

Abstract
Hepatitis C virus (HCV) is thought to be involved in the pathogenesis of autoimmune hepatitis (AIH) type 2, which is defined by the presence of type I antiliver kidney microsome autoantibodies directed mainly against cytochrome P450 (CYP)2D6 and by autoreactive liver infiltrating T cells. Virus-specific CD8+ cytotoxic T lymphocytes (CTLs) that recognize infected cells and contribute to viral clearance and tissue injury during HCV infection could be involved in the induction of AIH. To explore whether the antiviral cellular immunity may turn against self-antigens, we characterized the primary CTL response against an HLA-A*0201–restricted HCV-derived epitope, i.e., HCV core 178–187, which shows sequence homology with human CYP2A6 and CYP2A7 8–17. To determine the relevance of these homologies for the pathogenesis of HCV-associated AIH, we used synthetic peptides to induce primary CTL responses in peripheral blood mononuclear cells of healthy blood donors and patients with chronic HCV infection. We found that the naive CTL repertoire of both groups contains cross-reactive CTLs inducible by the HCV peptide recognizing both CYP2A6 and CYP2A7 peptides as well as endogenously processed CYP2A6 protein. Importantly, we failed to induce CTLs with the CYP-derived peptides that showed a lower capacity to form stable complexes with the HLA-A2 molecule. These findings demonstrate the potential of HCV to induce autoreactive CD8+ CTLs by molecular mimicry, possibly contributing to virus-associated autoimmunity.
Keywords: hepatitis C-like viruses; autoimmunity; hepatitis, autoimmune; HLA-A2 antigen; antigens, viral
Autoimmune hepatitis (AIH)1 is a chronic liver disease of unknown etiology characterized by a persistent inflammatory reaction in the liver. The hallmarks of AIH are high titers of serum autoantibodies against different autoantigens and the presence of a hepatic, predominantly mononuclear cell infiltrate associated with liver cell damage. Several reports have shown the presence of autoreactive T cells in AIH patients. CD4+ or CD8+ liver-infiltrating T cells proliferate in response to autologous hepatocytes and some of them express high cytolytic activity 1. Peripheral T cell clones from AIH patients also proliferate in response to liver-specific lipoprotein and the asialoglycoprotein receptor 2. Type 2 AIH is defined by the presence of type I antiliver kidney microsome antibodies (LKM-1) recognizing cytochrome P450 (CYP)2D6 3. In the liver and blood of patients with AIH type 2, CD4+ and CD8+ autoreactive T cells recognizing CYP2D6 have been detected, indicating a role of T cells in the pathogenesis of this disease 4 5. AIH type 2 appears to be epidemiologically linked to hepatitis C virus (HCV) infection. Some patients with AIH type 2 have been shown to be positive for anti-HCV antibodies 6 and also for HCV RNA 7, suggesting that HCV may cause a secondary autoimmune response. MHC class I–restricted CD8+ CTLs are a major defense mechanism in viral infections. It has been suggested that the CTL response may contribute to viral clearance as well as to liver cell injury during HCV infection and might therefore play a role in the induction of autoreactive antibodies and CD4+ helper T cells. CTLs recognize endogenously processed antigenic peptides in combination with MHC class I molecules presented on the cell surface. Several HCV-derived immunogenic CTL epitopes have been described thus far 8 9 10 11 12 13. Amino acid sequence comparison of HLA-A2–restricted HCV-derived CTL epitopes revealed the presence of two human CYP sequences related to HCV core 178–187, i.e., CYP2A6 8–17 and CYP2A7 8–17 (Table ). Compared with the HCV core 178 epitope, they display an eight amino acid sequence identity, one conservative substitution at the NH2-terminal anchor position (Leu to Val), and one nonconservative substitution at position 6 (Ser to Val or Ser to Ala). Both CYP sequences still contain the HLA-A2 binding motif, because both Leu and Val can serve as anchor residues 14 15. Therefore, the HCV core 178 peptide is a candidate for molecular mimicry, that is, similarity of infectious agents with host antigens. Such similarity may lead to an inability of the host immune system to recognize the foreign antigen or it may lead to an autoreactive immune response at the level of antibodies or T cells 16 17 18.
Table 1.
HCV Core 178–187 and Related Human Cytochrome P450 Amino Acid Sequences
| Origin | Residues | Sequence |
|---|---|---|
| HCV-1 core | 178–187 | LLALLSCLTV |
| Cytochrome P450 2A6 | 8–17 | ·V···V···· |
| Cytochrome P450 2A7 | 8–17 | ·V···A···· |
Molecular mimicry at the level of CD4+ T cells seems to play a role in several human autoimmune diseases such as multiple sclerosis, rheumatoid arthritis, myocarditis, and herpes stromal keratitis 19 20 21 22 23. A report describing a possible molecular mimicry at the level of CD8+ T cells has also been published 24.
To determine the relevance of the sequence similarities between an HCV-derived CTL epitope and members of the CYP family for pathogenesis of HCV-associated autoimmune hepatitis, we analyzed the primary CTL repertoire, inducing primary CTL responses in the PBMCs of HCV-negative, healthy blood donors as well as those of patients with chronic HCV infection without markers for AIH. For induction, we used the HCV core 178 epitope as well as both CYP-derived peptides. CTL were then tested for their ability to recognize all three different peptide ligands.
Materials and Methods
Study Population.
12 HLA-A2–positive healthy individuals and 10 HLA-A2–positive patients with chronic hepatitis C were studied. Samples of patients with chronic HCV infection were obtained at least 6 mo after diagnosis of HCV infection. All patients repeatedly tested positive for anti-HCV antibodies, and with one exception HCV RNA was detected by PCR. Most (8 out of 10) of the patients had not been treated with IFN-α before sample collection, and the other two patients finished an ineffective IFN-α treatment before sample collection. None of the HCV patients tested positive for autoimmune hepatitis serum markers. Patients and healthy blood donors were negative for antibodies to HIV and HBV and healthy donors were also negative for anti-HCV antibodies.
Cell Lines.
The EBV-transformed B cell line JY (HLA-A*0201, -B7, -Cw7, -DR4, -Drw6, and -Dpw2) and K562 cells were cultured in RPMI 1640 medium supplemented with l-glutamine (2 mM), penicillin (50 U/ml), streptomycin (50 μg/ml), and Hepes (5 mM) containing 10% (vol/vol) heat-inactivated FCS (FCS-medium). The human B lymphoblastoid cell line AHH-1 TK+/− derived from the RPMI 1788 cell line (HLA type: A2, Aw33, B7, B14) was maintained in FCS-medium. H2A3 and h2D6 cells (AHH-1 TK+/− cells transfected with vectors coding for human CYP2A6 and CYP2D6, respectively; reference 25) were cultured in selective RPMI 1640 medium with 2 mM l-histidinol without l-histidine (Gentest Corp.) supplemented with l-glutamine (2 mM), penicillin (50 U/ml), streptomycin (50 μg/ml), and Hepes (5 mM) containing 10% (vol/vol) heat-inactivated FCS (h2-medium). AHH-1 T/K+/−, h2A3, and h2D6 cells were a gift from Charles L. Crespi (Gentest Corp., Woburn, MA).
Induction of Primary CTLs.
PBMCs from HLA-A2–positive healthy donors and HCV patients were isolated on Ficoll-Paque density gradients and washed three times in PBS containing 10% FCS-medium. 4 × 106 PBMCs were incubated with synthetic peptide (10 μg/ml; Chiron Mimotopes) in RPMI 1640 medium supplemented with l-glutamine (2 mM), penicillin (50 U/ml), streptomycin (50 μg/ml), and Hepes (5 mM) containing 10% (vol/vol) heat-inactivated human AB serum (AB-medium) for 1 h, washed once in PBS containing 10% FCS-medium, and then plated in 24-well plates at 4 × 106 cells/well in AB-medium. On day 3 and weekly thereafter, 1 ml of complete medium supplemented with rIL-2 (20 U/ml; EuroCetus B.V.) was added to each well. On day 7 and weekly thereafter, the cultures were restimulated with 106 peptide-pulsed, irradiated (7,400 rads) autologous feeder cells in 1 ml of AB-medium containing rIL-2 (20 U/ml). The cultured PBMCs were tested for CTL activity against different peptides on day 35.
Generation of Peptide-specific Cell Lines.
Peptide-specific induction cultures were depleted of CD4+ cells using negative selection according to the manufacturer's instructions (Dynabeads®; DYNAL A.S.) and plated at 100 cells per well in 96-well plates. Cells were plated in FCS-medium in the presence of PHA (1 μg/ml), rIL-2 (30 U/ml), irradiated (10,400 rads) allogeneic PBMCs (106 cells/ml), and irradiated (22,000 rads), peptide-pulsed (10 μg/ml; 1 h) JY EBV-B cells (105 cells/ml). Peptide-specific wells were expanded by restimulation in a 24-well plate as described above.
Cytotoxicity Assay.
JY target cells were incubated with synthetic peptides (10 μg/ml) in FCS-medium overnight. Target cells (peptide-pulsed JY cells or AHH-1 TK+/−-derived cells) were labeled with 100 μCi of Na2[51Cr]O4 (Amersham Pharmacia Biotech) for 1 h and washed four times with PBS containing 10% FCS-medium. Cytolytic activity was determined in a standard 4-h 51Cr-release assay using U-bottomed 96-well plates containing 2,500 targets per well. Where indicated in the figure legends, 2,500 K562 cells per well were added to reduce unspecific lysis. Percentage of cytotoxicity was determined from the formula: 100 × [(experimental release − spontaneous release)/(maximum release − spontaneous release)]. Maximum release was determined by lysis of targets with HCl. Spontaneous release was <25% of maximal release in all assays. Specific lysis was calculated as difference between lysis of targets with peptide (or plasmid) and targets without peptide (or plasmid).
In peptide titration experiments, JY target cells were incubated with various peptide concentrations for 90 min after 51Cr labeling, washed once with PBS, and used as described above. In functional MHC binding assays, JY target cells were pulsed with synthetic peptide for 90 min at indicated times before the assay, then washed twice with PBS containing 10% FCS-medium, incubated in FCS-medium until 51Cr labeling, and used as described above. CD8 dependency of target recognition was tested by addition of 10 μg/ml anti-CD8 antibody OKT8 (Ortho Diagnostic Systems Inc.) during the cytotoxicity assay.
Peptide “Stripping” by Mild Acid Treatment.
JY cells were washed twice with PBS and then put on ice for 5 min. 107 cells were then treated for 90 s with 2 ml ice-cold citric acid–Na2HPO4 buffer (a mixture of an equal volume of 0.263 M citric acid and 0.123 M Na2HPO4), pH 3.2. Immediately thereafter, the eluted cells were buffered with cold IMDM, washed with IMDM, and resuspended at 5 × 105 cells in IMDM with 1 μg/ml β2-micro-globulin (Sigma Chemical Co.).
Competition-based MHC Class I Peptide Binding Assay.
Peptides were tested for their binding affinity using the previously described peptide binding assay 26. In brief, cells were stripped (see above) and resuspended at 7 × 105 cells/ml in IMDM plus 1.5 μg/ml β2-microglobulin. A fluorescein (FL)-labeled reference peptide (FLPSDC(FL) FPSV), 25 μl (end concentration, 150 nM), was incubated with 25 μl of competitor peptide (different end concentrations) in a 96-well V-bottomed plate. 100 μl of mild acid-treated JY cells was added to these wells. The mixture was incubated for 24 h at 4°C, washed twice with PBS containing 1% BSA (PBA1%), resuspended in PBA1% containing 0.5% paraformaldehyde, and analyzed by FACScan® (Becton Dickinson).
The mean fluorescence (MF) value obtained in the wells without competitor peptide was regarded as maximal binding and equated to 0% inhibition; the MF obtained from the wells without reference peptide was equated to 100% inhibition. Percentage of inhibition of binding was calculated using the formula: [1 − (MF 150 nM reference and competitor peptide − MF no reference peptide) / (MF 150 nM reference peptide − MF no reference peptide)] × 100%.
Measurement of MHC–Peptide Complex Stability.
JY cells at a concentration of 1–2 × 106 cells/ml were incubated with 10−4 M emetine (Sigma Chemical Co.) for 1 h at 37°C to stop protein synthesis and the subsequent emergence of de novo synthesized class I molecules at the cell surface. Cells were washed twice with PBS and peptide stripped (see above). 106 cells were added to 200 μg of peptide in 1 ml and incubated for 90 min at room temperature. Cells were washed twice with ice-cold IMDM and resuspended in 1 ml IMDM. Subsequently, the cells were incubated for 0, 2, 4, and 6 h at 37°C and thereafter stained with BB7.2, an HLA-A2 confirmation-specific mAb 27, and goat anti–mouse FITC. Thereafter, the cells were fixed by resuspension in PBA1% containing 0.5% paraformaldehyde and analyzed by FACScan®. The fluorescence index (FI) was calculated as FI = (mean fluorescence sample − mean fluorescence background) / mean fluorescence background without peptide. Samples were tested in duplicate and the variation between both samples was always <10%.
Results
HLA-A2 Binding Affinity and HLA-A2–Peptide Complex Stability.
To characterize the HLA-A2 binding properties of HCV core 178 and the homologous CYP peptides, we determined their affinity to HLA-A2 and the stability of the formed HLA-A2–peptide complexes. The HCV core 178 bound to HLA-A2 with intermediate affinity as previously described 28, whereas both CYP peptides bound with low affinity (Fig. 1 A). Similar results were obtained measuring the peptide-induced stabilization of HLA-A2 molecules at the surface of transporter-associated with antigen processing–deficient T2 cells (data not shown). Determination of MHC–peptide complex stability showed that the HCV core 178 peptide was able to form stable complexes with a half-life of ∼5 h. The CYP2A6 8–17 as well as the CYP2A7 8–17 peptides dissociated much faster with half-lives of ∼1 h (Fig. 1 B).
Figure 1.


HLA-A2 binding affinities and dissociation rates of HCV core 178–187, CYP2A6 8–17, and CYP2A7 8–17 peptides. HLA-A2 binding affinities (A) and HLA-A2–peptide complex stabilities (B) of HCV core 178 (•), CYP2A6 8 (▪), and CYP2A7 8 (▴) were determined as described in Materials and Methods. In B, the lines represent linear regression.
Induction of Primary CTLs.
To determine the effect of the different HLA-A2 binding properties on T cell activation and antigen recognition by CTLs, we first analyzed the naive CTL repertoire in healthy blood donors. PBMCs from 12 healthy HCV-seronegative, HLA-A2–positive blood donors were stimulated with synthetic HCV core 178–187, CYP2A6 8–17, or CYP2A7 8–17 peptide in four replica cultures. After 5 wk, the cultures were tested for CTL activity against target cells presenting each of the three peptides. Long-term stimulation with the HCV core 178 peptide induced HCV core 178–specific CTLs in nine HCV-seronegative blood donors (Table ). In five individuals, the HCV core 178–specific CTLs not only recognized the inducing HCV peptide but also CYP2A6 and/or CYP2A7 self-peptides. A higher specific lysis of targets presenting CYP2A7 was observed in most cases. Two donors recognized all three peptides, three individuals recognized HCV core 178 and CYP2A7 8–17, four recognized HCV core 178 only, and three did not show a CTL response after stimulation with HCV core 178. Representative results of three donors are shown in Fig. 2 a. These results as well as data obtained from CTL lines derived from positive cultures (data not shown) indicate the presence of three phenotypes of HCV core 178–specific CTLs: CTLs recognizing HCV core 178 only, CTLs cross-reactive with HCV core 178 and CYP2A7, and CTLs specific for HCV core 178, CYP2A7, and CYP2A6. We did not find CTLs recognizing CYP2A6 without recognition of CYP2A7, nor cells recognizing one of the CYP peptides without recognition of HCV core 178. Importantly, no CTL response could be induced with the self-peptides CYP2A6 8–17 (Fig. 2 b) and CYP2A7 8–17 (Fig. 2 c) in any of the 12 donors tested. This fits with our observation of different MHC–peptide interactions, because peptide-induced MHC stability and immunogenicity of the peptide are strongly correlated 28.
Table 2.
Phenotype Distribution of HCV Core 178–inducible CTLs in Healthy Donors and Patients with Chronic HCV Infection
| Peptides recognized | Healthy donors(n = 12) | Chronic HCV patients(n = 10) |
|---|---|---|
| HCV core + CYP2A7 + CYP2A6 | 2 (17%) | 2 (20%) |
| HCV core + CYP2A7 | 3 (25%) | 2 (20%) |
| HCV core | 4 (33%) | 4 (40%) |
| None | 3 (25%) | 2 (20%) |
Figure 2.
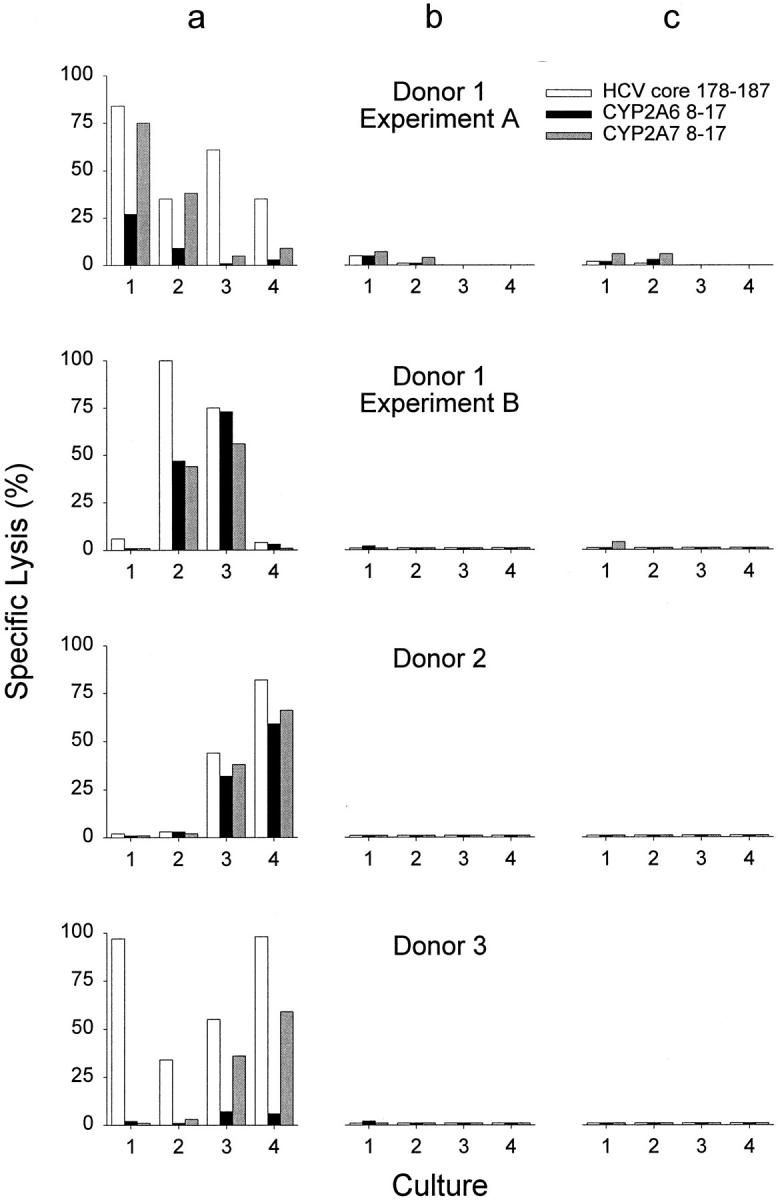
Peptide-specific cytotoxicity induced in vitro with HCV core 178–187, CYP2A6 8–17, or CYP2A7 8–17 peptides. PBMCs from healthy HCV-seronegative blood donors were stimulated with HCV core 178–187 (a), CYP2A6 8–17 (b), or CYP2A7 8-18 (c) in four replicas 1 2 3 4. After 5 wk of stimulation, peptide-specific lysis of each culture was determined on JY target cells pulsed with HCV core 178–187 (white bars), CYP2A6 8–17 (black bars), or CYP2A7 8–17 (gray bars). E/T cell ratios were fixed for each culture and ranged from 20:1 to 40:1 for the different cultures.
The same strategy was used to analyze the HCV core 178–specific CTL repertoire in patients with chronic HCV infection without markers for AIH. The percentage of patients having cross-reactive CTLs is comparable to that of healthy donors (Table ), suggesting that the peripheral pool of naive CTLs specific for HCV core 178 is comparable to the one of uninfected individuals and that the precursor frequency of cross-reactive CTLs is similar. This is not astonishing, as chronic HCV infection is associated with a low number of CTLs in the peripheral blood 29 30.
Peptide Recognition by Cross-reactive CTL Lines.
HCV core 178–induced CTL lines derived from donor 2 were used to test the biologic function of the peptides in cytotoxicity assays. Despite different MHC binding affinities and abilities to stabilize MHC complexes, all three peptides were recognized by CTLs with the same efficiency (Fig. 3 A). The higher off-rate of the CYP peptides had no effect on CTL recognition when the peptide-pulsed target cells were further incubated without peptide for up to 24 h before exposure to CTLs (Fig. 3 B). An explanation for this phenomenon could be the CD8 dependency of target recognition. Although lysis of target cells presenting HCV core 178 is markedly reduced by the anti-CD8 antibody OKT8, recognition of CYP2A7 is less affected and there is no effect on recognition of CYP2A6 (Fig. 3 C). These findings were unexpected but similar findings had been observed by al-Ramadi et al., demonstrating that the pattern of functional activities of variant peptides does not always correlate with MHC binding 31. In fact, other mechanisms involved in the interaction between the CTL and the target cell may be important, such as the TCR affinity for the MHC–peptide-complex, and CD8 binding to MHC class I, as well as other costimulatory and cell adhesion molecules 32. To assess the role of CD8 binding on CTL–target cell interaction, we used a panel of three different anti-CD8 antibodies and tested their effect on target cell lysis. A representative experiment is shown in Fig. 3 C using the OKT8 antibody. Although recognition of HCV core 178 depends in part on CD8 availability, self-peptide recognition of CYP2A6 does not. Further studies will be required to define the mechanism responsible for the discrepancy observed between peptide–MHC binding and CTL-mediated cytotoxicity.
Figure 3.



Peptide recognition by CTL lines. Cross-reactive CTL lines derived from donor 2 were tested for their ability to lyse (A) JY target cells preincubated at various concentrations of peptide and (B) JY target cells pulsed with 10 μM peptide and further incubated without peptide before exposure to CTL. Peptides were HCV core 178–187 (•), CYP2A6 8–17 (▪), and CYP2A7 8–17 (▴). (C) The effect of anti-CD8 antibodies on target cell recognition was tested by addition of OKT8 antibody (10 μg/ml) during the cytotoxicity assay. Shown are the mean ± SD of three to four replicas at an E/T ratio of 10:1.
Recognition of Endogenously Synthesized Antigen.
The ability of HCV core 178–induced CTLs to recognize endogenously synthesized CYP2A6 antigen was studied using the AHH-1 TK+/− cell line transfected with CYP2A6 (h2A3), and cells transfected with the unrelated CYP2D6 (h2D6) as target cells in 4- and 8-h cytotoxicity assays. Killing of CYP2A6-transfected cells by CTL lines from donor 2 was higher than lysis of the parental cell line AHH-1 TK+/− or control cells transfected with CYP2D6 (Fig. 4), suggesting that naturally processed CYP2A6 8–17 peptide is generated by the proteolytic machinery and presented on the HLA-A2 molecule.
Figure 4.

Recognition of endogenously processed cytochrome P450. Cross-reactive CTL lines derived from donor 2 were tested for their ability to lyse AHH-1 TK+/− target cells transfected with CYP2A6 (circles) or CYP2D6 (squares). Shown is the plasmid-specific lysis: lysis of transfected cells − lysis of untransfected cells in a 4- (filled symbols) or 8-h (open symbols) cytotoxicity assay. The 4-h assay was performed in the presence of K562 cells.
Discussion
The HCV core 176 peptide has been described as a target epitope for CTLs that are present in the peripheral blood of patients with chronic HCV infection 10. This peptide is processed and presented via the endogenous MHC class I pathway 11. We used PBMCs from healthy, HCV-seronegative individuals as well as from patients with chronic HCV infection to induce primary CTL responses against this epitope and two homologous CYP-derived peptides. CTLs induced with the HCV core 178 peptide not only recognized the inducing HCV peptide, but also showed autoreactivity, lysing targets presenting CYP-derived self-peptides and target cells stably transfected with a plasmid coding for the whole CYP2A6 protein, showing that the CYP epitope is also presented via the endogenous MHC class I pathway. Among the CTLs we could distinguish three different functional types in the same individual: cells recognizing HCV core 178 only, cells with cross-reaction between HCV core 178 and CYP2A7, and cells recognizing HCV core and both CYP epitopes, indicating a polyclonal response against HCV core 178 with a distinct hierarchy.
The induction of self-reactive CD8+ CTLs by a viral epitope is compatible with molecular mimicry (resemblance of pathogen and host antigens), a mechanism that has been described mainly at the level of antibodies and CD4+ T cells 16 17 18. Molecular mimicry at the level of T cells has been implicated in human autoimmune diseases such as multiple sclerosis 19 20, rheumatoid arthritis 21, and myocarditis 22, as well as in herpes stromal keratitis 23. A recent study also describes cross-reactivity of CD8+ T cells specific for a myelin-derived peptide with a Saccharomyces cerevisiae peptide 24. In this case, cross-reactive CTLs could only be induced with the self-peptide and not with the Saccharomyces antigen.
In our study we could not induce CTLs with the CYP-derived self-peptides, although the same self-peptides were recognized by CTLs induced with the HCV core 178 epitope. This indicates different T cell activation properties of the self-epitopes compared with the HCV peptide as described for altered peptide ligands (APLs), which are analogues of immunogenic peptides with amino acid substitutions inducing different effects in CTLs. APLs can act as agonists leading to full activation of T cells, partial agonists inducing only a reduced T cell response, or even antagonists inhibiting a response against the unaltered immunogenic epitope 33 34. The molecular mechanisms of APL-induced partial T cell activation are still a matter of debate 35. Complexes of the native ligand or APLs with MHC molecules binding to the TCR can induce different intracellular signals in T cells 36 37 38, either by different oligomerization of necessary molecules (CD3, CD8, or other molecules), or by a failure of the APL to induce a required conformational change in the TCR. Other studies state that the level of T cell activation is dependent on the number of TCRs triggered in a process of serial engagement of many TCRs by a few peptide–MHC complexes, allowing a single CTL to generate different biological responses 39. Although specific cytotoxicity is already detectable at very low peptide concentrations, IFN-γ production and proliferation require higher concentrations corresponding to higher numbers of TCRs being triggered 40. This is in accordance with the finding that the immunogenicity of antigenic peptides strongly correlates with the stability of MHC–peptide complexes formed 28, indicating that a high number and a high stability of MHC–peptide complexes is essential to trigger a sufficient number of TCRs to fully activate naive T cells. Despite this, the final effect of affinity differences or kinetic changes on the multimolecular interactions during antigen recognition cannot be predicted, as there is no strict correlation between functional activity of the various peptides and their MHC binding efficiency and the affinity of the MHC–peptide complexes for the TCR 31.
We have shown that the signal induced by the unstable complexes of HLA-A2 and CYP-derived self-peptides is not sufficient to activate naive CTLs. In the same way there is evidence that the CYP peptides neither induce negative selection during thymic development nor lead to anergy, as we detected cross-reactive CTL precursors in the peripheral blood. On the other hand, the HCV core 178 peptide that forms stable MHC–peptide complexes is able to induce full activation including maturation of CTLs. In the activated cross-reactive CTLs, cytolytic functions can then be induced by the HCV peptide, and also by the CYP-derived APLs, as cytotoxicity requires a lower threshold of activation than proliferation. Once activated, a subpopulation of cross-reactive CTLs that shows no differences in recognition of the three peptide ligands, because they recognize the HCV core 178 and the CYP peptides at similar peptide concentrations, can be found.
The clinical observation that HCV infection is preceding the development of LKM-1–positive AIH is in accordance with our hypothesis that antiviral immune response has to predate autoimmunity 41 42. Moreover, the presence of autoreactive T cells in the blood and the liver of AIH type 2 patients has been demonstrated, indicating a role for both CD4+ and CD8+ T cells in the pathogenesis of this disease 4 5. Cross-reactive CTLs may contribute to liver cell damage by lysis of infected and uninfected hepatocytes during ongoing viral infection and by lysis of uninfected autoantigen-expressing hepatocytes after viral clearance. However, the presence of virus-inducible autoreactive CTLs alone is not sufficient to lead to typical AIH, as none of the tested HCV patients had markers for AIH (LKM-1 autoantibodies). Probably the presence of autoreactive CD8+ as well as autoreactive CD4+ helper T cells is required for the induction of AIH. LKM-1 antigen (CYP2D6)–specific CD4+ T cells detected in AIH patients would be needed for the activation of B cells secreting the AIH marker LKM-1 autoantibodies. Furthermore, autoreactive CD4+ T cells could maintain the autoreactive CD8 T cell response after viral clearance, comparable with chronic viral infections where CD4+ T cells are essential for maintaining the CTL response 43 44 45 46.
In summary, several arguments underscore the biological relevance of our findings. First, the viral epitope as well as the self-epitopes are naturally processed and presented by human host cells. Second, the self-epitopes and the viral epitope are coexpressed and colocalize to the liver in natural HCV infection, and expression of CYP2A6 is even enhanced in HCV-infected livers 47. Third, the absence of CYP-inducible CTLs in the peripheral blood suggests that central or peripheral tolerance mechanisms are operational, indicating that the self-epitopes are presented to T cells in vivo. Fourth, the data presented show a high level of cross-recognition of virally induced CTLs against the self-peptides, suggesting that they can mediate liver cell damage to uninfected cells. Fifth, another line of evidence links HCV infection to autoimmune hepatitis type 2 6. This disease has been described to occur subsequent to HCV infection 42 and, interestingly, in association with HLA-A2 48. Typically, it is associated with the presence of autoantibodies directed against cytochrome P450 2D6 3 6, as well as autoreactive CYP2D6-specific T cells, suggesting the parallel occurrence of autoreactivity against cytochrome P450 at the level of B and T cells 4 5. It can be hypothesized that liver cell damage mediated by virus-specific CTLs leads to the release of intracellular proteins like CYP2D6, uptake of these autoantigens by professional APCs, and autoantibody formation in the presence of autoreactive CD4+ T and B cells. After viral clearance, the autoimmune disease is upheld by ongoing hepatocyte lysis by cross-reactive, HCV-induced CTLs maintained by autoreactive helper T cells. The coincidence of HCV, autoreactive B cells, and autoreactive CD8+ and CD4+ T cells is thus required for the induction of HCV-associated AIH. This would explain the relatively low frequency of AIH among HCV patients despite the high number of individuals with cross-reactive CTLs.
Our findings demonstrate the potential of HCV to induce autoreactive CD8+ CTLs by a molecular mimicry of CYP2A6/2A7 by the core protein and therefore show a possible mechanism by which HCV may trigger AIH. Further studies analyzing the cross-reactivity pattern of HCV core–specific CTLs derived from untreated patients with active HCV-associated autoimmune hepatitis are required in order to establish this mechanism.
Acknowledgments
We thank C.L. Crespi for providing CYP transfected and control cell lines; and F.V. Chisari, W.J. Pichler, R.M. Zinkernagel, and M. Baggiolini for helpful discussions.
This work was supported by the Swiss National Science Foundation (SNF 31-37291.93 and 32-52915.97); the Bonizzi-Theler Foundation; the Sandoz Foundation; the Helmut Horten Foundation; the Niels and Desirée Yde Foundation; the University of Bern; and the Central Laboratory Blood Transfusion Service of the Swiss Red Cross.
Footnotes
1used in this paper: AIH, autoimmune hepatitis; APL, altered peptide ligand; CYP, cytochrome P450; HCV, hepatitis C virus; LKM-1, type I antiliver kidney microsome antibodies; MF, mean fluorescence; PBA1%, PBS containing 1% BSA
This work was presented in part at the 5th International Meeting on Hepatitis C Virus and Related Viruses in Venice, Italy, June 25–28, 1998.
References
- Franco A., Barnaba V., Ruberti G., Benvenuto R., Balsano C., Musca A. Liver-derived T cell clones in autoimmune chronic active hepatitisaccessory cell function of hepatocytes expressing class II major histocompatibility complex molecules. Clin. Immunol. Immunopathol. 1990;54:382–394. doi: 10.1016/0090-1229(90)90052-r. [DOI] [PubMed] [Google Scholar]
- Wen L., Peakman M., Lobo-Yeo A., McFarlane B.M., Mowat A.P., Mieli-Vergani G., Vergani D. T-cell-directed hepatocyte damage in autoimmune chronic active hepatitis. Lancet. 1990;336:1527–1530. doi: 10.1016/0140-6736(90)93306-a. [DOI] [PubMed] [Google Scholar]
- Manns M.P., Johnson E.F., Griffin K.J., Tan E.M., Sullivan K.F. Major antigen of liver kidney microsomal autoantibodies in idiopathic autoimmune hepatitis is cytochrome P450db1. J. Clin. Invest. 1989;83:1066–1072. doi: 10.1172/JCI113949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr H., Manns M., Kyriatsoulis A., Lohse A.W., Trautwein C., Meyer zum Buschenfelde K.H., Fleischer B. Clonal analysis of liver-infiltrating T cells in patients with LKM-1 antibody-positive autoimmune chronic active hepatitis. Clin. Exp. Immunol. 1991;84:297–302. doi: 10.1111/j.1365-2249.1991.tb08164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohr H.F., Schlaak J.F., Lohse A.W., Bocher W.O., Arenz M., Gerken G., Meyer Zum Buschenfelde K.H. Autoreactive CD4+ LKM-specific and anticlonotypic T-cell responses in LKM-1 antibody-positive autoimmune hepatitis. Hepatology. 1996;24:1416–1421. doi: 10.1002/hep.510240619. [DOI] [PubMed] [Google Scholar]
- Lenzi M., Ballardini G., Fusconi M., Cassani F., Selleri L., Volta U., Zauli D., Bianchi F.B. Type 2 autoimmune hepatitis and hepatitis C virus infection. Lancet. 1990;335:258–259. doi: 10.1016/0140-6736(90)90070-l. [DOI] [PubMed] [Google Scholar]
- Garson J.A., Lenzi M., Ring C., Cassani F., Ballardini G., Briggs M., Tedder R.S., Bianchi F.B. Hepatitis C viraemia in adults with type 2 autoimmune hepatitis. J. Med. Virol. 1991;34:223–226. doi: 10.1002/jmv.1890340405. [DOI] [PubMed] [Google Scholar]
- Koziel M.J., Dudley D., Wong J.T., Dienstag J., Houghton M., Ralston R., Walker B.D. Intrahepatic cytotoxic T lymphocytes specific for hepatitis C virus in persons with chronic hepatitis. J. Immunol. 1992;149:3339–3344. [PubMed] [Google Scholar]
- Kita H., Moriyama T., Kaneko T., Harase I., Nomura M., Miura H., Nakamura I., Yazaki Y., Imawari M. HLA B44-restricted cytotoxic T lymphocytes recognizing an epitope on hepatitis C virus nucleocapsid protein. Hepatology. 1993;18:1039–1044. [PubMed] [Google Scholar]
- Cerny A., McHutchison J.G., Pasquinelli C., Brown M.E., Brothers M.A., Grabscheid B., Fowler P., Houghton M., Chisari F.V. Cytotoxic T lymphocyte response to hepatitis C virus-derived peptides containing the HLA A2.1 binding motif. J. Clin. Invest. 1995;95:521–530. doi: 10.1172/JCI117694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battegay M., Fikes J., Di Bisceglie A.M., Wentworth P.A., Sette A., Celis E., Ching W.M., Grakoui A., Rice C.M., Kurokohchi K. Patients with chronic hepatitis C have circulating cytotoxic T cells which recognize hepatitis C virus-encoded peptides binding to HLA-A2.1 molecules. J. Virol. 1995;69:2462–2470. doi: 10.1128/jvi.69.4.2462-2470.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai M., Arichi T., Nishioka M., Nomura T., Ikeda K., Kawanishi K., Engelhard V.H., Feinstone S.M., Berzofsky J.A. CTL responses of HLA-A2.1-transgenic mice specific for hepatitis C viral peptides predict epitopes for CTL of humans carrying HLA-A2.1. J. Immunol. 1995;154:2733–2742. [PubMed] [Google Scholar]
- Wentworth P.A., Sette A., Celis E., Sidney J., Southwood S., Crimi C., Stitely S., Keogh E., Wong N.C., Livingston B. Identification of A2-restricted hepatitis C virus-specific cytotoxic T lymphocyte epitopes from conserved regions of the viral genome. Int. Immunol. 1996;8:651–659. doi: 10.1093/intimm/8.5.651. [DOI] [PubMed] [Google Scholar]
- Ruppert J., Sidney J., Celis E., Kubo R.T., Grey H.M., Sette A. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell. 1993;74:929–937. doi: 10.1016/0092-8674(93)90472-3. [DOI] [PubMed] [Google Scholar]
- del Guercio M.F., Sidney J., Hermanson G., Perez C., Grey H.M., Kubo R.T., Sette A. Binding of a peptide antigen to multiple HLA alleles allows definition of an A2-like supertype. J. Immunol. 1995;154:685–693. [PubMed] [Google Scholar]
- Oldstone M.B. Molecular mimicry and autoimmune disease. Cell. 1987;50:819–820. doi: 10.1016/0092-8674(87)90507-1. [DOI] [PubMed] [Google Scholar]
- von Herrath M.G., Oldstone M.B. Virus-induced autoimmune disease. Curr. Opin. Immunol. 1996;8:878–885. doi: 10.1016/S0952-7915(96)80019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J.M. Molecular mimicrycan epitope mimicry induce autoimmune disease? Immunol. Cell. Biol. 1997;75:113–126. doi: 10.1038/icb.1997.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wucherpfennig K.W., Strominger J.L. Molecular mimicry in T cell-mediated autoimmunityviral peptides activate human T cell clones specific for myelin basic protein. Cell. 1995;80:695–705. doi: 10.1016/0092-8674(95)90348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot P.J., Paquette J.S., Ciurli C., Antel J.P., Ouellet F. Myelin basic protein and human coronavirus 229E cross-reactive T cells in multiple sclerosis. Ann. Neurol. 1996;39:233–240. doi: 10.1002/ana.410390213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eden W., Hogervorst E.J., Hensen E.J., van der Zee R., van Embden J.D., Cohen I.R. A cartilage-mimicking T-cell epitope on a 65K mycobacterial heat-shock proteinadjuvant arthritis as a model for human rheumatoid arthritis. Curr. Top. Microbiol. Immunol. 1989;145:27–43. doi: 10.1007/978-3-642-74594-2_3. [DOI] [PubMed] [Google Scholar]
- Malkiel S., Kuan A.P., Diamond B. Autoimmunity in heart diseasemechanisms and genetic susceptibility. Mol. Med. Today. 1996;2:336–342. doi: 10.1016/1357-4310(96)81799-0. [DOI] [PubMed] [Google Scholar]
- Zhao Z.S., Granucci F., Yeh L., Schaffer P.A., Cantor H. Molecular mimicry by herpes simplex virus-type 1autoimmune disease after viral infection. Science. 1998;279:1344–1347. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- Honma K., Parker K.C., Becker K.G., McFarland H.F., Coligan J.E., Biddison W.E. Identification of an epitope derived from human proteolipid protein that can induce autoreactive CD8+ cytotoxic T lymphocytes restricted by HLA-A3evidence for cross-reactivity with an environmental microorganism. J. Neuroimmunol. 1997;73:7–14. doi: 10.1016/s0165-5728(96)00161-0. [DOI] [PubMed] [Google Scholar]
- Crespi C.L., Langenbach R., Penman B.W. Human cell lines, derived from AHH-1 TK+/− human lymphoblasts, genetically engineered for expression of cytochromes P450. Toxicology. 1993;82:89–104. doi: 10.1016/0300-483x(93)90062-w. [DOI] [PubMed] [Google Scholar]
- van der Burg S.H., Ras E., Drijfhout J.W., Benckhuijsen W.E., Bremers A.J., Melief C.J., Kast W.M. An HLA class I peptide-binding assay based on competition for binding to class I molecules on intact human B cells. Identification of conserved HIV-1 polymerase peptides binding to HLA-A*0301. Hum. Immunol. 1995;44:189–198. doi: 10.1016/0198-8859(95)00105-0. [DOI] [PubMed] [Google Scholar]
- Parham P., Brodsky F.M. Partial purification and some properties of BB7.2. A cytotoxic monoclonal antibody with specificity for HLA-A2 and a variant of HLA-A28. Hum. Immunol. 1981;3:277–299. doi: 10.1016/0198-8859(81)90065-3. [DOI] [PubMed] [Google Scholar]
- van der Burg S.H., Visseren M.J., Brandt R.M., Kast W.M., Melief C.J. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J. Immunol. 1996;156:3308–3314. [PubMed] [Google Scholar]
- Koziel M.J., Dudley D., Afdhal N., Choo Q.L., Houghton M., Ralston R., Walker B.D. Hepatitis C virus (HCV)-specific cytotoxic T lymphocytes recognize epitopes in the core and envelope proteins of HCV. J. Virol. 1993;67:7522–7532. doi: 10.1128/jvi.67.12.7522-7532.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehermann B., Chang K.M., McHutchison J.G., Kokka R., Houghton M., Chisari F.V. Quantitative analysis of the peripheral blood cytotoxic T lymphocyte response in patients with chronic hepatitis C virus infection. J. Clin. Invest. 1996;98:1432–1440. doi: 10.1172/JCI118931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al-Ramadi B.K., Jelonek M.T., Boyd L.F., Margulies D.H., Bothwell A.L. Lack of strict correlation of functional sensitization with the apparent affinity of MHC/peptide complexes for the TCR. J. Immunol. 1995;155:662–673. [PubMed] [Google Scholar]
- de Vries J.E., Yssel H., Spits H. Interplay between the TCR/CD3 complex and CD4 or CD8 in the activation of cytotoxic T lymphocytes. Immunol. Rev. 1989;109:119–141. doi: 10.1111/j.1600-065x.1989.tb00022.x. [DOI] [PubMed] [Google Scholar]
- Klenerman P., Rowland-Jones S., McAdam S., Edwards J., Daenke S., Lalloo D., Koppe B., Rosenberg W., Boyd D., Edwards A. Cytotoxic T-cell activity antagonized by naturally occurring HIV-1 Gag variants. Nature. 1994;369:403–407. doi: 10.1038/369403a0. [DOI] [PubMed] [Google Scholar]
- Bertoletti A., Sette A., Chisari F.V., Penna A., Levrero M., De Carli M., Fiaccadori F., Ferrari C. Natural variants of cytotoxic epitopes are T-cell receptor antagonists for antiviral cytotoxic T cells. Nature. 1994;369:407–410. doi: 10.1038/369407a0. [DOI] [PubMed] [Google Scholar]
- Sloan-Lancaster J., Allen P.M. Altered peptide ligand-induced partial T cell activationmolecular mechanisms and role in T cell biology. Annu. Rev. Immunol. 1996;14:1–27. doi: 10.1146/annurev.immunol.14.1.1. [DOI] [PubMed] [Google Scholar]
- Hollsberg P., Weber W.E., Dangond F., Batra V., Sette A., Hafler D.A. Differential activation of proliferation and cytotoxicity in human T-cell lymphotropic virus type I Tax-specific CD8 T cells by an altered peptide ligand. Proc. Natl. Acad. Sci. USA. 1995;92:4036–4040. doi: 10.1073/pnas.92.9.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrenas J., Wange R.L., Wang J.L., Isakov N., Samelson L.E., Germain R.N. Zeta phosphorylation without ZAP-70 activation induced by TCR antagonists or partial agonists. Science. 1995;267:515–518. doi: 10.1126/science.7824949. [DOI] [PubMed] [Google Scholar]
- Reis e Sousa C., Levine E.H., Germain R.N. Partial signaling by CD8+ T cells in response to antagonist ligands. J. Exp. Med. 1996;184:149–157. doi: 10.1084/jem.184.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valitutti S., Muller S., Cella M., Padovan E., Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide-MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- Valitutti S., Muller S., Dessing M., Lanzavecchia A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J. Exp. Med. 1996;183:1917–1921. doi: 10.1084/jem.183.4.1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie F.D., Peakman M., Yun M., Sallie R., Smith H., Davies E.T., Mieli-Vergani G., Vergani D. Primary and secondary liver/kidney microsomal autoantibody response following infection with hepatitis C virus. Gastroenterology. 1994;106:1672–1675. doi: 10.1016/0016-5085(94)90426-x. [DOI] [PubMed] [Google Scholar]
- Vento S., Cainelli F., Renzini C., Ercole C. Autoimmune hepatitis type 2 induced by HCV and persisting after viral clearance. Lancet. 1997;350:1298–1299. doi: 10.1016/S0140-6736(05)62476-2. [DOI] [PubMed] [Google Scholar]
- Battegay M., Moskophidis D., Rahemtulla A., Hengartner H., Mak T.W., Zinkernagel R.M. Enhanced establishment of a virus carrier state in adult CD4+ T-cell-deficient mice. J. Virol. 1994;68:4700–4704. doi: 10.1128/jvi.68.7.4700-4704.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M., Concepcion R.J., Ahmed R. CD4+ T cells are required to sustain CD8+ cytotoxic T-cell responses during chronic viral infection. J. Virol. 1994;68:8056–8063. doi: 10.1128/jvi.68.12.8056-8063.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardin R.D., Brooks J.W., Sarawar S.R., Doherty P.C. Progressive loss of CD8+ T cell-mediated control of a gamma-herpesvirus in the absence of CD4+ T cells. J. Exp. Med. 1996;184:863–871. doi: 10.1084/jem.184.3.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zajac A.J., Blattman J.N., MuraliKrishna K., Sourdive D.J.D., Suresh M., Altman J.D., Ahmed R. Viral immune evasion due to persistence of activated T cells without effector function. J. Exp. Med. 1998;188:2205–2213. doi: 10.1084/jem.188.12.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby G.M., Batist G., Alpert L., Lamoureux E., Cameron R.G., Alaoui-Jamali M.A. Overexpression of cytochrome P-450 isoforms involved in aflatoxin B1 bioactivation in human liver with cirrhosis and hepatitis. Toxicol. Pathol. 1996;24:458–467. doi: 10.1177/019262339602400408. [DOI] [PubMed] [Google Scholar]
- Vento S., Guella L., Concia E. Discordant manifestations of hepatitis C in monozygotic twins. N. Engl. J. Med. 1995;333:1224–1225. doi: 10.1056/NEJM199511023331817. [DOI] [PubMed] [Google Scholar]