the nitric oxide (NO) synthase inhibitors as well as polyethylene-glycolated superoxide dismutase abolished the vasospasm by preventing the inactivation and nitration of PGI2 synthase, suggesting that peroxynitrite was implicated. Moreover, concomitant administration to the organ baths of the two precursors of peroxynitrite, superoxide, and NO mimicked the effects of hypoxia–reoxygenation, although none of them were effective when given separately. We conclude that hypoxia–reoxygenation elicits the formation of superoxide, which causes loss of the vasodilatory action of NO and at the same time yields peroxynitrite. Subsequently, peroxynitrite nitrates and inactivates PGI2 synthase, leaving unmetabolized prostaglandin H2, which causes vasospasm, platelet aggregation, and thrombus formation via the TxA2/prostaglandin H2 receptor.
Keywords: peroxynitrite, nitric oxide, superoxide anions, prostaglandin, thromboxane A2/prostaglandin H2 receptor
Under physiological conditions, the endothelium provides vasodilatory and antiaggregatory properties to the cardiovascular system and also prevents growth of the underlying vascular smooth muscle cells by releasing nitric oxide (NO), endothelium-derived hyperpolarizing factor, and prostacyclin (PGI2) 1 2 3. Each of these mediators acts by a different mechanism, NO by the stimulation of guanylyl cyclase 2 and PGI2 by activating adenylyl cyclase, so that these mediators can act synergistically and also serve as backup systems for each other 1 2 3. It was interesting to study this interdependence after a selective inhibition of PGI2 synthase by peroxynitrite had been described 4. As peroxynitrite is generated in vivo by a rapid combination of NO and superoxide radicals (O2 . −) 5 6, it seemed that O2 . − not only can neutralize NO but subsequently, through its product peroxynitrite, could suppress PGI2 formation. The underlying mechanism for PGI2 synthase blockade has been suggested as the nitration of a tyrosine residue at the active site through a reaction catalyzed by the ferric iron of this heme-thiolate (P450) protein 7. In spite of the cellular antioxidant potential, peroxynitrite causes PGI2 synthase nitration in whole cells 8 as well as in intact coronary arteries 9.
Endothelial dysfunction (impaired vasorelaxation and/or increased vasoconstriction) is an important early-recurring phenomenon in virtually all forms of ischemia/reperfusion diseases, including a variety of shock states 10. This dysfunction appears to be triggered simultaneously with the endothelial generation of O2 . −. Therefore, the formation of peroxynitrite during hypoxia–reoxygenation is likely because of a simultaneous generation of NO and O2 . − 11. In this study, we investigated a role of peroxynitrite in hypoxia–reoxygenation-induced coronary vasospasm by monitoring the pattern of angiotensin II–triggered vasoconstriction/vasorelaxation, PGI2 release, and the nitration of PGI2 synthase. Our results suggest that hypoxia–reoxygenation elicits peroxynitrite formation, leading to a subsequent nitration and inhibition of PGI2 synthase. Most importantly, the unmetabolized PGH2 triggers vasospasm by acting upon the thromboxane (Tx)A2/PGH2 receptor before being converted to PGE2.
Materials and Methods
Materials.
All chemicals, if not indicated otherwise, were obtained from the same sources as previously described 9. Nω-mono-methyl-l-arginine (L-NMMA), Nω-nitro-l-arginine methyl ester (L-NAME), and diethylamine NONoate (DEA-NO) were obtained from Cayman SPI. Polyethylene-glycolated superoxide dismutase (PEG-SOD) was obtained from Sigma Chemical Co. 14C-PGH2 was prepared from 14C-arachidonic acid as previously described 4 7. Rabbit anti–PGI2 synthase antisera were produced in this laboratory as previously described 12.
Experimental Protocol.
As previously described 9, bovine coronary arteries (BCA) of the left ventricle were isolated and resuspended in a tissue bath with 15 ml of Krebs buffer, gassed with 95% O2/5% CO2 (37°C; pH 7.4), and attached to a force–displacement TFT6V5 transducer coupled to a polygraph for the measurement of isometric tension. Passive tension was adjusted to ∼1 g over a 30-min equilibrating period, and then coronary arteries were preconstricted by addition of the Tx mimetic U46619 (0.001–0.01 μmol/liter). The vasorelaxation after acetylcholine (0.01–1 μmol/liter) was used to demonstrate the presence of endothelium-dependent relaxation. Throughout the experiment, care was taken to avoid any injury to the endothelium.
Experiments were started by obtaining from each spiral a reference response of vasoconstriction–relaxation with angiotensin II (50 nM) for 30 min. After removal of the agonist from the chambers and return of tone to basal levels, the oxygen tension was then reduced abruptly from 95% O2 /5% CO2 to 95% N2 /5% CO2
 and was maintained for 10-, 30-, or 40-min hypoxia. After this phase of hypoxia, 95% O2/5% CO2 was resumed (reoxygenation), and the tension was allowed to stabilize for 40 min before addition of agonists. If required, pharmacological agents were added in the organ chamber 40 min before hypoxia and kept during hypoxia–reoxygenation and the second stimulation with the agonists. Tissues were kept for immunohistochemistry or immunoprecipitation and Western blots. The media from the first and second stimulations with angiotensin II were collected and stored at −20°C for prostanoid analysis, as previously described 9, using ELISA kits according to the manufacturer's instructions.
and was maintained for 10-, 30-, or 40-min hypoxia. After this phase of hypoxia, 95% O2/5% CO2 was resumed (reoxygenation), and the tension was allowed to stabilize for 40 min before addition of agonists. If required, pharmacological agents were added in the organ chamber 40 min before hypoxia and kept during hypoxia–reoxygenation and the second stimulation with the agonists. Tissues were kept for immunohistochemistry or immunoprecipitation and Western blots. The media from the first and second stimulations with angiotensin II were collected and stored at −20°C for prostanoid analysis, as previously described 9, using ELISA kits according to the manufacturer's instructions.
Activity Assay of PGI2 Synthase.
BCA spirals with or without hypoxia–reoxygenation were incubated with 100 μM 14C-PGH2 for 3 min as previously described 4 7 9. The reaction was stopped by acidification with 1 N HCl to pH 3.5. The incubation media were extracted with ethyl acetate (3 vol). After centrifugation, the organic phases were evaporated to dryness under nitrogen. Samples were then resuspended in 60 μl of ethyl acetate and subsequently separated by TLC (ethyl acetate/water/iso-octane/acetic acid, 90:100:50:20). Prostanoids were quantified with a PhosphorImager system (ImageQuant; Molecular Dynamics).
Immunohistochemistry, Immunoprecipitation, and Western Blots.
Immunohistochemistry, immunoprecipitation, and Western blots of 3-nitrotyrosine–containing protein in hypoxia–reoxygenation-treated BCA were performed as previously described 9.
Results and Discussion
In BCA, angiotensin II elicited a biphasic response consisting of a primary vasoconstriction that was selectively blocked by losartan, a selective angiotensin 1 receptor blocker, and a secondary vasorelaxation that was mainly PGI2 dependent 9.
Abrupt decrease of oxygen tension from 95% O2/5% CO2 to 95% N2 /5% CO2 caused a slight decrease in tension after a transient rise. Reoxygenation (from 95% N2 /5% CO2 to 95% O2 /5% CO2) did not alter the tension of unstimulated arteries. Although hypoxia–reoxygenation did not affect the initial constriction of angiotensin II, it selectively blunted the angiotensin II–triggered relaxation phase after 30 min of hypoxia. Along with this suppression of the relaxation phase, a second constriction phase developed in parallel, with a decrease of 6-keto-PGF1α (Fig. 1 A) that closely resembled the pattern seen previously after peroxynitrite pretreatment 9.
Figure 1.


Hypoxia–reoxygenation on angiotensin II–triggered relaxation and eicosanoid metabolism in BCA. (A) Effects of indomethacin, SQ 29548, CGS13080, SOD, L-NMMA, and L-NAME on angiotensin II–triggered relaxation (white bars) and the release of 6-keto-PGF1α (black bars) and PGE2 (hatched bars) in hypoxia–reoxygenated BCA. After having obtained a reference response to angiotensin II, the coronary strip was exposed to hypoxia for 40 min after 40-min reoxygenation in presence of indomethacin (10 μM), CGS13080 (10 μM), SQ-29548 (10 μM), PEG-SOD (500 U/ml), L-NMMA (10−4 M), or L-NAME (10−4 M). PGE2 and 6-keto-PGF1α in the media were analyzed by ELISA. Data represent means ± SEM from 10 experiments. (B) Effects of superoxide, NO, and concurrent administration of superoxide and NO on angiotensin II–induced vasorelaxation and prostaglandin release in BCA. After having obtained a reference response to angiotensin II, the coronary strip was exposed to superoxide generated from 10 mU/ml xanthine oxidase/100 μM hypoxanthine or to NO generated from 20 μM DEA-NO or superoxide plus NO. PGE2 (hatched bars) and 6-keto-PGF1α (black bars) in the media were analyzed by ELISA. Data represent means ± SEM from 12 experiments. White bars, relaxation.
Both the COX (cyclo-oxygenase) inhibitor indomethacin and the TxA2/PGH2 receptor blocker SQ29548 restored hypoxia–reoxygenation-impaired relaxation without affecting 6-keto-PGF1α release (Fig. 1 A). This suggests that a COX-derived product, TxA2 or PGH2, caused vasoconstriction via the TxA2/PGH2 receptor (Fig. 1 A). A role of TxA2 was excluded, as CGS13080, a TxA2 synthase inhibitor, did not affect hypoxia–reoxygenation-triggered vasospasm (Fig. 1 A), and the levels of TxB2 remained low and unaffected after hypoxia–reoxygenation treatment (97 ± 11 vs. 99 ± 15 pg/30 min). The level of 8-iso-PGF2α, which also could act on the TxA2/PGH2 receptor 14, was low and remained unchanged after hypoxia–reoxygenation (43 ± 12 vs. 47 ± 17 pg/30 min).
PGH2 is known to cause vasoconstriction via the TxA2/PGH2 receptor 13. In arterial vessels, PGI2 is metabolized primarily by PGI2 synthase to yield PGI2. Therefore, we postulated that an inhibition on PGI2 synthase could be the primary cause for the accumulation of PGH2, which then caused vasoconstriction by stimulating the TxA2/PGH2 receptor. Parallel measurements of prostaglandins in the incubation medium further supported this hypothesis. Hypoxia–reoxygenation lowered 6-keto-PGF1α formation but raised the level of PGE2, an enzymatic or nonenzymatic metabolite of PGH2 (Fig. 1 A). As COX activity was slightly decreased after hypoxia–reoxygenation, an excess formation of PGH2 after hypoxia–reoxygenation could be excluded.
Conclusive evidence for an inactivation of PGI2 synthase came from the experiments with 14C-PGH2 as a substrate for PGI2 synthase. A significant inhibition on the conversion of 14C-PGH2 into 6-keto-PGF1α (−89 ± 8%) with a concomitant increase of PGE2 (135 ± 21%) was observed, confirming the selective inhibition of PGI2 synthase and the reorientation of PGH2 metabolism toward PGE2 after hypoxia–reoxygenation.
The sustained vasoconstriction after hypoxia–reoxygenation could also have been the consequence of a decreased sensitivity of vascular smooth muscle to NO. However, NO generated from DEA-NO in arteries with or without hypoxia–reoxygenation resulted in a similar potency to induce vasorelaxation. Similarly, an increased sensitivity of the TxA2/PGH2 receptor was excluded, as pD2 (the negative logarithm of the molar concentration of agonist that elicits a half- maximal response) of U46619, an agonist for the TxA2/PGH2 receptor, was identical before and after exposure to hypoxia–reoxygenation (7.7 ± 0.5 vs. 7.8 ± 0.5). A correspondent dose of PGE2 (20 ng/ml) applied to the bath solution did not cause vasoconstriction (data not shown), indicating that the higher levels of PGE2 after hypoxia–reoxygenation were not responsible for the second constriction phase.
According to our working hypothesis, hypoxia–reoxygenation could induce peroxynitrite formation, which inactivates PGI2 synthase. To test whether peroxynitrite was indeed responsible for the hypoxia–reoxygenation-induced vasospasm, the NO synthase inhibitors, both L-NMMA (10−4 M) and L-NAME (10−4 M) were concurrently administered during hypoxia–reoxygenation. L-NMMA, which reduced the angiotensin II–induced relaxation phase by about 25% without affecting 6-keto-PGF1α formation in normal vessels (data not shown), prevented hypoxia–reoxygenation-induced suppression of angiotensin II–induced vasorelaxation and 6-keto-PGF1α release (Fig. 1 A); L-NAME was as effective as L-NMMA (data not shown).
Alternatively, concurrent administration of PEG-SOD (500 U/ml) abolished hypoxia–reoxygenation-induced secondary vasoconstriction and restored angiotensin II–stimulated vasorelaxation by blunting the hypoxia–reoxygenation-mediated inhibition on 6-keto-PGF1α formation (Fig. 1 A).
It was important to show that neither NO nor O2 . − alone could cause vascular dysfunction and inhibition on PGI2 release. Therefore, preincubation of BCA with an O2 . −-generating system (10 mU/ml xanthine oxidase/100 μM hypoxanthine) did not significantly affect either angiotensin II–triggered vasorelaxation or 6-keto-PGF1α release (Fig. 1 B). Alternatively, the incubation of BCA with DEA-NO (20 μM) as an NO-releasing system also failed to affect angiotensin II–induced relaxation and 6-keto-PGF1α release (Fig. 1 B). However, concomitant administration to the organ baths of both O2 . −- and NO-generating agents for 40 min caused a loss of PGI2-dependent vasorelaxation, an indomethacin- and SQ29548-sensitive vasospasm, and a nitration of PGH2 synthase (Fig. 1 B).
As previously described, the exposure of BCA to peroxynitrite produced a nitrated protein that colocalized with PGI2 synthase 9. The same technique was applied to hypoxia–reoxygenation-exposed BCA segments after their mechanical responses had been established. Staining with antinitrotyrosine Ab was weakly visible in control tissue (Fig. 2 A), but clearly enhanced stainings emerged in endothelium and smooth muscle after hypoxia–reoxygenation (Fig. 2 B), where the stainings with Ab against PGI2 synthase were intensively presented (Fig. 1 C). A computer-generated overlay of the stainings with antinitrotyrosine (green) and anti–PGI2 synthase (red) resulted in the yellow colocalizing patches in vessels after hypoxia–reoxygenation (Fig. 2 D). The presence of L-NMMA or PEG-SOD abolished the increased stainings with antinitrotyrosine Ab (Fig. 2F and Fig. G) but not those with anti–PGI2 synthase Ab (data not shown). The specificity of the staining with antinitrotyrosine Ab was deduced from its suppression by 10 mM 3-nitrotyrosine (Fig. 2 E) and the lack of staining when antinitrotyrosine Ab was omitted (Fig. 2 H). 3-chloro- or 3-aminotyrosine or phosphotyrosine were ineffective in blocking the stainings with antinitrotyrosine Ab (data not shown).
Figure 2.
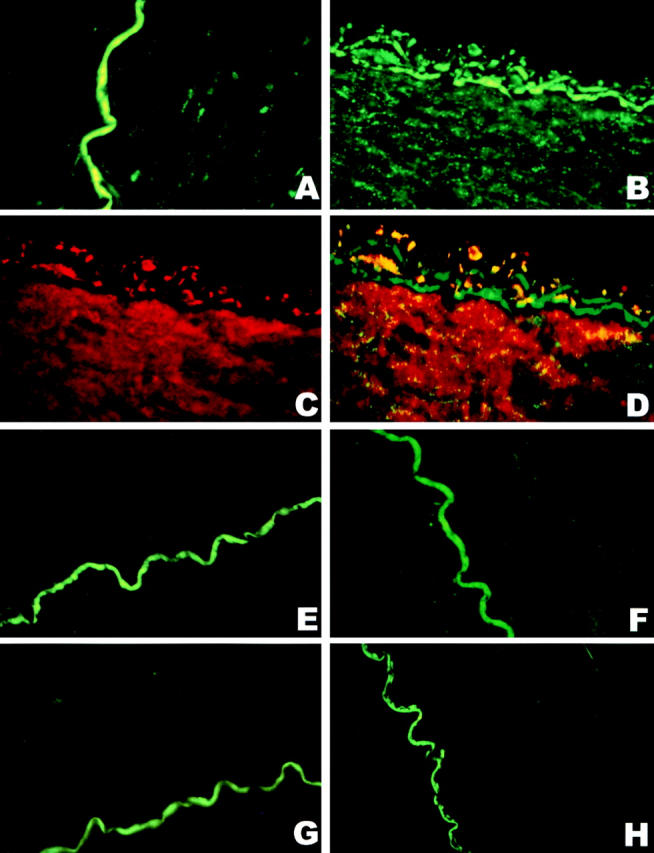
Immunohistochemical colocalization of a polyclonal anti–PGI2 synthase Ab and an mAb against 3-nitrotyrosine in hypoxia-reoxygenated BCA. The yellow coloring resulting from a computer-generated overlay of green (3-nitrotyrosine) and red (PGI2 synthase) fluorescence indicates areas of the colocalization of antinitrotyrosine and anti–PGI2 synthase Abs in BCA with or without hypoxia–reoxygenation treatment. All pictures were obtained under 400-fold magnification with identical camera and print settings. (A) 3-nitrotyrosine staining in a sham-treated artery (green), where 3-nitrotyrosine staining is very weak and the endothelium is intact. The green wiggly line is due to endogenous fluorescence of the lamina and not specific immunostaining for 3-nitrotyrosine. (B) 3-nitrotyrosine staining in a hypoxia-reoxygenated artery; both endothelium and vascular smooth muscle cells are strongly immunopositive for 3-nitrotyrosine (green). (C) The staining of PGI2 synthase Ab in a hypoxia-reoxygenated artery. Dense staining with anti–PGI2 synthase Ab was visible in both endothelium and smooth muscle (red). (D) A computer-generated overlay of the stainings with the Abs against 3-nitrotyrosine (B) and PGI2 synthase (C) in a hypoxia-reoxygenated artery. Yellow indicates the colocalization of the binding with both Abs. (E) An hypoxia-reoxygenated artery was stained for antinitrotyrosine Ab in the presence of 10 mM free 3-nitrotyrosine. Only the autofluorescence of the lamina is visible. (F) 3-nitrotyrosine stainings in a hypoxia-reoxygenated artery in the presence of L-NMMA, where the staining for 3-nitrotyrosine is only weakly visible in both endothelium and vascular smooth muscle. (G) 3-nitrotyrosine stainings in a hypoxia-reoxygenated artery in the presence of PEG-SOD, where 3-nitrotyrosine staining is weakly visible in vascular smooth muscle. (H) An hypoxia-reoxygenated artery was stained for 3-nitrotyrosine when the Ab against 3-nitrotyrosine was omitted, where only the autofluorescence of the lamina is visible.
Further evidence for hypoxia–reoxygenation-triggered tyrosine nitration of PGI2 synthase came from the immunoprecipitation with both antibodies against 3-nitrotyrosine and PGI2 synthase. Immunoprecipitation with anti–PGI2 synthase Ab yielded similar amounts of protein in both hypoxia–reoxygenation-treated arteries and control tissues (Fig. 3 A). In Western blot analysis, a dense band was detected by antinitrotyrosine Ab in the immunoprecipitates with anti–PGI2 synthase Ab from hypoxia–reoxygenation-treated vessel homogenates against a weakly visible signal in those from the control tissues (Fig. 3 A). In the same homogenates, 3-nitrotyrosine–containing proteins were precipitated with antinitrotyrosine Ab in parallel. Higher amounts of these proteins were recovered in hypoxia–reoxygenation-treated arteries than in those without hypoxia–reoxygenation. Moreover, when these precipitates were blotted and probed with anti–PGI2 synthase Ab, a dense staining appeared in arteries with hypoxia–reoxygenation (Fig. 3 B). In agreement with the immunohistochemistry results, both PEG-SOD and L-NMMA effectively abolished the increased stainings with both antibodies in the immunoprecipitates with either antinitrotyrosine or anti–PGI2 synthase (Fig. 3a and Fig. b).
Figure 3.



Immunoprecipitation of nitrated proteins and PGI2 synthase in hypoxia-reoxygenated BCA. (A) Proteins from normal or hypoxia-reoxygenated BCA were immunoprecipitated with a polyclonal Ab against PGI2 synthase (α-PGI2 synthase) after hypoxia–reoxygenation. As described in Materials and Methods, proteins precipitated by α-PGI2 synthase were separated electrophoretically and immunostained with a monoclonal α-PGI2 synthase (left) or a monoclonal α-nitrotyrosine (right). Lane A, hypoxia–reoxygenation + L-NMMA; lane B, hypoxia–reoxygenation + SOD; lane C, hypoxia–reoxygenation; lane D, control. (B) 3-nitrotyrosine–positive proteins were precipitated by an mAb against 3-nitrotyrosine. Proteins were separated similarly and analyzed by immunoblots with polyclonal Ab against PGI2 synthase. Lane A, control; lane B, hypoxia–reoxygenation; lane C, hypoxia–reoxygenation + L-NMMA; lane D, hypoxia–reoxygenation + SOD.
In summary, our results indicate that hypoxia–reoxygenation elicits the formation of O2 . −, which neutralize NO to form peroxynitrite. Subsequently, peroxynitrite nitrates and inactivates PGI2 synthase, leaving PGH2 unmetabolized, which then causes vasospasm and platelet aggregation via the TxA2/PGH2 receptor. This finding might offer a new mechanism for coronary vasospasm during hypoxia–reoxygenation, especially in atherosclerotic arteries where PGI2 synthase is partially nitrated 15.
Acknowledgments
We dedicate this paper to Prof. Ullrich on the occasion of his 60th birthday for his generous support. We thank Ms. Nie Hong for technical support and Dr. Leist for help with the confocal images.
This work was supported by the Deutsche Forschungsgemeinschaft Forschergruppe We686/18-1.
References
- Vane J.R., Anggard E.E., Botting R.M. Regulatory function of the vascular endothelium. N. Engl. J. Med. 1990;323:27–36. doi: 10.1056/NEJM199007053230106. [DOI] [PubMed] [Google Scholar]
- Moncada S., Palmer R.M., Higgs E.A. Nitric oxidephysiology, pathophysiology, and pharmacology. Pharmacol. Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- Moncada S., Vane J.R. Pharmacology and endogenous roles of endoperoxide prostaglandins, thromboxane A2 and prostacyclin. N. Engl. J. Med. 1979;300:1142–1147. [Google Scholar]
- Zou M.H., Ullrich V. Peroxynitrite formed by simultaneous generation of nitric oxide and superoxide selectively inhibits bovine aortic prostacyclin synthase. FEBS Lett. 1996;382:101–104. doi: 10.1016/0014-5793(96)00160-3. [DOI] [PubMed] [Google Scholar]
- Beckman J.S., Beckman T.W., Chen J., Marshall P.A., Freeman B. Apparent hydroxyl radical production by peroxynitriteimplications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman J.S., Koppenol W.H. Nitric oxide, superoxide, and peroxynitritethe good, the bad, and the ugly. Am. J. Physiol. 1995;271:C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- Zou M.H., Martin C., Ullrich V. Tyrosine nitration as a mechanism of selective inactivation of prostacyclin synthase by peroxynitrite. Biol. Chem. 1997;378:707–713. doi: 10.1515/bchm.1997.378.7.707. [DOI] [PubMed] [Google Scholar]
- Zou M.H., Klein T., Pasquet J.P., Ullrich V. Interleukin 1β decreases prostacyclin synthase in rat mesangial cells via endogenous peroxynitrite formation. Biochem. J. 1998;336:507–512. doi: 10.1042/bj3360507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M.H., Jendral M., Ullrich V. Peroxynitrite induces prostaglandin endoperoxide-dependent vasospasm in bovine coronary arteries via tyrosine nitration of prostacyclin synthase. Br. J. Pharmacol. 1999;126:1283–1292. doi: 10.1038/sj.bjp.0702434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefer A.M., Lefer D.J. Pharmacology of the endothelium in ischemia-reperfusion and circulatory shock. Annu. Rev. Pharmacol. Toxicol. 1993;33:71–91. doi: 10.1146/annurev.pa.33.040193.000443. [DOI] [PubMed] [Google Scholar]
- Huie R.E., Padmaja S. The reaction of nitric oxide with superoxide. Free Radic. Res. Commun. 1993;18:195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- Siegle I., Nüsing R., Brugger R., Sprenger R., Zecher R., Ullrich V. Characterization of monoclonal antibodies generated against bovine and porcine prostacyclin synthase and quantitation of bovine prostacyclin synthase. FEBS Lett. 1994;347:221–225. doi: 10.1016/0014-5793(94)00504-4. [DOI] [PubMed] [Google Scholar]
- Mais D.E., Saussy D.L., Chaikouni A., Kochel P.J., Knapp D.R., Hamanaka N., Halushka P.V. Pharmacological characterization of human and canine thromboxane A2/prostaglandin H2 receptors in platelets and blood vesselsevidence for different receptors. J. Pharmacol. Exp. Ther. 1985;233:418–424. [PubMed] [Google Scholar]
- Takahashi K., Nammour T.M., Fukunaga M., Enbert J., Morrow J.D., Roberts L.J., Hoover R.L., Badr K. Glomerular actions of a free radical–generated novel prostaglandin, 8-iso-prostaglandin F2α, in the rat. Evidence for interaction with thromboxane A2 receptor. J. Clin. Invest. 1992;90:136–141. doi: 10.1172/JCI115826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou M.H., Leist M., Ullrich V. Selective nitration of prostacyclin synthase and defective vasorelaxation in atherosclerotic bovine coronary arteries. Am. J. Pathol. 1999;154:1559–1565. doi: 10.1016/S0002-9440(10)65390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]