

Abstract
Agents that restore vascular patency in stroke also increase the risk of intracerebral hemorrhage (ICH). As Factor IXa is a key intermediary in the intrinsic pathway of coagulation, targeted inhibition of Factor IXa–dependent coagulation might inhibit microvascular thrombosis in stroke without impairing extrinsic hemostatic mechanisms that limit ICH. A competitive inhibitor of native Factor IXa for assembly into the intrinsic Factor X activation complex, Factor IXai, was prepared by covalent modification of the Factor IXa active site. In a modified cephalin clotting time assay, in vivo administration of Factor IXai caused a dose-dependent increase in time to clot formation (3.6-fold increase at the 300 μg/kg dose compared with vehicle-treated control animals, P < 0.05). Mice given Factor IXai and subjected to middle cerebral artery occlusion and reperfusion demonstrated reduced microvascular fibrin accumulation by immunoblotting and immunostaining, reduced 111In-labeled platelet deposition (42% decrease, P < 0.05), increased cerebral perfusion (2.6-fold increase in ipsilateral blood flow by laser doppler, P < 0.05), and smaller cerebral infarcts than vehicle-treated controls (70% reduction, P < 0.05) based on triphenyl tetrazolium chloride staining of serial cerebral sections. At therapeutically effective doses, Factor IXai was not associated with increased ICH, as opposed to tissue plasminogen activator (tPA) or heparin, both of which significantly increased ICH. Factor IXai was cerebroprotective even when given after the onset of stroke, indicating that microvascular thrombosis continues to evolve (and may be inhibited) even after primary occlusion of a major cerebrovascular tributary.
Keywords: Factor IX, thrombosis, anticoagulation, intrinsic coagulation pathway, cerebral ischemia
Timely reestablishment of blood flow to ischemic brain using thrombolytic agents represents the current treatment paradigm for acute stroke 1 2 3. However, this approach is limited by an increased risk of intracerebral hemorrhage (ICH)1 and an increase in early mortality 1 2 3 4 5. Furthermore, even if the best available agent, recombinant tissue plasminogen activator (tPA), is given promptly, within 3 h of symptom onset, there is no improvement in overall mortality 1 2 3 5. Even though there is an improvement in the composite end point of death and morbidity with tPA treatment, it is likely that efficacy could be substantially improved if the risk of ICH were lowered. In addition, recent reports indicate that tPA may have a direct role in neuronal injury in the setting of stroke 6 7 8 9 10 11. Limited anticoagulant trials of heparin in stroke showed heparin to be ineffective and/or associated with an unacceptably high incidence of hemorrhagic conversion 12 13 14 15 16 17. In a murine model of stroke, a platelet glycoprotein IIb/IIIa receptor antagonist, although effective at reducing cerebral infarct volumes, caused a dose-dependent increase in ICH 18. Consequently, there remains a clear need to identify new agents that can promote reperfusion without increasing the risk of ICH.
After an ischemic event, the vascular wall is modified from its quiescent, antiadhesive, antithrombotic state to one which promotes leukocyte adhesion and thrombosis. In acute stroke, active recruitment of leukocytes by adhesion receptors expressed in the ipsilateral microvasculature, such as intercellular adhesion molecule 1 (ICAM-1 [19]) and P-selectin 20, potentiates postischemic hypoperfusion. However, experiments with mice deletionally mutant for each of these genes demonstrate that even in their absence, postischemic cerebral blood flow (CBF) returns only partially to baseline after removal of an intraluminal middle cerebral artery occluding suture. These observations imply the existence of additional mechanisms responsible for postischemic cerebrovascular no-reflow, especially the possibility that local thrombosis occurs at the level of the microvasculature (distal to the site of primary occlusion) in stroke. Furthermore, if the ischemic insult is particularly severe, reflow continues to worsen over time subsequent to withdrawal of the occluding suture, suggesting ongoing vascular obstructive processes (such as de novo thrombosis) in the distal microvasculature. Recent data in a murine model of stroke implicate glycoprotein IIb/IIIa receptor–dependent platelet recruitment as a mechanism which amplifies thrombosis in the postischemic microvasculature 18.
These observations provide the rationale for identifying new strategies to selectively limit thrombosis in stroke without increasing ICH. We hypothesized that anticoagulant strategies that do not impair tissue factor–mediated hemostatic events might reduce thrombosis in the microvascular lumen yet not impair the ability of friable postischemic cerebral microvessels to form effective hemostatic plugs to limit ICH. Heparin or hirudin, both of which interfere with the final common pathway of coagulation, or thrombolytic agents such as tPA, which lyse fibrin, do not offer the theoretical advantage offered by targeting earlier points in the coagulation cascade. These experiments test whether selective blockade of IXa/VIIIa/X activation complex assembly using a competitive inhibitor of Factor IXa (active site–blocked IXa, Factor IXai) can limit intravascular thrombosis while preserving mechanisms of extravascular hemostasis in stroke.
Materials and Methods
Murine Stroke Model.
Transient focal cerebral ischemia was induced in mice by intraluminal occlusion of the middle cerebral artery (45 min) and reperfusion (24 h) as reported previously 21. Serial measurements of relative CBF were recorded via laser doppler flowmetry at previously defined neuroanatomic landmarks 21, and infarct volumes (percentage of ipsilateral hemisphere) were determined by planimetric/volumetric analysis of triphenyl tetrazolium chloride (TTC)-stained serial cerebral sections 21.
111Indium-platelet Studies.
Platelet accumulation was determined using 111In-labeled platelets, collected and prepared as described previously 22. Immediately before surgery, mice were given 5 × 106 111In-labeled platelets intravenously; deposition was quantified after 24 h as ipsilateral cpm/contralateral cpm.
Fibrin Immunoblotting/Immunostaining.
The accumulation of fibrin was measured after killing (of fully heparinized animals) using immunoblotting/immunostaining procedures that have been recently described and validated 21. Because fibrin is extremely insoluble, hemispheric brain tissue extracts were prepared by plasmin digestion, then applied to a standard SDS-polyacrylamide gel for electrophoresis, followed by immunoblotting using a polyclonal rabbit anti–human antibody prepared to gamma-gamma chain dimers present in cross-linked fibrin which can detect murine fibrin, with relatively little cross-reactivity with fibrinogen 22. In additional experiments to localize sites of fibrin accumulation, brains were embedded in paraffin, sectioned, and immunostained using the same antifibrin antibody.
Spectrophotometric Hemoglobin Assay and Visual ICH Score.
ICH was quantified by a spectrophotometry-based assay which we have developed and validated 23. In brief, mouse brains were homogenized, sonicated, and centrifuged, and methemoglobin in the supernatants was converted (using Drabkin's reagent) to cyanomethemoglobin, the concentration of which was assessed by measuring OD at 550 nm. For each experiment, the OD relative to that obtained from a group of control brains is reported.
Preparation and Purification of Human IXa/IXai.
Factor IXai was prepared by applying Proplex (a mixture of human vitamin K–dependent coagulation factors [Factors II, VII, IX, and X] supplied by Dr. Roger Lundblad, Baxter, Duarte, CA), reconstituted in Tris-buffered saline (TBS) containing CaCl2, to a column of calcium-dependent anti–human Factor IX mAb (CaFIX-1) coupled to Affi-Gel 10 (BioRad Laboratories) equilibrated at 4°C with TBS containing CaCl2 (0.01 M). After sample application, the column was washed extensively with TBS containing CaCl2 (0.01 M) and NaCl (0.5 M), and Factor IX was subsequently eluted in Tris-HCl (0.1 M; pH 8.0) containing EDTA (0.03 M). Minimal residual contaminants were then removed using Q-Sepharose Fast Flow chromatography. Factor IX thus purified migrated as a single band on SDS-PAGE in the absence and presence of mercaptoethanol (10%) with an apparent M r of ≈68 kD. Factor IX was then activated at 37°C by incubation with purified human Factor XIa (Haematologic Technologies, Inc.) at a 1:1,000 enzyme to substrate ratio in Tris-HCl (0.05 M; pH 7.5) containing NaCl (0.1 M) and CaCl2 (0.005 M) for 1 h. Purified Factor IXa migrated as a single band in nonreduced SDS-PAGE gels (M r ≈ 45 kD), and as two bands, corresponding to the heavy and light chains of Factor IXaβ, on reduced gels. The latter material was reacted with a 100-fold molar excess of dansyl-glu-gly-arg chloromethylketone (Calbiochem) for 3 h at 37°C, and the mixture was dialyzed overnight at 4°C versus 20,000 volumes of PBS. The final product, Factor IXai, was devoid of procoagulant activity, migrated identically to untreated Factor IXa on SDS-PAGE, and had no effect on the clotting time of plasma initiated by Factor Xa or thrombin. Factor IXai was used for experiments after filtration (0.2 μm) and chromatography on DeToxi-gel columns (Pierce Chemical Co.). These preparations had no detectable LPS at a protein concentration of 1–2 mg/ml, using the limulus amebocyte assay (Sigma Chemical Co.). For experiments in which Factor IXai was used, it was given as a single intravenous bolus at the indicated times and at the indicated doses.
Modified Cephalin Clotting Time.
Equal volumes of Factor IX–deficient plasma (American Diagnostica Inc.) and 0.144 g/100 ml celite in 0.05 M barbital buffer (Sigma Chemical Co.) were combined in silicone-coated glass tubes (Sigma Chemical Co.) for 2 min at 37°C. To this mixture, an equal volume of 1:16 (vol/vol) cephalin (10 mg/ml; Sigma Chemical Co.) in 0.05 M barbital buffer was added, followed by a one-half volume of sample plasma. After the addition of calcium chloride to a final concentration of 0.001 M, the time required for clot formation was determined.
Results
Using a murine model of middle cerebral artery occlusion (MCAO) with an intraluminal vascular suture, which is removed after 45 min to initiate reperfusion, the occurrence of microvascular thrombosis distal to the site of primary occlusion was examined. Platelet-rich thrombotic foci occur within the ischemic cerebral hemisphere, as shown by experiments in which 111In-labeled platelets were administered to mice immediately before ischemia and their accumulation in the ipsilateral hemisphere measured at 24 h. In animals not subjected to the surgical procedure to create stroke, the presence of platelets was approximately equal between the right and left hemispheres, as would be expected (Fig. 1 A, left bar). However, when animals were subjected to stroke (and received only saline vehicle for control), radiolabeled platelets preferentially accumulated in the ischemic (ipsilateral) hemisphere, compared with significantly less deposition in the contralateral (nonischemic) hemisphere (Fig. 1 A, middle bar). These data support the occurrence of platelet-rich thrombi in the ischemic territory. Another line of evidence also supports the occurrence of microvascular thrombosis in stroke. These data come from the immunodetection of fibrin, using an antibody directed against a neoepitope on the gamma-gamma chain dimer of cross-linked fibrin. Immunoblots demonstrate a band of increased intensity in the ipsilateral (right) hemisphere of vehicle-treated animals subjected to focal cerebral ischemia and reperfusion (Fig. 1 B, Vehicle). To demonstrate that fibrin accumulation was due to the deposition of intravascular fibrin (rather than due to nonspecific permeability changes and exposure to subendothelial matrix), fibrin immunostaining clearly localized the increased fibrin to the lumina of ipsilateral intracerebral microvessels (Fig. 1 C, top panels). As an in vivo physiological correlate of microvascular thrombosis, relative CBF was measured by laser doppler during the occlusive period as well as after stroke. These data (Fig. 1 D, bars labeled Vehicle) show that the intraluminal suture technique significantly reduces ipsilateral CBF during the occlusive period (Fig. 1 D, middle). Blood flow remains depressed even 24 h after removing the intraluminal occluding suture (Fig. 1 D, right), corresponding to the platelet, fibrin immunoblot, and fibrin immunostaining data indicating the presence of postischemic microvascular thrombosis.
Figure 1.




(A) The effect of stroke on the accumulation of radiolabeled platelets, and the inhibitory effects of Factor IXai on platelet accumulation. 111In-labeled platelets were administered either to control animals not subjected to stroke 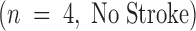 or to animals immediately before stroke treated preoperatively with vehicle
or to animals immediately before stroke treated preoperatively with vehicle 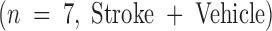 or with Factor IXai
or with Factor IXai 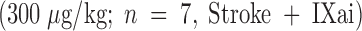 . Platelet accumulation at 24 h is expressed as the ipsilateral cpm/contralateral cpm. Means ± SEM are shown. *P < 0.05 vs. No Stroke and vs. Stroke + IXai. (B) Accumulation of fibrin in infarcted cerebral tissue. After 45 min of right MCAO and 24 h of reperfusion, brains were harvested from representative mice which had been treated before surgery with either vehicle (left lanes) or Factor IXai (300 μg/kg, right lanes). The brains were divided into ipsilateral (R) and contralateral (L) hemispheres, and plasmin digestion was performed to solubilize accumulated fibrin. Immunoblotting was performed using a primary antibody directed against a neoepitope expressed on the gamma-gamma chain dimer of cross-linked fibrin. (C) Immunohistochemical identification of sites of fibrin formation in stroke. Using the same procedures described in B, brains were harvested at 24 h, formalin fixed/paraffin embedded, and fibrin was detected immunohistochemically using the antifibrin antibody. Cerebral microvessels, shown in the center of each field, stained prominently for fibrin (sepia) in the ipsilateral hemisphere of vehicle-treated animals (top right). In contrast, microvessels from the ipsilateral hemisphere of Factor IXai–treated mice rarely demonstrated intravascular fibrin (bottom right). (D) Effect of Factor IXai on CBF in a murine stroke model. Serial measurements of relative CBF were made using a laser doppler over precisely defined neuroanatomic landmarks (reference 21), expressed as ipsilateral/contralateral CBF. Experiments were performed as described in B;
. Platelet accumulation at 24 h is expressed as the ipsilateral cpm/contralateral cpm. Means ± SEM are shown. *P < 0.05 vs. No Stroke and vs. Stroke + IXai. (B) Accumulation of fibrin in infarcted cerebral tissue. After 45 min of right MCAO and 24 h of reperfusion, brains were harvested from representative mice which had been treated before surgery with either vehicle (left lanes) or Factor IXai (300 μg/kg, right lanes). The brains were divided into ipsilateral (R) and contralateral (L) hemispheres, and plasmin digestion was performed to solubilize accumulated fibrin. Immunoblotting was performed using a primary antibody directed against a neoepitope expressed on the gamma-gamma chain dimer of cross-linked fibrin. (C) Immunohistochemical identification of sites of fibrin formation in stroke. Using the same procedures described in B, brains were harvested at 24 h, formalin fixed/paraffin embedded, and fibrin was detected immunohistochemically using the antifibrin antibody. Cerebral microvessels, shown in the center of each field, stained prominently for fibrin (sepia) in the ipsilateral hemisphere of vehicle-treated animals (top right). In contrast, microvessels from the ipsilateral hemisphere of Factor IXai–treated mice rarely demonstrated intravascular fibrin (bottom right). (D) Effect of Factor IXai on CBF in a murine stroke model. Serial measurements of relative CBF were made using a laser doppler over precisely defined neuroanatomic landmarks (reference 21), expressed as ipsilateral/contralateral CBF. Experiments were performed as described in B; 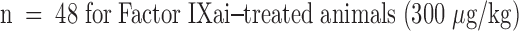 ,
, 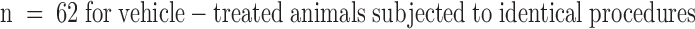 . Means ± SEM are shown. *P < 0.05.
. Means ± SEM are shown. *P < 0.05.
To help establish a functionally deleterious role of microvascular thrombosis in stroke, experiments were performed to test the effect of inhibiting assembly of the Factor IXa/VIIIa/X activation complex in vivo. This particular strategy was selected based on the hypothesis that inhibition of Factor IXa participation in coagulation might inhibit intravascular thrombosis yet not impair tissue Factor VIIa/Xa–mediated extravascular hemostasis (and hence, may not increase ICH at clinically effective doses). An estimate of the antithrombotic potency of Factor IXai was obtained by testing mouse plasma in a modified cephalin clotting time assay (MCCT assay, in which the activity of Factor IXa is a rate-limiting step in thrombus formation) at timed intervals after bolus administration of Factor IXai or control agents. Because of the limited quantity of murine plasma obtained from each sacrificial bleed, plasma was obtained from individual control mice each day this assay was performed (rather than using pooled plasma). Although MCCT control values in mice varied slightly from day to day, the approximate mean control MCCT (for the 15-min postadministration time point) was 150 ± 6 s (range 108–200 s). After administration, Factor IXai demonstrated antithrombotic potency similar to heparin, prolonging the time to clot formation in this assay compared with control animals that had received a normal saline bolus (Fig. 2 A). The effect of Factor IXai to prolong clotting time in this assay was dose dependent (Fig. 2 B). To test the in vivo efficacy of Factor IXai in the setting of stroke, Factor IXai was administered to mice immediately before stroke, and effects on cerebral microvascular thrombosis, infarct volume, and ICH were examined. When Factor IXai (300 μg/kg) is administered to animals before introduction of the intraluminal occluding suture, there is a significant reduction in the accumulation of radiolabeled platelets in the ipsilateral hemisphere (Fig. 1 A, right bar), no apparent increase in the ipsilateral accumulation of fibrin (Fig. 1 B, Factor IXai), and decreased evidence of intravascular fibrin by immunostaining (Fig. 1 C). In addition, there is a significant increase in postischemic blood flow by this treatment, albeit not completely to preischemic levels (Fig. 1 D).
Figure 2.


MCCT to evaluate the antithrombotic effects of intravenous Factor IXai. (A) Antithrombotic effects of Factor IXai compared with heparin. Vehicle  , Factor IXai
, Factor IXai 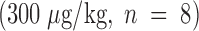 , or heparin
, or heparin 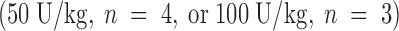 was administered to mice as an intravenous bolus, plasma was obtained at various time points after administration, and the time to clot formation was measured in the MCCT assay described in Materials and Methods. Relative time to clot formation was determined as the ratio of the time to clot formation of a given experimental condition relative to the mean time to clot formation of vehicle-treated controls. Means ± SEM are shown. (B) Dose-dependent antithrombotic effects of Factor IXai. Mice were given an intravenous single bolus of either vehicle
was administered to mice as an intravenous bolus, plasma was obtained at various time points after administration, and the time to clot formation was measured in the MCCT assay described in Materials and Methods. Relative time to clot formation was determined as the ratio of the time to clot formation of a given experimental condition relative to the mean time to clot formation of vehicle-treated controls. Means ± SEM are shown. (B) Dose-dependent antithrombotic effects of Factor IXai. Mice were given an intravenous single bolus of either vehicle  or Factor IXai at doses of 150 μg/kg
or Factor IXai at doses of 150 μg/kg  , 300 μg/kg
, 300 μg/kg  , 600 μg/kg
, 600 μg/kg  , or 1,200 μg/kg
, or 1,200 μg/kg  , and plasma was obtained at 45 min after administration. Time to clot formation was measured, and data were expressed as in A. *P < 0.05, ***P < 0.001 vs. vehicle-treated animals.
, and plasma was obtained at 45 min after administration. Time to clot formation was measured, and data were expressed as in A. *P < 0.05, ***P < 0.001 vs. vehicle-treated animals.
The clinical relevance of these observations is underscored by the striking ability of Factor IXai to reduce cerebral infarct volumes (3.3-fold reduction in infarct volumes at the 300 μg/kg dose, P < 0.05; Fig. 3 A). To test whether this infarct size-reducing property of Factor IXai was unique to this compound, or whether a nonspecific anticoagulant would also demonstrate efficacy in this regard, intravenous heparin was also examined at two doses. Only at the highest dose tested (100 U/kg) did heparin reduce cerebral infarct volumes; however, this was at the cost of a significant increase in ICH, measured with a recently validated spectrophotometric assay (23; Fig. 3 B). In sharp contrast, Factor IXai caused an increase in ICH only at the highest dose tested, but did not do so at doses that demonstrated striking efficacy to reduce cerebral infarct volumes (Fig. 3 B). Because a desirable therapeutic agent in stroke will not only reduce cerebral infarction volumes, but will also minimize ICH, the data shown in Fig. 3a and Fig. b, are displayed with infarct volumes plotted along the ordinate and ICH plotted along the abscissa (Fig. 3 C). As can be seen in the figure, Factor IXai is able to minimize both infarction volumes and ICH (bottom left corner), whereas only the high dose heparin is able to reduce infarct volumes, but at the cost of increasing ICH.
Figure 3.



Effect of Factor IXai on infarct volume and ICH in a murine stroke model. (A) Effect of Factor IXai on cerebral infarct volumes, measured by TTC staining of serial coronal sections of brain. Before stroke, animals were given either vehicle  , Factor IXai at 150 μg/kg
, Factor IXai at 150 μg/kg  , 300 μg/kg
, 300 μg/kg  , 600 μg/kg
, 600 μg/kg  , or 1,200 μg/kg
, or 1,200 μg/kg  , or heparin at 50 U/kg
, or heparin at 50 U/kg  or 100 U/kg
or 100 U/kg  . Means ± SEM are shown. *P < 0.05 vs. vehicle-treated animals. (B) Effect of Factor IXai on ICH 24 h after stroke, as measured by a quantitative spectrophotometric hemoglobin assay (reference 23), in which OD at 550 nm is linearly related to brain hemoglobin content. Relative OD was determined as the ratio of the OD of a given experimental condition relative to the mean OD of vehicle-treated animals. Before stroke, animals were given either vehicle
. Means ± SEM are shown. *P < 0.05 vs. vehicle-treated animals. (B) Effect of Factor IXai on ICH 24 h after stroke, as measured by a quantitative spectrophotometric hemoglobin assay (reference 23), in which OD at 550 nm is linearly related to brain hemoglobin content. Relative OD was determined as the ratio of the OD of a given experimental condition relative to the mean OD of vehicle-treated animals. Before stroke, animals were given either vehicle  , Factor IXai at 150 μg/kg
, Factor IXai at 150 μg/kg  , 300 μg/kg
, 300 μg/kg  , 600 μg/kg
, 600 μg/kg  , or 1,200 μg/kg
, or 1,200 μg/kg  , or heparin at 50 U/kg
, or heparin at 50 U/kg  or 100 U/kg
or 100 U/kg  . Means ± SEM are shown. *P < 0.05 vs. vehicle-treated animals. (C) Infarct volume to ICH plot of data shown in A and B. Infarct volumes were plotted against ICH to display the effects on infarct volume and ICH simultaneously for a given agent at specified doses. V, vehicle; H, heparin; IXai, Factor IXai; doses are shown. Significant values are shown in A and B, but are omitted here for clarity.
. Means ± SEM are shown. *P < 0.05 vs. vehicle-treated animals. (C) Infarct volume to ICH plot of data shown in A and B. Infarct volumes were plotted against ICH to display the effects on infarct volume and ICH simultaneously for a given agent at specified doses. V, vehicle; H, heparin; IXai, Factor IXai; doses are shown. Significant values are shown in A and B, but are omitted here for clarity.
To compare these results with a current therapy for clinical stroke in humans, tPA, experiments were performed in which tPA was administered to mice subjected to stroke and reperfusion. Intravenous tPA at doses of 0.5, 1.0, or 2.0 mg/kg 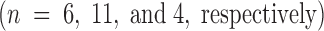 or vehicle
or vehicle  were administered to mice in the postocclusion period immediately after withdrawal of the occluding suture. Data were not collected for animals treated with tPA before occlusion because of excessive bleeding associated with the operative procedure mandated by the stroke model. At the three doses examined, tPA demonstrated only trends towards reductions in infarct size compared with vehicle-treated control animals (1.9-, 1.6-, and 1.3-fold reductions for the 0.5, 1.0, and 2.0 mg/kg doses, respectively); however, none of these reductions was statistically significant. On the other hand, administration of tPA at all doses caused statistically significant increases in ICH
were administered to mice in the postocclusion period immediately after withdrawal of the occluding suture. Data were not collected for animals treated with tPA before occlusion because of excessive bleeding associated with the operative procedure mandated by the stroke model. At the three doses examined, tPA demonstrated only trends towards reductions in infarct size compared with vehicle-treated control animals (1.9-, 1.6-, and 1.3-fold reductions for the 0.5, 1.0, and 2.0 mg/kg doses, respectively); however, none of these reductions was statistically significant. On the other hand, administration of tPA at all doses caused statistically significant increases in ICH 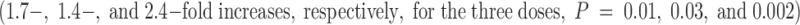 . Therefore, these data showed no significant reductions in cerebral infarction volumes (although there were trends in this direction) and increased ICH with tPA. These data are in concordance with the recent report that tPA given to tPA−/− or wild-type mice does not improve and may exacerbate cerebral injury in stroke 24.
. Therefore, these data showed no significant reductions in cerebral infarction volumes (although there were trends in this direction) and increased ICH with tPA. These data are in concordance with the recent report that tPA given to tPA−/− or wild-type mice does not improve and may exacerbate cerebral injury in stroke 24.
Because therapies directed at improving outcome from acute stroke must be given after clinical presentation, and because fibrin continues to form after the initial ischemic event in stroke, we tested whether Factor IXai might be effective when given after initiation of cerebral ischemia. Factor IXai given after MCAO (after removal of the occluding suture) provided significant cerebral protection judged by its ability to significantly reduce cerebral infarction volumes compared with vehicle-treated controls (Fig. 4).
Figure 4.

Effect of timing of Factor IXai administration on cerebral infarct volumes. Mice were either pretreated with intravenous vehicle  or Factor IXai
or Factor IXai 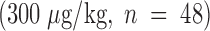 before focal cerebral ischemia and reperfusion, or immediately upon withdrawal of the intraluminal middle cerebral arterial occluding suture
before focal cerebral ischemia and reperfusion, or immediately upon withdrawal of the intraluminal middle cerebral arterial occluding suture 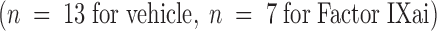 . Cerebral infarct volumes were determined from TTC-stained serial cerebral sections. Means ± SEM are shown. *P < 0.05 vs. similarly vehicle-treated animals. (The preocclusion administration data are the same data as those shown in Fig. 3 A for the 300 μg/kg dose, but is repeated here to facilitate comparison with the postreperfusion data.)
. Cerebral infarct volumes were determined from TTC-stained serial cerebral sections. Means ± SEM are shown. *P < 0.05 vs. similarly vehicle-treated animals. (The preocclusion administration data are the same data as those shown in Fig. 3 A for the 300 μg/kg dose, but is repeated here to facilitate comparison with the postreperfusion data.)
Discussion
The data in these studies demonstrate clear evidence of intravascular thrombus formation (both platelets and fibrin) within the postischemic cerebral microvasculature. In fact, the ability of an anticoagulant such as Factor IXai to improve outcome even when given after the onset of the reperfusion phase suggests that the process of microvascular thrombosis is not limited to that which occurs during the major occlusive event. Rather, microvascular thrombosis appears to be a dynamic process that continues to evolve even after recanalization of the major vascular tributary. The pathophysiological relevance of microvascular thrombosis in stroke is underscored by the ability of Factor IXai to reduce microvascular thrombosis (both platelet and fibrin accumulation are reduced, with an attendant increase in postischemic CBF) and to improve stroke outcome. At clinically relevant doses, treatment with Factor IXai does not cause an increase in ICH, in sharp contrast to tPA in this same model of stroke, in which tPA did not significantly reduce infarct volumes and also increased the degree of ICH. These data suggest that selective inhibition of the Factor IXa/VIIIa/X activation complex assembly with Factor IXai is a logical target for stroke therapy in humans. In addition, the potent antithrombotic actions of Factor IXai are likely to be clinically significant in the setting of stroke, because Factor IXai reduces infarct volumes even when given after the onset of stroke.
There are several reasons why targeted anticoagulant strategies might be superior to the current use of thrombolytic agents in the management of acute stroke, which have had checkered success in clinical trials. Theoretically, an ideal treatment for acute stroke would prevent the formation or induce dissolution of the fibrin-platelet mesh that causes microvascular thrombosis in the ischemic zone without increasing the risk of ICH. However, thrombolytic agents that have been studied in clinical trials of acute stroke have consistently increased the risk of ICH 1 2 3 4 5. Streptokinase, given in the first several (<6) hours after stroke onset, was associated with an increased rate of hemorrhagic transformation (up to 67%); although there was increased early mortality, surviving patients suffered less residual disability. A recent meta-analysis of evidence on thrombolytic therapy for acute ischemic stroke shows that when the major tPA trials are considered, there was a 2.99-fold increase in symptomatic ICH, and when all thrombolytic trials were analyzed, there is a 3.62-fold increase in symptomatic ICH 5. In addition to the potential increased hemorrhagic risk with tPA, there is also the risk of therapeutic failure; platelets continue to be activated during administration of tPA, which may account for some of the therapeutic failures observed with tPA administration. In addition, tPA is short-lived, which may limit its usefulness if microvascular thrombus continues to accrue well beyond its therapeutic half-life. Although tPA is the best among available thrombolytic agents in terms of improving morbidity in clinical stroke, there remains the concern that tPA has been shown to directly mediate excitotoxic neuronal cell injury via extracellular tPA-catalyzed proteolysis of nonfibrin substrates 6 7 8 9 10 11.
Because of the usually precipitous onset of ischemic stroke, therapy has been targeted primarily towards lysing the major fibrinous/atheroembolic debris that occludes a major vascular tributary to the brain. However, the current work reinforces the previous observation 18 that there is an important component of microvascular thrombosis that occurs downstream from the site of original occlusion. This is likely to be of considerable pathophysiological significance for postischemic hypoperfusion (no-reflow) and cerebral injury in evolving stroke. These data are in excellent agreement with those previously reported, in which microthrombi have been topographically localized to the ischemic region in fresh brain infarcts 25. These data, along with those in the current manuscript, show a critical role for platelet accumulation at these downstream sites in cerebral microvascular thrombosis. It is not surprising that Factor IXai inhibits platelet accumulation in stroke, because Factor IXa has an integral role in promoting coagulation via the intrinsic pathway: Factor IXai competes with native Factor IXa for assembly into the tenase complex, and therefore causes competitive inhibition of tenase complex formation. Although this mechanism theoretically should not interfere directly with platelet adhesion, in vivo, coagulation reactions, platelet activation, and leukocyte recruitment all occur in close proximity (as well as in proximity to the vessel wall) and are highly interdependent. This is especially likely to be true in cerebral microvessels after ischemia, where blood flow and dissipation of activated products will be sluggish. Therefore, it is likely that local generation of thrombin (by Factor IXa–dependent coagulation) will locally activate and recruit platelets, as thrombin is a potent activator of platelets.
The studies shown here demonstrate that Factor IXa–mediated coagulation does participate in platelet recruitment, because when Factor IXa–dependent coagulation is inhibited, platelet recruitment is reduced by nearly half. Our data do not allow us to extrapolate that initial platelet activation is the sole cause of postischemic microvascular thrombosis; rather, it is likely that the phenotype of the endovascular wall changes, perhaps by diminution of nitric oxide levels, perhaps by tissue factor expression in recruited mononuclear phagocytes, perhaps by alterations in the fibrinolytic balance, all of which lead to a prothrombotic phenotype. Under these circumstances, even inactivated platelets passing by may become activated and deposit locally. Regardless of the relative importance of platelet accumulation versus fibrin formation in the development of microvascular thrombosis in stroke, the data are clear that the use of an agent that inhibits assembly of the Factor IXa/VIIIa/X activation complex represents an effective approach to limiting thrombosis that occurs within microvascular lumena. In the setting of murine stroke, treatment with Factor IXai reduces microvascular platelet and fibrin accumulation, improves postischemic CBF, and reduces cerebral infarct volumes. This approach is even more salient in stroke because it is effective without impairing extravascular hemostasis, the maintenance of which may be critical for preventing ICH.
The potency of Factor IXai as an inhibitor of coagulation stems from the integral role of activated Factor IX in the coagulation cascade. Patients with hemophilia B (“Christmas disease”) are deficient in Factor IX and exhibit hemorrhagic tendencies 26. However, inhibition of Factor IXa–mediated coagulation may be therapeutically useful in discrete circumstances. For the studies shown here, active site–blocked Factor IXa was shown to be a competitive inhibitor of Factor IXa–mediated coagulation in vitro using an MCCT assay instead of the standard activated partial thromboplastin time (APTT). The MCCT assay was used because the sensitivity of the APTT is not sufficient to detect the anticoagulant effect of IXai; for example, administration of Factor IXai (300 μg/kg) did not significantly alter the APTT (79.9 ± 8.9 vs. 70.6 ± 8.9 s APTT for IXai-treated 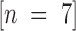 and vehicle-treated
and vehicle-treated 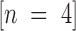 mice, respectively;
mice, respectively; 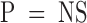 ). To increase the sensitivity of the standard APTT, the amount of “phospholipid” (cephalin) in the incubation mixture in the MCCT was decreased; theoretically, this resulted in a limiting amount of phospholipid. Using the MCCT, studies showed that increased levels of IXai prolonged the clotting time in a Factor IXai dose-dependent manner. The fact that there is a dose-dependent inhibition of Factor IXa–mediated coagulation by Factor IXai is not unexpected, because Factor IXai acts as a competitive inhibitor of assembly of the tenase complex. We would expect that after a point (that at which all Factor IXa activity is inhibited), we would see no further anticoagulant effect; however, the in vivo dose–response curves show that up to a dose of 1,200 μg/kg, we are not at that point.
). To increase the sensitivity of the standard APTT, the amount of “phospholipid” (cephalin) in the incubation mixture in the MCCT was decreased; theoretically, this resulted in a limiting amount of phospholipid. Using the MCCT, studies showed that increased levels of IXai prolonged the clotting time in a Factor IXai dose-dependent manner. The fact that there is a dose-dependent inhibition of Factor IXa–mediated coagulation by Factor IXai is not unexpected, because Factor IXai acts as a competitive inhibitor of assembly of the tenase complex. We would expect that after a point (that at which all Factor IXa activity is inhibited), we would see no further anticoagulant effect; however, the in vivo dose–response curves show that up to a dose of 1,200 μg/kg, we are not at that point.
In addition to its clear-cut efficacy in stroke, active site–blocked Factor IXa has also been shown to be useful in several other quite different in vivo models. In cardiopulmonary bypass, administration of Factor IXai alone (without heparin) was sufficient to maintain patency of the circuit 27. Factor IXai also appears to be effective at preventing progressive coronary artery occlusion induced after the initial application of electric current to the left circumflex coronary artery in dogs 28. This is consistent with the high thrombotic potency of Factor IXa in a Wessler stasis model 29. On the other hand, any new therapy for stroke should be greeted with cautious enthusiasm. Although the therapeutic window for Factor IXai is high (doses that increase ICH are substantially higher than those required for therapeutic efficacy), there is a potential for excessive inhibition of Factor IXa to promote ICH. For instance, protease nexin-2/amyloid beta protein precursor is a potent inhibitor of Factor IXa which accumulates extensively in the cerebral blood vessels of patients with amyloidosis Dutch-type with hereditary cerebral hemorrhage and may be a factor in the development of spontaneous ICH in these patients 30.
The data that demonstrate that IXai given after the onset of stroke is effective lead to another interesting hypothesis, that the formation of thrombus represents a dynamic equilibrium between the processes of ongoing thrombosis and ongoing fibrinolysis. Even under normal (nonischemic) settings, this dynamic equilibrium has been shown to occur in humans 31. The data in the current studies, which show that Factor IXai is effective even when administered after the onset of stroke, suggest that this strategy restores the dynamic equilibrium, which is shifted after cerebral ischemia to favor thrombosis, back towards a more quiescent (antithrombotic) vascular wall phenotype.
As a final consideration, even if thrombolysis successfully removes the major occluding thrombus, and/or anticoagulant strategies are effective to limit progressive microcirculatory thrombosis, blood flow usually fails to return to preischemic levels. This is exemplified by data in this study, in which although CBF is considerably improved by Factor IXai (which limits fibrin/platelet accumulation), CBF still does not return to preischemic levels. These data support the existence of multiple effector mechanisms for postischemic cerebral hypoperfusion, including postischemic neutrophil accumulation and consequent microvascular plugging with enhanced P-selectin and ICAM-1 expression by cerebral microvascular endothelial cells 19 20. Even when these adhesion receptors are absent, as is the case in mice deletionally mutant for these receptors, CBF levels are improved after stroke compared with controls but do not return to preischemic levels. These data show that both leukocytes and thrombosis play a role in postischemic cerebral no-reflow, although the interactions between leukocyte and platelet recruitment and thrombosis in vivo are likely to be highly complex, with both positive 32 33 34 and negative 35 interactions.
In summary, administration of a competitive inhibitor of Factor IXa, active site–blocked Factor IXa, represents a novel therapy for the treatment of stroke. This therapy not only reduces microcirculatory thrombosis, improves postischemic CBF, and reduces cerebral tissue injury after stroke, it can do so even if given after the onset of cerebral ischemia and without increasing the risk of ICH. This combination of infarct size reduction and relatively low downside risk of ICH makes this an extremely attractive approach for further testing and potential clinical trials in human stroke.
Acknowledgments
We thank Danny Batista and Hui Liao for their expert technical assistance.
This study was supported in part by the U.S. Public Health Service (grants R01 HL59488, R01 HL55397, and K08 NS02038), the American Heart Association (New York City Affiliate Grant-in-Aid, National American Heart Association Medical Student Research Fellowship to B.L. Hoh, and Clinician Scientist Award to D.J. Pinsky), the New York Academy of Medicine (Glorney-Raisbeck Medical Student Award to B.L. Hoh), and the American Association of Neurological Surgeons (Young Clinician Investigator Award to E.S. Connolly, Jr.).
Footnotes
1used in this paper: APTT, activated partial thromboplastin time; CBF, cerebral blood flow; Factor IXai, active site–blocked Factor IXa; ICH, intracerebral hemorrhage; MCAO, middle cerebral artery occlusion; MCCT, modified cephalin clotting time; tPA, tissue plasminogen activator; TTC, triphenyl tetrazolium chloride
References
- The National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- Hacke W., Kaste M., Fieschi C., Toni D., Lesaffre E., von Kummer R., Boysen G., Bluhmki E., Hoxter G., Mahagne M.H., Hennerici M. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study. JAMA (J. Am. Med. Assoc.). 1995;274:1017–1025. [PubMed] [Google Scholar]
- del Zoppo G.J. Acute stroke—on the threshold of a therapy? N. Engl. J. Med. 1995;333:1632–1633. doi: 10.1056/NEJM199512143332410. [DOI] [PubMed] [Google Scholar]
- The Multicenter Acute Stroke Trial—Europe Study Group Thrombolytic therapy with streptokinase in acute ischemic stroke. N. Engl. J. Med. 1996;335:145–150. doi: 10.1056/NEJM199607183350301. [DOI] [PubMed] [Google Scholar]
- Wardlaw J.M., Warlow C.P., Counsell C. Systematic review of evidence on thrombolytic therapy for acute ischaemic stroke. Lancet. 1997;350:607–614. doi: 10.1016/s0140-6736(97)03022-5. [DOI] [PubMed] [Google Scholar]
- Tsirka S.E., Bugge T.H., Degen J.L., Strickland S. Neuronal death in the central nervous system demonstrates a non-fibrin substrate for plasmin. Proc. Natl. Acad. Sci. USA. 1997;94:9779–9781. doi: 10.1073/pnas.94.18.9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka S.E., Rogove A.D., Bugge T.H., Degen J.L., Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J. Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korninger C., Collen D. Studies on the specific fibrinolytic effect of human extrinsic (tissue-type) plasminogen activator in human blood and in various animal species in vitro. Thromb. Haemost. 1981;46:561–565. [PubMed] [Google Scholar]
- Tsirka S.E., Rogove A.D., Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- Sappino A.-P., Madani R., Huarte J., Belin D., Kiss J.Z., Wohiwend A., Vassalli J.-D. Extracellular proteolysis in the adult murine brain. J. Clin. Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsirka S.E., Gualandris A., Amaral D.G., Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Duke R.J., Bloch R.F., Turpie A.G.G., Trebilcock R., Bayer N. Intravenous heparin for the prevention of stroke progression in acute partial stable strokea randomized controlled trial. Ann. Intern. Med. 1996;105:825–828. doi: 10.7326/0003-4819-105-6-825. [DOI] [PubMed] [Google Scholar]
- Haley E.C., Jr., Kassell N.F., Torner J.C. Failure of heparin to prevent progression in progressing ischemic infarction. Stroke. 1988;19:10–14. doi: 10.1161/01.str.19.1.10. [DOI] [PubMed] [Google Scholar]
- Slivka A., Levy D. Natural history of progressive ischemic stroke in a population treated with heparin. Stroke. 1990;21:1657–1662. doi: 10.1161/01.str.21.12.1657. [DOI] [PubMed] [Google Scholar]
- Ramirez-Lassepas M., Quifiones M.R., Nino H.H. Treatment of acute ischemic strokeopen trial with continuous intravenous heparinization. Arch. Neurol. 1986;43:386–390. doi: 10.1001/archneur.1986.00520040066022. [DOI] [PubMed] [Google Scholar]
- Bogousslavsky J., Regli F. Anticoagulant-induced intracerebral bleeding in brain ischemiaevaluation in 200 patients with TIAs, emboli from the heart, and progressing stroke. Acta Neurol. Scand. 1985;71:464–471. [PubMed] [Google Scholar]
- Cerebral Embolism Study Group Immediate anticoagulation of embolic strokebrain hemorrhage and management options. Stroke. 1984;15:779–789. doi: 10.1161/01.str.15.5.779. [DOI] [PubMed] [Google Scholar]
- Choudhri T.F., Hoh B.L., Zerwes H.-G., Prestigiacomo C.J., Kim S.C., Connolly E.S., Jr., Kottirsch G., Pinsky D.J. Reduced microvascular thrombosis and improved outcome in acute murine stroke by inhibiting GP IIb/IIIa receptor–mediated platelet aggregation. J. Clin. Invest. 1998;102:1301–1310. doi: 10.1172/JCI3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E.S., Jr., Winfree C.J., Springer T.A., Naka Y., Liao H., Yan S.D., Stern D.M., Solomon R.A., Gutierrez-Ramos J.-C., Pinsky D.J. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Invest. 1996;97:209–216. doi: 10.1172/JCI118392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly E.S., Jr., Winfree C.J., Prestigiacomo C.J., Kim S.C., Choudhri T.F., Hoh B.L., Naka Y., Solomon R.A., Pinsky D.J. Exacerbation of cerebral injury in mice which express the P-selectin geneidentification of P-selectin blockade as a new target for the treatment of stroke. Circ. Res. 1997;81:304–310. doi: 10.1161/01.res.81.3.304. [DOI] [PubMed] [Google Scholar]
- Connolly E.S., Jr., Winfree C.J., Stern D.M., Solomon R.A., Pinsky D.J. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–532. doi: 10.1097/00006123-199603000-00021. [DOI] [PubMed] [Google Scholar]
- Naka Y., Chowdhury N.C., Liao H., Roy D.K., Oz M.C., Micheler R.E., Pinsky D.J. Enhanced preservation of orthotopically transplanted rat lungs by nitroglycerin but not hydralazinerequirement for graft vascular homeostasis beyond harvest vasodilation. Circ. Res. 1995;76:900–906. doi: 10.1161/01.res.76.5.900. [DOI] [PubMed] [Google Scholar]
- Choudhri T.F., Hoh B.L., Solomon R.A., Connolly E.S., Jr., Pinsky D.J. Use of a spectrophotometric hemoglobin assay to objectively quantify intracerebral hemorrhage in mice. Stroke. 1997;28:2296–2302. doi: 10.1161/01.str.28.11.2296. [DOI] [PubMed] [Google Scholar]
- Wang Y.F., Tsirka S.E., Strickland S., Steig P.E., Soriano S.G., Lipton S.A. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat. Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Heye N., Paetzold C., Steinberg R., Cervos-Navarro J. The topography of microthrombi in ischemic brain infarct. Acta Neurol. Scand. 1992;86:450–454. doi: 10.1111/j.1600-0404.1992.tb05122.x. [DOI] [PubMed] [Google Scholar]
- Taran L.D. Factor IX of the blood coagulation systema review. Biochemistry. 1997;62:685–693. [PubMed] [Google Scholar]
- Spanier T.B., Chen J.M., Oz M.C., Edwards N.M., Kisiel W., Rose E.A., Schmidt A.M. Selective anticoagulation with active site-blocked Factor IXa suggests separate roles for intrinsic and extrinsic coagulation pathways in cardiopulmonary bypass. J. Thorac. Cardiovasc. Surg. 1998;116:860–869. doi: 10.1016/S0022-5223(98)00437-1. [DOI] [PubMed] [Google Scholar]
- Benedict C.R., Ryan J., Wolitzky B., Ramos R., Gerlach M., Tijburg P., Stern D. Active site-blocked Factor IXa prevents intravascular thrombus formation in the coronary vasculature without inhibiting extravascular coagulation in a canine thrombosis model. J. Clin. Invest. 1991;88:1760–1765. doi: 10.1172/JCI115495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray E., Tubbs J., Thomas S., Oates A., Boisclair M., Kemball-Cook G., Barrowcliffe T.W. Measurement of activated factor IX in factor IX concentratescorrelation with in vivo thrombogenicity. Thromb. Haemost. 1995;73:675–679. [PubMed] [Google Scholar]
- Schmaier A.H., Dahl L.D., Rozemuller A.J., Roos R.A., Wagner S.L., Chung R., Van Nostrand W.E. Protease nexin-2/amyloid beta protein precursor. A tight-binding inhibitor of coagulation factor IXa. J. Clin. Invest. 1993;92:2540–2545. doi: 10.1172/JCI116863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossel H.L. Relative proteolysis of the fibrinogen B beta chain by thrombin and plasmin as a determinant of thrombosis. Nature. 1981;291:165–167. doi: 10.1038/291165a0. [DOI] [PubMed] [Google Scholar]
- Furie B., Furie B.C. The molecular basis of platelet and endothelial cell interaction with neutrophils and monocytesrole of P-selectin and the P-selectin ligand, PSGL-1. Thromb. Haemost. 1995;74:224–227. [PubMed] [Google Scholar]
- Palabrica T., Lobb R., Furie B.C., Aronovitz M., Benjamin C., Hsu Y.-M., Sajer S.A., Furie B. Leukocyte accumulation promoting fibrin deposition is mediated in vivo by P-selectin. Nature. 1992;359:848–851. doi: 10.1038/359848a0. [DOI] [PubMed] [Google Scholar]
- Larsen E., Celi A., Gilbert G., Furie B.C., Erban J., Bonfanti R., Wagner D.D., Furie B. PADGEM proteina receptor that mediates the interaction of activated platelets with neutrophils and monocytes. Cell. 1989;59:305–312. doi: 10.1016/0092-8674(89)90292-4. [DOI] [PubMed] [Google Scholar]
- Valles J., Santos M.T., Marcus A.J., Safier L.B., Broekman M.J., Islam N., Ullman H.L., Aznar J. Downregulation of human platelet reactivity by neutrophilsparticipation of lipoxygenase derivatives and adhesive proteins. J. Clin. Invest. 1993;92:1357–1365. doi: 10.1172/JCI116709. [DOI] [PMC free article] [PubMed] [Google Scholar]