

Abstract
The Src family tyrosine kinases Lck and Fyn are critical for signaling via the T cell receptor. However, the exact mechanism of their activation is unknown. Recent crystal structures of Src kinases suggest that an important mechanism of kinase activation is via engagement of the Src homology (SH)3 domain by proline-containing sequences. To test this hypothesis, we identified several T cell membrane proteins that contain potential SH3 ligands. Here we demonstrate that Lck and Fyn can be activated by proline motifs in the CD28 and CD2 proteins, respectively. Supporting a role for Lck in CD28 signaling, we demonstrate that CD28 signaling in both transformed and primary T cells requires Lck as well as proline residues in CD28. These data suggest that Lck plays an essential role in CD28 costimulation.
Keywords: Lck, CD28, costimulation, tyrosine kinase, SH3 domain
Engagement of the TCR by antigen triggers a cascade of biochemical signaling events that culminates in T cell activation. Work over the last 10 years has demonstrated that the initial TCR signals are mediated by the activation of the Src family tyrosine kinases Lck and Fyn (for a review see reference 1). Lck and Fyn phosphorylate a conserved tyrosine motif known as the ITAM (immunoreceptor tyrosine-based activation motif),1 which is present multiple times in the TCR complex. Upon phosphorylation of the ITAM, another tyrosine kinase, ZAP-70, is recruited to the TCR complex and phosphorylates a variety of downstream substrates, leading to T cell activation.
Surprisingly, the mechanism of Lck and Fyn activation by TCR engagement is not currently known. Until recently, Src family tyrosine kinases were thought to be regulated mainly by phosphorylation of a critical COOH-terminal tyrosine residue 1 2 3. Phosphorylation of this tyrosine residue inhibits kinase activity, whereas dephosphorylation stimulates activity. However, in resting T cells that express the tyrosine phosphatase CD45, most of the Lck and Fyn molecules are already dephosphorylated at the COOH-terminal tyrosine and should therefore be in an active state 4 5. Thus, it is unclear how Lck and Fyn could be further activated during T cell activation.
The recently solved crystal structures for the Src kinases Src and Hck demonstrate how phosphorylation of the COOH terminus inhibits kinase activity and also suggests an additional mechanism of kinase regulation 6 7. The phosphorylated COOH-terminal tyrosine interacts intramolecularly with the Src homology (SH)2 domain, which restricts movement of the lower lobe of the kinase domain. These crystal structures also demonstrate that the SH3 domain is tethered to the upper lobe of the kinase domain via an intramolecular interaction with a proline motif contained in a protein segment that links the SH2 domain with the kinase domain (SH2 linker). This suggests that release of both the SH3 and SH2 domains will be important in achieving full kinase activity. Indeed, Moarefi et al. 8 showed that incubation of Hck with a peptide ligand for the SH3 and/or the SH2 domain could stimulate kinase activity. This increase in kinase activity is likely to be an important physiological mechanism for Src kinase regulation, because binding of HIV-Nef to the SH3 domain of Hck can activate kinase activity to levels sufficient to transform cells 9. Furthermore, c-Src kinase activity can be induced by coexpression with an SH3 binding protein, SIN, in 293 cells 10.
SH3-mediated kinase regulation has significant implications for our understanding of T cell activation. As most of the Lck and Fyn molecules in resting T cells lack COOH-terminal tyrosine phosphorylation, SH3 interactions could represent the major mechanism of Src kinase activation during TCR engagement. Thus, it will be important to identify proteins containing ligands for the SH3 domains of Lck and Fyn and determine whether they can regulate kinase activation.
Here, we demonstrate that sequences from the T cell accessory proteins CD2 and CD28 can activate Fyn and Lck via interactions with their SH3 domains. CD2 has been mainly implicated in adhesion, whereas CD28 is thought to provide a second signal, termed costimulation, important for the activation of naive T cells. Our data suggest that one function of CD28 may be to activate Lck via an SH3-mediated interaction. In support of this hypothesis, we show that the proline motif of CD28 that can activate Lck is also essential for CD28 function in both T cell lines and primary T cells. CD28 signaling is also defective in cells lacking or deficient in Lck expression. T cell costimulation, therefore, involves the activation of Lck by CD28.
Materials and Methods
Peptides.
Peptides were synthesized, purified, and analyzed as previously described 11. The peptides were synthesized on either an ABI (model 432A; Perkin-Elmer Corp.) or a Symphony/Multiplex synthesizer (Rainin Instruments Co.) using standard FMOC (fluorenylmethoxycarbonyl) chemistry. Reagents for peptide synthesis were purchased from ABI, Rainin, and Advanced ChemTech. The peptides were purified by C18 reversed-phase HPLC, and their identity was confirmed and their concentration determined by amino acid analysis (Beckman model 6300; Beckman Instruments, Inc.). All peptides were shown to consist of a single species of the correct molecular mass by mass spectrometry (Washington University Mass Spectrometry Facility).
Protein Expression.
The full length tyrosine kinases Lck and Fyn were expressed as glutathione S transferase (GST) fusion proteins using baculovirus expression in Sf9 cells. Proteins were purified by glutathione chromatography.
In Vitro Kinase Assays.
Kinase assays were performed as described by Moarefi et al. 8. In brief, peptides (1 mM) were preincubated with purified kinases (1 μM) in a buffer containing 50 mM Hepes, pH 7.4, 10 mM MnCl2, 100 μM sodium ortho vanadate, and 50 μM ATP in 10 μl for 20 min on ice. [γ-32P]ATP (10 μCi) and 500 μM peptide Src substrate (RRLEIDAHYAARG) were added to the reaction to a final volume of 20 μl. Reactions were run in triplicate at 30°C for 20 min and terminated with 10% cold TCA. The terminated reactions were centrifuged, blotted onto phosphocellulose paper, washed with 0.425% phosphoric acid, and quantitated using a PhosphorImager® (Molecular Dynamics).
Lck Binding Assays.
The DNA encoding the mouse CD28 cytoplasmic tail was generated using PCR and subcloned into pGEX-KT at the BamHI site. The ΔC mutant, which lacks the sequences encoding the last 16 residues, was generated using inverse PCR. Purified GST proteins were incubated with cell lysates prepared from Sf9 cells infected with an Lck-containing baculovirus or with a control Sf9 lysate. Lysates were incubated at 4°C for 2–3 h before glutathione agarose beads were added. Beads were washed three times in lysis buffer (1% NP-40, 300 mM NaCl, 25 mM Hepes, pH 8.0, 25 mM NaF, and 100 μM Na3Vo4) and then subjected to in vitro kinase reactions with or without exogenous substrate. Kinase reactions were performed as described above using 10 μCi [γ-32P]ATP. The reactions were terminated in 10% TCA. Peptide substrate phosphorylation was determined after blotting onto phosphocellulose paper; Lck and GST-CD28 phosphorylation was assessed after SDS-PAGE. Phosphorylation was quantitated using PhosphorImaging.
DNA Constructs and Mutants.
Mouse CD28 cDNA was modified with NotI and XbaI restriction sties at the 5′ and 3′ ends, respectively, using PCR and subcloned into the pCDNA3.1 vector (Invitrogen Corp.). COOH-terminal deletions (Δ16) and point mutants (P187,190A) were generated by inverse PCR. All mutated cDNAs were sequenced to verify their identities. The c-fos promoter driving luciferase was provided by Dr. Philip Stork (Vollum Institute, Portland, OR).
Transient Transfections.
Cycling Jurkat cells were resuspended at 2 × 107 cells/ml in RPMI plus 10% fetal bovine serum (FBS), and 0.5 ml was placed into a 4-mm cuvette. Cells were incubated at room temperature for 10 min with 5 μg vector plus 25 μg CD28 construct or empty vector and electroporated in a BTX ECM 600 at 300 V, 960 μF, and R9. Cells were allowed to recover for 10 min at room temperature and then placed into prewarmed RPMI with 10% FBS. 10–12 h later, bulk cultures were split and stimulated with antibodies to mCD28 (1 μg/ml) or PMA (5 ng/ml). Cells were harvested 24 h later, lysed in hypotonic lysis buffer (1 mM EDTA, pH 8.0, and 10 mM KH2PO4), and assayed for luciferase activity. Transfection efficiency was normalized to cytomegalovirus renilla luciferase activity (coelenterazine purchased from SeaLite Sciences, Inc.) and by immunoblotting and flow cytometry. JCaM1.6 cells lacking functional Lck were electroporated under the following conditions: 300 V, 1050 μF, R10. All other aspects of the transfection were performed as above.
Retroviral Infections and Proliferation.
Full length or mutant mCD28 cDNA constructs were cloned into the retroviral vector GFP-RV (provided by Dr. W. Sha, University of California Berkeley, Berkeley, CA) and transiently transfected into the Phoenix E packaging cell line (provided by G. Nolan, Stanford University, Palo Alto, CA) by chloroquine-mediated CaPO4 transfection. 48 h after transfection, the retroviral supernatant was harvested and used to infect lymph node cells from either wild-type C57Bl/6 mice (The Jackson Laboratory) or CD28-deficient mice in the C57Bl/6 background 12 that had been activated 48 h previously with PMA (5 ng/ml) and ionomycin (0.1 μg/ml). In addition, the primary activation of CD28-deficient cells included 100 U/ml IL-2 to facilitate proliferation. T cells were cocultured with the retroviral supernatant for 72 h and then washed in fresh media and rested overnight. Infection efficiency was determined by flow cytometric measurement of GFP expression and varied from 10 to 20% between experiments but was similar for each construct within a given experiment. CD28 expression was confirmed by surface staining with a PE-conjugated anti-CD28 mAb (PharMingen).
CD28-mediated costimulation was determined by a standard proliferation assay. In brief, 7.5 × 104 cells were plated in each well of a round-bottom 96-well plate (Costar Corp.) and stimulated with media, PMA (5 ng/ml), or PMA plus anti-CD28 (1.0 μg/ml; mAb PV-1 provided by Dr. C. June, Naval Medical Research Institute, Bethesda, MD). The cultures were pulsed with 1.0 μCi tritiated thymidine (ICN Biomedicals, Inc.) for the final 12 h of a 36-h culture and harvested with a Skatron 96-well plate harvester onto glass microfiber filtermats. The counts of incorporated thymidine were determined by liquid scintillation counting on a Wallach 1205 Betaplate.
Results
Peptides Based on Proline Motifs Contained in CD2 and CD28 Can Activate Fyn and Lck In Vitro.
The crystal structures of Src and Hck show that in the inactive conformation of the kinase, the SH3 domain forms an intramolecular interaction with the SH2 linker region and the kinase domain 6 7. In solution, full kinase activation of Src and Hck only occurs when both the COOH-terminal tyrosine is dephosphorylated and the SH3 domain is displaced 8. This observation led us to hypothesize that cellular proteins containing SH3 binding motifs might be important to activate tyrosine kinases during TCR signaling.
The sequences of transmembrane proteins known to be associated with the TCR were examined for the consensus SH3 binding motif, PXXP (P, proline and X, any amino acid) 13 14. CD2, CD28, and CD3∈ were found to contain one or more PXXP sequences. CD3∈ is an essential component of the TCR complex and is critically involved in T cell activation. CD2 has been mainly implicated in adhesion, whereas CD28 provides a second signal, termed costimulation, important for the activation of naive T cells.
Peptides of 15–20 residues were generated for each of the potential SH3 binding sites in CD2, CD28, and CD3∈. CD2 contains five proline-rich regions, and CD28 and CD3∈ have two and one sites, respectively (Fig. 1 A). In the case of the CD3∈ peptide, the tyrosine in the peptide was changed to phenylalanine to prevent it from being used as a substrate in the kinase reaction, as this tyrosine is the first tyrosine of the CD3∈ ITAM.
Figure 1.

Specific activation of Lck and Fyn tyrosine kinases by CD2 and CD28 peptides in vitro. (A) Sequences of peptides based on the cytoplasmic domain sequences of CD2, CD28, and CD3∈ are shown using the one-letter amino acid code. Phenylalanine was substituted for tyrosine in the CD3∈ peptide to prevent phosphorylation of this peptide in vitro. (B) Activation of Lck in vitro by a CD28 peptide. Purified Lck was preincubated with each of the peptides listed in A for 20 min on ice, and kinase activity was measured using a Src substrate peptide and [γ-32P]ATP as described by Moarefi et al. 8. Bar graphs represent fold activation of kinases incubated with peptides as compared with the activity of kinase incubated with buffer and substrate alone. (C) Activation of Fyn in vitro by a CD2 peptide. Kinase reactions were performed as in B except that purified Fyn was used instead of Lck. All data are representative of at least three independent trials.
Each of the peptides was tested for its ability to stimulate the kinase activity of Lck and Fyn using in vitro kinase reactions. Lck and Fyn proteins were purified from baculovirus-infected Sf9 cells as GST fusion proteins. Because Lck and Fyn were expressed in the absence of Csk, the kinase that phosphorylates the COOH-terminal tyrosine, the regulatory COOH-terminal tyrosine is not phosphorylated 6 7. Thus, the only known mechanism for increasing kinase activity, based on the crystal structures of Src and Hck, is by displacement of the SH3 domain.
Enzymatic activity of purified Lck and Fyn was measured after the kinases were preincubated with each of the peptides. One of the CD28 peptides (CD28-2) maximally stimulated Lck kinase activity (Fig. 1 B). This threefold magnitude of activation is in agreement with the maximum level of activation predicted by Moarefi et al. 8. Thus, COOH-terminally dephosphorylated Lck is fully activated by a CD28 peptide.
The ability of the CD28-2 peptide to activate Lck was specific, as the same peptide had no effect on the activity of Fyn (Fig. 1 C). Conversely, a CD2 peptide (CD2-5) was able to strongly stimulate Fyn kinase activity but had no effect on Lck (Fig. 1 C). This peptide, murine residues 294–311, corresponds to the most highly conserved portion of the CD2 cytoplasmic domain, suggesting that this interaction may be physiologically relevant. In addition, these data are consistent with previous results demonstrating that CD2 and CD28 can interact via their proline-rich tails with the SH3 domains of Fyn and Lck 15 16 17 18. Our data extend these previous findings by demonstrating that interactions between CD2 and CD28 with Fyn and Lck, respectively, can function to specifically activate these kinases.
Substitution of the Proline Residues with Alanine Abrogates Kinase Activation.
To confirm that the proline residues are responsible for regulating kinase activity, peptides were synthesized containing alanine residues substituted for the proline residues. Peptides were then retested for their ability to stimulate kinase activity (Fig. 2 A). Substitution of proline 187 and proline 190 (P187,190A) in the CD28-2 peptide completely abrogated the ability of the peptide to activate Lck. Thus, the prolines in the CD28–2 peptide are required for kinase activation (Fig. 2 B).
Figure 2.

Proline residues are required to activate Lck and Fyn. (A) Sequences of mutated CD2 and CD28 peptides used are depicted using the one-letter amino acid code. The mutated residues are shown in bold. (B) Activation of Lck kinase activity by CD28 peptide requires proline residues. Kinase reactions were performed as described in Fig. 1 using wild-type and mutated CD28 peptides. (C) Activation of Fyn kinase activity by CD2 peptide requires proline residues. Kinase reactions were performed as described in Fig. 1 using wild-type and mutated CD2 peptides. All data are representative of at least three independent trials.
We also tested the role of prolines in the CD2-5 peptide for Fyn activation. Because the CD2-5 peptide contains two potential SH3-binding proline motifs, we synthesized two peptides with each of the pairs of prolines substituted with alanine residues. Although both mutant peptides were significantly impaired in their ability to stimulate Fyn kinase activity, the P306,309A peptide demonstrated a greater impairment than the P297,300A peptide (Fig. 2 C). This data suggests that the proline motif between residues 306 and 309 is the primary Fyn-activating motif.
Lck Binds to the Sequence Corresponding to the CD28-2 Peptide.
The sequence of the CD28-2 peptide corresponds to the last 20 residues of the CD28 cytoplasmic domain. To determine whether Lck can bind to this portion of CD28, two GST fusion proteins were generated that contain either the complete cytoplasmic domain of CD28 (GST-CD28) or the cytoplasmic domain lacking the last 16 residues (GST-ΔC). Both proteins were incubated with Sf9 cell lysate containing exogenously expressed Lck. GST proteins were isolated and Lck binding was assessed by in vitro kinase assay measuring Lck autophosphorylation (Fig. 3 A) or by using a specific tyrosine kinase peptide as a substrate (Fig. 3 B). As shown in Fig. 3, Lck tyrosine kinase activity coprecipitated with the full length but not the truncated form of CD28. Partial V8 protease mapping confirmed the identity of the putative autophosphorylated band as Lck (data not shown). In addition, no tyrosine kinase activity was detected after the fusion proteins were incubated in cell lysates lacking Lck (data not shown). Thus, Lck interacts with CD28 and, specifically, the sequence corresponding to the CD28-2 peptide.
Figure 3.
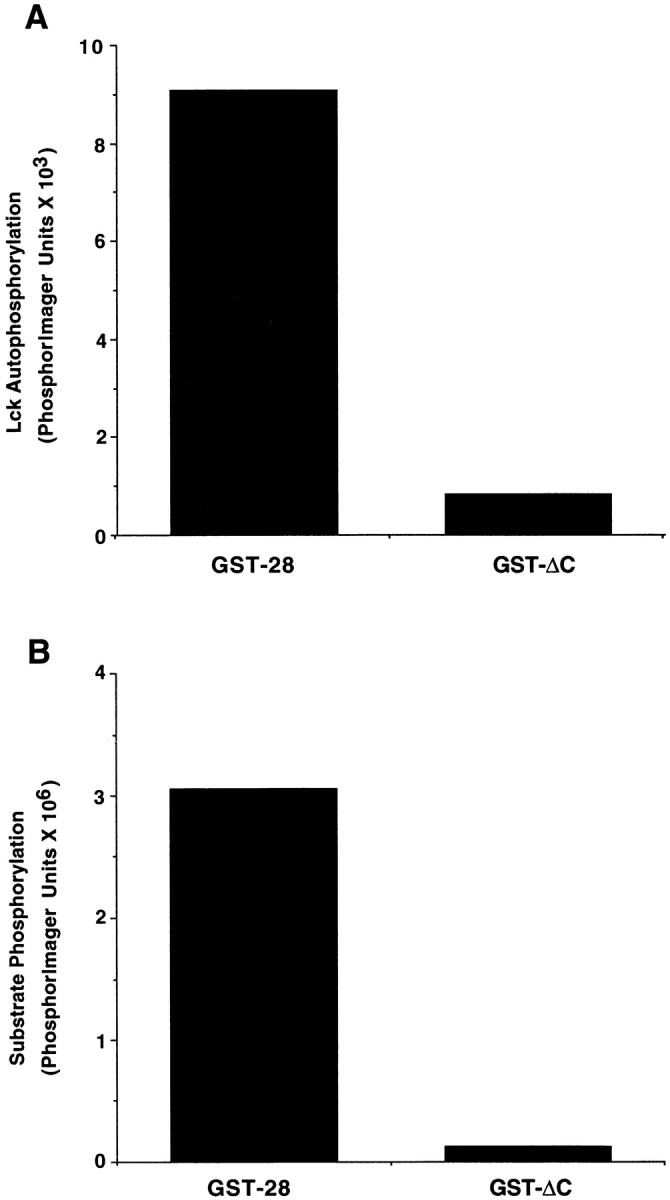
Lck interacts with the last 16 residues in the CD28 tail. (A) Chimeric GST molecules containing the full length CD28 tail or lacking the last 16 residues of CD28 (ΔC) were incubated with Sf9 cell lysates with or without exogenously expressed Lck. GST fusion proteins were isolated with glutathione agarose beads and washed three times. In vitro kinase assays were performed in the presence of [γ-32P]ATP. Kinase reactions were analyzed by SDS-PAGE, and Lck autophosphorylation was quantitated by PhosphorImage analysis. This data is representative of three independent trials. (B) Procedure was the same as described in A, except that in vitro kinase assays were performed in the presence of a specific tyrosine kinase substrate peptide. Kinase reactions were incubated with 10% TCA to precipitate phosphorylated proteins. Peptide phosphorylation was quantitated after binding to phosphocellulose paper using a PhosphorImager. Background kinase activity, determined by measuring activity in reactions prepared with GST alone, was subtracted from all samples. The data is representative of three independent trials.
CD28 Molecules Lacking Prolines 187 and 190 Are Defective in Their Ability to Stimulate a c-fos Reporter Construct.
Upon binding of a ligand, CD80 or CD86, CD28-mediated signals cooperate with signals transduced by the TCR, resulting in IL-2 production, CD25 expression, cell cycle progression, and cell survival (for review see references 19 20). As activation of Lck results in mitogen-activated protein (MAP) kinase induction, and because c-fos is stimulated in response to MAP kinase activation 21, we reasoned that if CD28 activates Lck, CD28 engagement should induce expression of a c-fos reporter plasmid. Furthermore, Lck-dependent MAP kinase activation in T cells has been shown to be dependent on the SH3 domain of Lck 22.
Wild-type mCD28 or the mutated mCD28 construct were transiently cotransfected into Jurkat cells with the c-fos reporter plasmid. Flow cytometry was used to verify equivalent mCD28 surface expression, and a control expression plasmid (renilla luciferase) was used to normalize transfection efficiency (data not shown). In the absence of antibody ligation, overexpression of wild-type mCD28 stimulated the activity of the c-fos promoter approximately four–fivefold. Treatment with soluble anti-CD28 monoclonal antibodies, however, strongly stimulated the c-fos reporter plasmid 10–12-fold (Fig. 4 A).
Figure 4.

CD28-mediated activation of the c-fos promoter requires prolines 187 and 190 and Lck. (A) CD28 requires prolines 187 and 190 to activate the c-fos promoter. Wild-type and mutated mCD28 (P187,190A) was transiently cotransfected into Jurkat cells with a c-fos reporter plasmid. Anti-CD28 mAb (37.51) was added to the media 10–12 h after electroporation, and cells were harvested and assayed for luciferase activity 24 h later. CD28 surface expression was verified by flow cytometry, and transfections were normalized using a renilla luciferase reporter plasmid. Data is shown as fold activation compared with unstimulated cells transfected with the c-fos reporter plasmid alone. (B) CD28 stimulation of the c-fos reporter requires Lck. Wild-type mCD28 was cotransfected with the c-fos reporter plasmid into JCaM1.6 cells, which lack Lck expression. Experiments were performed as described in A. In this experiment, PMA stimulation of the fos-luciferase (fos-luc) reporter was used to control for transfection efficiency. All data are representative of at least three independent trials.
We next focused on determining whether prolines 187 and 190 were critical for CD28 function. A mutated form of CD28 was generated with alanines substituted for prolines 187 and 190. The mutated form of mCD28 was then tested for its ability to stimulate the c-fos reporter. P187, 190A was significantly impaired in its ability to induce the c-fos promoter, both in the presence and absence of CD28 antibodies (Fig. 4 A). In repeated experiments, P187,190A always induced at significantly lower levels, even when expression was as high or higher than wild-type CD28. We suspect that some of the activity measured with this mutant may be due to formation of heterodimers with wild-type CD28 molecules. Nevertheless, these data clearly demonstrate that the proline residues required to activate Lck kinase activity are important for CD28 signaling in Jurkat cells.
CD28 Induction of c-fos Requires Lck.
To determine whether CD28 induction of c-fos required Lck, wild-type mCD28 was cotransfected with the c-fos reporter into JCaM1.6 cells, which lack functional Lck 23. No detectable c-fos reporter activity was measured both in the presence or absence of anti-CD28 antibodies (Fig. 4 B). The induction of c-fos was specific for Lck, as reconstitution of JCaM1.6 cells with wild-type Lck but not Fyn reconstituted CD28 stimulation of c-fos to levels similar to those of the parental Jurkat cells (Fig. 5A and Fig. C). This demonstrates that CD28-mediated stimulation of the c-fos reporter requires Lck.
Figure 5.
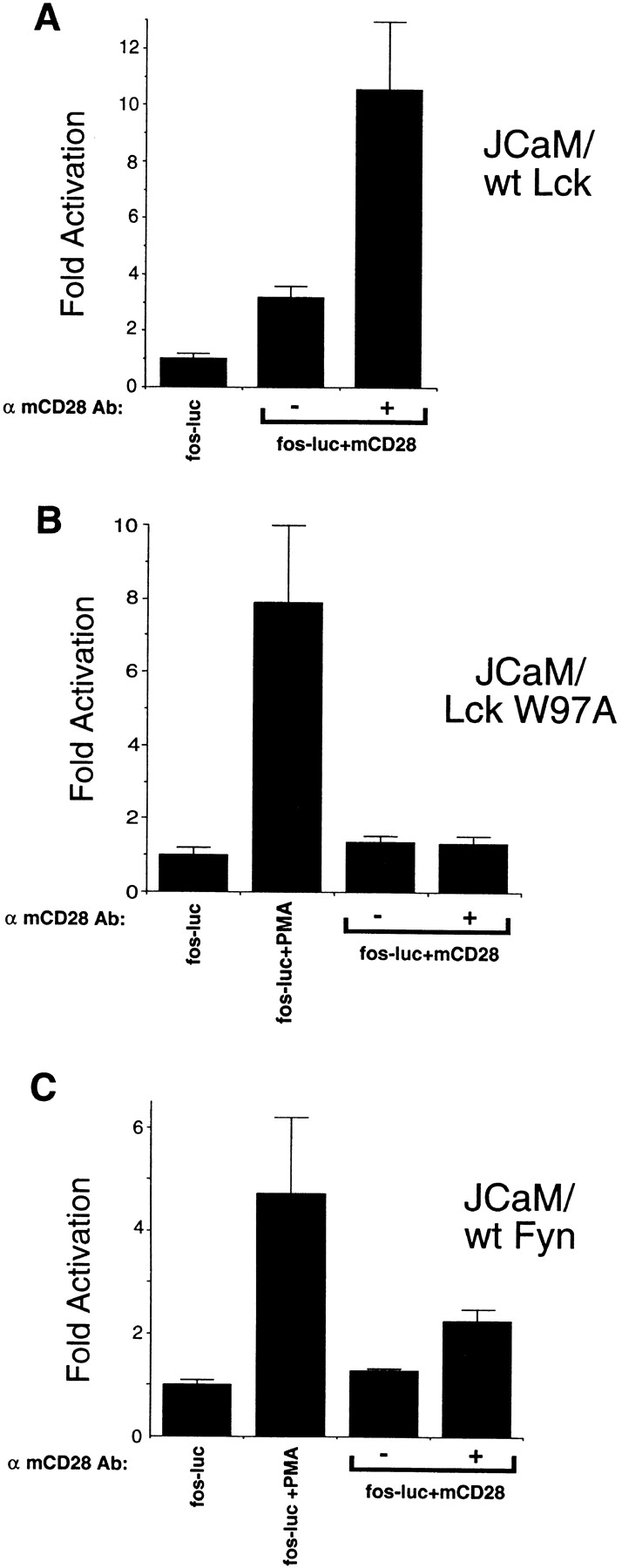
CD28-mediated activation of c-fos requires the SH3 domain of Lck and cannot be reconstituted by Fyn. (A) Reconstitution of Lck-deficient Jurkat cells with wild-type Lck rescues CD28 stimulation of the c-fos reporter. Wild-type mCD28 was cotransfected with the c-fos reporter plasmid into JCaM1.6 cells stably transfected with Lck. Experiments were performed as described for Fig. 3 A. (B) Lck-deficient Jurkat cells reconstituted with Lck SH3 point mutants (W97A) do not allow CD28 to drive the c-fos reporter. Wild-type mCD28 was cotransfected with the c-fos reporter plasmid into JCaM1.6 cells stably transfected with Lck W97A SH3 mutant. Experiments were performed as for Fig. 3 A. In this experiment, PMA stimulation of the fos-luciferase (fos-luc) reporter was used to control transfection efficiency. (C) Overexpression of Fyn in Lck-deficient Jurkat cells cannot reconstitute CD28 c-fos induction. JCaM1.6 cells stably overexpressing wild-type Fyn were transiently cotransfected with the c-fos reporter as in Fig. 3 A. In this experiment, PMA stimulation of the fos-luciferase reporter was used to control transfection efficiency. All data are representative of three independent trials.
To test whether the SH3 domain of Lck was involved in CD28-mediated c-fos induction, JCaM1.6 cells stably transfected with a form of Lck containing a mutation in the SH3 domain (W97A) were tested for the ability of CD28 to stimulate c-fos. mCD28 did not induce the c-fos reporter in this cell line, even in the presence of cross-linking antibodies (Fig. 5 B). Thus, an intact Lck SH3 domain is required for CD28 induction of c-fos.
Reconstitution of T Cells from CD28-deficient Mice Confirms the Requirement for Prolines 187 and 190 of CD28.
As CD28 costimulation is most important for the activation of naive T cells, our current studies in transformed cell lines may not accurately reflect the true biological role of CD28 in primary cells. To examine the importance of the proline residues in CD28 signaling in primary T cells, we reconstituted T cells from CD28 knockout animals using retroviruses expressing either wild-type or mutated mCD28.
Retroviruses were prepared encoding either wild-type mCD28 or mutated forms of mCD28 either lacking the COOH-terminal 16 residues (Δ16) or with prolines 187 and 190 substituted with alanines (P187,190A). Lymph node T cells from CD28 knockout mice were infected with wild-type or mutated forms of CD28 using retroviral infection. Flow cytometry demonstrated similar levels of expression and similar mean fluorescence intensities of all the mCD28 constructs, with ∼10–15% of T cells positive for CD28 expression (data not shown).
To test the function of CD28, infected T cells were stimulated with anti-CD3 antibodies, PMA, or a combination of anti-CD3 or PMA and antibodies to CD28. As expected, cells infected with the control retrovirus proliferated weakly upon treatment with anti-CD3 or PMA alone, and this effect was not enhanced by coculture with antibodies to CD28 (Fig. 6). T cells infected with the retrovirus encoding wild-type mCD28 proliferated strongly with both the combination of anti-CD3 or PMA and antibodies to CD28, whereas cells expressing the truncated form of CD28 or the P187,190A construct completely failed to respond to the addition of CD28 antibodies (Fig. 6). We suspect that the enhanced proliferation of anti-CD3 in cells expressing wild-type mCD28 is due to the presence of B7-positive cells in our cultures. Thus, the ability of CD28 to signal in primary T cells requires the COOH terminus of CD28, and specifically prolines 187 and 190.
Figure 6.

Reconstitution of CD28 signaling in T cells from CD28-deficient mice requires proline residues at positions 187 and 190. Lymph node T cells from CD28-deficient animals were infected with retroviruses encoding either wild-type mCD28 or mCD28 lacking the 16 COOH-terminal residues (Δ16) or with the P187,190A mutated construct. 3 d after infection, T cells were stimulated with anti-CD3 antibodies, PMA, or anti-CD3 and PMA in combination with antibodies to mCD28. T cell proliferation was measured by [3H]thymidine incorporation. Retroviral transduction efficiency was measured by analyzing mCD28 surface expression using flow cytometry. Values are the means of quadruplicate samples. The data is representative of three independent experiments.
Primary T Cells Lacking Lck Are Unable to Proliferate in Response to PMA and CD28.
To verify that Lck is indeed required for CD28-mediated proliferation of primary T cells, we also examined CD28 signaling in T cells from Lck knockout mice 24. Peripheral T cells from the Lck-deficient mice were harvested and tested for their ability to proliferate in response to anti-murine CD28 antibodies and PMA. As expected, wild-type cells strongly proliferated in response to PMA and anti-CD28 (Fig. 7). T cells from homozygous Lck-deficient mice did not respond to PMA and anti-CD28 (Fig. 7). Importantly, mice heterozygous for Lck expression showed a decreased response to PMA and anti-CD28 (Fig. 7), proliferating about half as well as wild-type cells. As T cells from Lck-heterozygous mice are phenotypically normal, the decreased response to CD28 signaling is not related to abnormalities in T cell development. These findings confirm our results obtained in Jurkat cell lines and demonstrate clearly that Lck is required for CD28 signaling in primary T cells.
Figure 7.

T cells from the Lck-deficient mouse are defective in their response to PMA and anti-CD28. Peripheral T cells from wild-type C57BL/6, Lck heterozygotes, or Lck-deficient animals were harvested and plated at 7.5 × 104 cells per well in a 96-well round-bottom plate. Cells were stimulated with antibodies to CD28 alone (l μg/ml) or anti-CD28 plus 5 ng/ml PMA. 24 h later, cells were pulsed with [3H]thymidine and harvested after 12 h of incubation. Lck−/− A and B represent two different Lck-deficient mice. Values are the means of triplicate samples. These results are representative of two independent experiments.
Discussion
To date, the role of SH3 domain–mediated activation of Src kinases has not been tested as a model for Src kinase activation in vivo. Previous studies used an artificial, high affinity SH3-binding peptide or a viral protein, HIV-Nef, to study Hck kinase activation 8 9. Here we have demonstrated that sequences from cellular proteins known to be involved in T cell activation can function as activators of Fyn and Lck kinases via their interactions with SH3 domains.
These data support a model where engagement of the SH3 domains of Lck and Fyn by T cell accessory molecules plays an important role in kinase activation during T cell activation. Recruitment and clustering of CD2 and CD28 with the TCR into the T cell contact cap 25 could potentially bring together Lck (associated with CD4 and CD8) and Fyn (associated with the TCR) with their respective activating ligands contained in CD2 and CD28. Although the millimolar concentrations of peptide required to activate the Src kinases in vitro seem high, these concentrations are probably in the physiological range. As both CD28 and Lck are membrane-bound proteins, both molecules are concentrated in two-dimensional space and rotationally constrained. It has been estimated that a 1-mM concentration of CD28 corresponds to a density of ∼600 molecules/μm2 26. Given that the resting density of CD28 is ∼100–200 molecules/μm2, clustering of membrane proteins could easily attain concentrations sufficient to activate Lck and Fyn. These calculations can also explain why overexpression of CD28 in our transient expression is able to stimulate the c-fos reporter. Thus, the concentration of peptide required to activate Lck and Fyn is in the appropriate physiological range.
Although the magnitude of SH3-mediated activation seems low, ∼2.5–3-fold, it is similar to the level of activation induced by COOH-terminal tyrosine dephosphorylation, which is sufficient to transform cells 1 2 3. Indeed, engagement of the SH3 domain is sufficient by itself to induce transformation of fibroblast cells 9. This low magnitude of activation can also potentially explain difficulties in demonstrating Lck and Fyn activation induced by TCR, CD2, or CD28 engagement. Although several groups have reported activation of Lck and Fyn after cross-linking of various T cell membrane proteins 15 16 17 18 27 28 29 30, the levels of activation are low and have been difficult to reproduce. However, as only a fraction of kinase molecules are likely to be activated by specific transmembrane proteins, it is not surprising that a relatively small increase in kinase activity would be difficult to be detect.
The observation that Lck and Fyn are specifically activated by proline residues in CD28 and CD2, respectively, raises the question of whether the SH3 domains of Lck and Fyn have different affinities for the proline regions of CD28 and CD2. To address this issue, we measured the affinities of purified Lck and Fyn SH3 domains to the CD28-2 and CD2-5 peptides by both surface plasmon resonance and fluorescence anisotropy. Similar results were obtained with both SH3 domains for both peptides (12–20 μM; data not shown). These values are in the range of other published studies for SH3 domain–proline interactions 14 31 and suggest that the activation of these tyrosine kinases is not due to differential affinities of the SH3 domains for the proline regions. Rather, activation may be dependent on the ability of a specific peptide to displace the SH2 linker region from the kinase lobe.
Others have previously demonstrated association of CD28 with Lck in T cells and activation of Lck after CD28 cross-linking 15 28. The mechanism of activation, however, was unknown. Previous mapping studies in Jurkat cells showed that truncating 10 or 17 residues of the CD28 tail, but not 5 residues, could abrogate CD28 function 32 33. This suggests that a critical sequence for CD28 function lies between the last 6 to 17 residues of the CD28 cytoplasmic domain. Our studies extend previous mapping data by demonstrating that Lck can bind to this sequence and implicate two proline residues (P187 and P190), which are contained between the last 6–17 residues of CD28, as critical for CD28 function.
To verify the physiological relevance of this interaction, we demonstrated that CD28 engagement by itself could stimulate a c-fos reporter. This reporter induction was dependent on the presence of wild-type Lck. CD28 in Jurkat T cells lacking Lck (JCaM1.6) 23 or expressing an SH3-mutated form of Lck 22 were unable to stimulate the c-fos reporter. This observation clearly demonstrates a requirement for Lck in this pathway. These studies demonstrate that c-fos induction by CD28 is specific to Lck, as JCaM1.6 cells express abundant amounts of Fyn 34. Furthermore, overexpression of Fyn in JCaM1.6 cells did not reconstitute the ability of CD28 to induce c-fos expression (data not shown).
Previous studies had shown that JCaM1.6 cells lacking Lck can signal normally through CD28 35. These studies, however, focused on a different readout for CD28 function, namely the production of IL-2 by coengagement of CD28. Here we used a readout that allows us to measure signals mediated by CD28 engagement by itself; in our studies, we did not require coengagement with the TCR. The requirement for Lck is strongly supported by our observation that primary T cells lacking Lck could not respond to anti-CD28 plus PMA. More importantly, CD28 stimulation was reduced by half in mice that are heterozygous for Lck expression. As T cells from heterozygous animals are otherwise phenotypically normal 24, this reduction in stimulation demonstrates genetically that there is a strong dose effect for Lck in CD28 costimulation.
Although we demonstrated that Lck can bind to the last 16 residues of CD28, Lck may interact with other domains of CD28. For example, CD28 is known to be tyrosine phosphorylated during T cell activation 36 37 and could therefore interact with the Lck SH2 domain. Such an interaction could facilitate recruitment of Lck to CD28. This seems plausible, as the SH2 affinities are generally much higher than the typical SH3 interaction. In addition, as resting T cells mostly lack Lck COOH-terminal tyrosine phosphorylation, the SH2 domain should be unliganded and available for binding. It will be interesting to determine whether tyrosine phosphorylation of CD28 cooperates with the proline motif to enhance Lck activity during T cell activation.
CD28 and T Cell Costimulation.
Although extensively studied, the exact nature of T cell costimulation by CD28 remains unclear. Costimulation was originally defined as a distinct signal required in conjunction with the signal transduced by the TCR for the initial activation of naive T cells 38. As engagement of CD28 by antibodies or its ligand, B7, can block the induction of T cell anergy 38, CD28 is thought to transduce unique signals that are required for T cell activation.
Extensive studies on CD28 signaling have demonstrated potential involvement of CD28 in a variety of signaling pathways. Many studies have focused on transcription factors induced by CD28. These include the c-fos/c-jun heterodimer, activator protein 1 33 39, as well as the latent transcription factor, nuclear factor κB 33 40 41. However, in most of these cases, signals detected by CD28 engagement require coengagement with the TCR. However, several groups have shown that CD28 cross-linking by itself can induce tyrosine phosphorylation 15 28 42 43 44. In support of this, tyrosine kinase inhibitors can block CD28 costimulation 45 46. The interactions between CD28 and Lck described here are consistent with this data.
Some of the difficulty in understanding the role of CD28 in costimulation may be due to the fact that most studies of CD28 were performed in transformed T cell lines such as Jurkat, which do not require costimulation for activation and cannot be anergized. Thus, the requirements for CD28 function defined in Jurkat cells may not be the same as in primary T cells. Our reconstitution of CD28-deficient T cells with wild-type and mutated CD28 molecules is the first study, to our knowledge, to define structural features of CD28 in primary nontransformed T cells.
Our data are consistent with the idea that CD28 does not transduce a unique signal. Rather, our data supports the idea that CD28 costimulation functions mainly to enhance TCR signaling. By binding to its ligand, B7, on the APC, CD28 could strengthen adhesion of T cells with APCs, potentiating TCR engagement with antigen. By activating Lck, CD28 could also enhance and amplify tyrosine phosphorylation events induced by TCR engagement. In support of a model where CD28 functions mainly to potentiate signals transduced by the TCR, two recent studies demonstrate that CD28 stimulates a transport mechanism that recruits lipid rafts to the contact surface between the T cell and the APC 47 48. One consequence of this recruitment is enhanced tyrosine phosphorylation and increased consumption of Lck 48. These data are consistent with the role for Lck that we propose here. Our data, however, cannot rule out the possibility that CD28 transduces other signals in addition to Lck activation.
Here we have shown that SH3 binding motifs contained in the cytoplasmic domain of T cell transmembrane proteins can potentially activate Src family tyrosine kinases in T cells. This finding suggests that the formation of the immunological synapse, a key event in T cell activation, may have effects in addition to stabilizing cell–cell contacts 49. The recruitment and concentration of small molecules like CD2 and CD28 may also result in the activation of the tyrosine kinases involved in transducing the earliest signals mediated by the TCR. Focusing on the costimulation protein CD28, we demonstrated that CD28 engagement activates Lck based on the ability of CD28 to induce c-fos activity in a proline and Lck SH3–dependent manner. More importantly, we confirmed these findings in primary T cells by showing that CD28 signaling required prolines 187 and 190 in CD28 as well as the presence of Lck. This data provides novel mechanistic insights into CD28 function and may lead to a better understanding of how Src kinases are regulated in other cell types.
Acknowledgments
We are extremely grateful to Kenneth Murphy, Gary Noland, William Sha, and Deepta Bhattarcharya for providing the retroviral expression system. We thank Andy Chan, Mike Dustin, and Philip Stork for providing essential reagents and advice and Helen Piwnica-Worms, Joseph Shih, Ancho Nguyen, and Bob Schreiber for critical reading of the manuscript.
Support was provided by National Institutes of Health grants AI07163 (to A.D. Holdorf), KO8HL03408 (to J.M. Green), and AI54094 (to A. Shaw). V. Link was supported by a scholarship from the Deutscher Akademischer Austauschdienst.
Footnotes
1used in this paper: GST, glutathione S transferase; ITAM, immunoreceptor tyrosine-based activation motif; MAP, mitogen-activated protein; SH, Src homology
References
- Cartwright C.A., Eckhart W., Simon S., Kaplan P.L. Cell transformation by pp60c-src mutated in the carboxy-terminal regulatory domain. Cell. 1987;49:83–91. doi: 10.1016/0092-8674(87)90758-6. [DOI] [PubMed] [Google Scholar]
- Kmiecik T.E., Shalloway D. Activation and suppression of pp60c-src transforming ability by mutation of its primary sites of tyrosine phosphorylation. Cell. 1987;49:65–73. doi: 10.1016/0092-8674(87)90756-2. [DOI] [PubMed] [Google Scholar]
- Piwnica-Worms H., Saunders K.B., Roberts T.M., Smith A.E., Cheng S.H. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 1987;49:75–82. doi: 10.1016/0092-8674(87)90757-4. [DOI] [PubMed] [Google Scholar]
- McFarland E.D., Hurley T.R., Pingel J.T., Sefton B.M., Shaw A., Thomas M.L. Correlation between Src family member regulation by the protein-tyrosine-phosphatase CD45 and transmembrane signaling through the T-cell receptor. Proc. Natl. Acad. Sci. USA. 1993;90:1402–1406. doi: 10.1073/pnas.90.4.1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostergaard H.L., Trowbridge I.S. Coclustering CD45 with CD4 or CD8 alters the phosphorylation and kinase activity of p56lck. J. Exp. Med. 1990;172:347–350. doi: 10.1084/jem.172.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheri F., Moarefi I., Kuriyan J. Crystal structure of the Src family tyrosine kinase Hck. Nature. 1997;385:602–609. doi: 10.1038/385602a0. [DOI] [PubMed] [Google Scholar]
- Xu W., Harrison S.C., Eck M.J. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 1997;385:595–602. doi: 10.1038/385595a0. [DOI] [PubMed] [Google Scholar]
- Moarefi I., LaFevre B.M., Sicheri F., Huse M., Lee C.H., Kuriyan J., Miller W.T. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 1997;385:650–653. doi: 10.1038/385650a0. [DOI] [PubMed] [Google Scholar]
- Briggs S.D., Sharkey M., Stevenson M., Smithgall T.E. SH3-mediated Hck tyrosine kinase activation and fibroblast transformation by the Nef protein of HIV-1. J. Biol. Chem. 1997;272:17899–17902. doi: 10.1074/jbc.272.29.17899. [DOI] [PubMed] [Google Scholar]
- Alexandropoulos K., Baltimore D. Coordinate activation of c-Src by SH3- and SH2-binding sites on a novel p130Cas-related protein, Sin. Genes Dev. 1996;10:1341–1355. doi: 10.1101/gad.10.11.1341. [DOI] [PubMed] [Google Scholar]
- Lorenz R.G., Tyler A.N., Allen P.M. Reconstruction of the immunogenic peptide RNase(43-56) by identification and transfer of the critical residues into an unrelated peptide backbone. J. Exp. Med. 1989;170:203–215. doi: 10.1084/jem.170.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J.M., Noel P.J., Sperling A.I., Walunas T.L., Gray G.S., Bluestone J.A., Thompson C.B. Absence of B7-dependent responses in CD28-deficient mice. Immunity. 1994;1:501–508. doi: 10.1016/1074-7613(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Ren R., Mayer B.J., Cicchetti P., Baltimore D. Identification of a ten-amino acid proline-rich SH3 binding site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- Feng S., Chen J.K., Yu H., Simon J.A., Schreiber S.L. Two binding orientations for peptides to the Src SH3 domaindevelopment of a general model for SH3-ligand interactions. Science. 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- August A., Dupont B. Activation of src family kinase lck following CD28 crosslinking in the Jurkat leukemic cell line. Biochem. Biophys. Res. Commun. 1994;199:1466–1473. doi: 10.1006/bbrc.1994.1396. [DOI] [PubMed] [Google Scholar]
- Bell G.M., Bolen J.B., Imboden J.B. Association of Src-like protein tyrosine kinases with the CD2 cell surface molecule in rat T lymphocytes and natural killer cells. Mol. Cell. Biol. 1992;12:5548–5554. doi: 10.1128/mcb.12.12.5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell G.M., Fargnoli J., Bolen J.B., Kish L., Imboden J.B. The SH3 domain of p56lck binds to proline-rich sequences in the cytoplasmic domain of CD2. J. Exp. Med. 1996;183:169–178. doi: 10.1084/jem.183.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H., Hutchcroft J.E., Andoniou C.E., Kamoun M., Band H., Bierer B.E. Association of p59(fyn) with the T lymphocyte costimulatory receptor CD2. Binding of the Fyn Src homology (SH) 3 domain is regulated by the Fyn SH2 domain. J. Biol. Chem. 1998;273:19914–19921. doi: 10.1074/jbc.273.31.19914. [DOI] [PubMed] [Google Scholar]
- June C.H., Bluestone J.A., Nadler L.M., Thompson C.B. The B7 and CD28 receptor families. Immunol. Today. 1994;15:321–331. doi: 10.1016/0167-5699(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Rudd C.E. Upstream-downstreamCD28 cosignaling pathways and T cell function. Immunity. 1996;4:527–534. doi: 10.1016/s1074-7613(00)80479-3. [DOI] [PubMed] [Google Scholar]
- Shibuya H., Kohu K., Yamada K., Barsoumian E.L., Perlmutter R.M., Taniguchi T. Functional dissection of p56lck, a protein tyrosine kinase which mediates interleukin-2-induced activation of the c-fos gene. Mol. Cell. Biol. 1994;14:5812–5819. doi: 10.1128/mcb.14.9.5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny M.F., Kaufman H.C., Chan A.C., Straus D.B. The Lck SH3 domain is required for activation of the MAP kinase pathway, but not the initiation of TCR signaling. J. Biol. Chem. 1999;274:5146–5152. doi: 10.1074/jbc.274.8.5146. [DOI] [PubMed] [Google Scholar]
- Straus D.B., Weiss A. Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell. 1992;70:585–593. doi: 10.1016/0092-8674(92)90428-f. [DOI] [PubMed] [Google Scholar]
- Molina T.J., Kishihara K., Siderovski D.P., van Ewijk W., Narendran A., Timms E., Wakeham A., Paige C.J., Hartmann K.U., Veillette A. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- Shaw A.S., Dustin M.L. Making the T cell receptor go the distancea topological view of T cell activation. Immunity. 1997;6:361–369. doi: 10.1016/s1074-7613(00)80279-4. [DOI] [PubMed] [Google Scholar]
- Dustin M.L., Golan D.E., Zhu D.M., Miller J.M., Meier W., Davies E.A., van der Merwe P.A. Low affinity interaction of human or rat T cell adhesion molecule CD2 with its ligand aligns adhering membranes to achieve high physiological affinity. J. Biol. Chem. 1997;272:30889–30898. doi: 10.1074/jbc.272.49.30889. [DOI] [PubMed] [Google Scholar]
- Danielian S., Alcover A., Polissard L., Stefanescu M., Acuto O., Fischer S., Fagard R. Both T cell receptor (TcR)-CD3 complex and CD2 increase the tyrosine kinase activity of p56lck. CD2 can mediate TcR-CD3-independent and CD45-dependent activation of p56lck. Eur. J. Immunol. 1992;22:2915–2921. doi: 10.1002/eji.1830221124. [DOI] [PubMed] [Google Scholar]
- Gibson S., Truitt K., Lu Y., Lapushin R., Khan H., Imboden J.B., Mills G.B. Efficient CD28 signalling leads to increases in the kinase activities of the TEC family tyrosine kinase EMT/ITK/TSK and the SRC family tyrosine kinase LCK. Biochem. J. 1998;330:1123–1128. doi: 10.1042/bj3301123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K.X., Sefton B.M. Cross-linking of T-cell surface molecules CD4 and CD8 stimulates phosphorylation of the lck tyrosine protein kinase at the autophosphorylation site. Mol. Cell. Biol. 1990;10:5305–5313. doi: 10.1128/mcb.10.10.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsygankov A.Y., Spana C., Rowley R.B., Penhallow R.C., Burkhardt A.L., Bolen J.B. Activation-dependent tyrosine phosphorylation of Fyn-associated proteins in T lymphocytes. J. Biol. Chem. 1994;269:7792–7800. [PubMed] [Google Scholar]
- Yu H., Chen J.K., Feng S., Dalgarno D.C., Brauer A.W., Schreiber S.L. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 1994;76:933–945. doi: 10.1016/0092-8674(94)90367-0. [DOI] [PubMed] [Google Scholar]
- Pages F., Ragueneau M., Klasen S., Battifora M., Couez D., Sweet R., Truneh A., Ward S.G., Olive D. Two distinct intracytoplasmic regions of the T-cell adhesion molecule CD28 participate in phosphatidylinositol 3-kinase association. J. Biol. Chem. 1996;271:9403–9409. doi: 10.1074/jbc.271.16.9403. [DOI] [PubMed] [Google Scholar]
- Shapiro V.S., Truitt K.E., Imboden J.B., Weiss A. CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol. Cell. Biol. 1997;17:4051–4058. doi: 10.1128/mcb.17.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszowy M.W., Leuchtmann P.L., Veillette A., Shaw A.S. Comparison of p56lck and p59fyn protein expression in thymocyte subsets, peripheral T cells, NK cells, and lymphoid cell lines. J. Immunol. 1995;155:4236–4240. [PubMed] [Google Scholar]
- Stein P.H., Fraser J.D., Weiss A. The cytoplasmic domain of CD28 is both necessary and sufficient for costimulation of interleukin-2 secretion and association with phosphatidylinositol 3′-kinase. Mol. Cell. Biol. 1994;14:3392–3402. doi: 10.1128/mcb.14.5.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King P.D., Sadra A., Teng J.M., Xiao R.L., Han A., Selvakumar A., August A., Dupont B. Analysis of CD28 cytoplasmic tail tyrosine residues as regulators and substrates for the protein tyrosine kinases, EMT and LCK. J. Immunol. 1997;158:580–590. [PubMed] [Google Scholar]
- Sadra A., Cinek T., Arellano J.L., Shi J., Truitt K.E., Imboden J.B. Identification of tyrosine phosphorylation sites in the CD28 cytoplasmic domain and their role in the costimulation of Jurkat T cells. J. Immunol. 1999;162:1966–1973. [PubMed] [Google Scholar]
- Harding F.A., McArthur J.G., Gross J.A., Raulet D.H., Allison J.P. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356:607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- Su B., Jacinto E., Hibi M., Kallunki T., Karin M., Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736. doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Bryan R.G., Li Y., Lai J.H., Van M., Rice N.R., Rich R.R., Tan T.H. Effect of CD28 signal transduction on c-Rel in human peripheral blood T cells. Mol. Cell. Biol. 1994;14:7933–7942. doi: 10.1128/mcb.14.12.7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P., Tan T.H., Rice N.R., Sica A., Young H.A. The interleukin 2 CD28-responsive complex contains at least three members of the NF κB familyc-Rel, p50, and p65. Proc. Natl. Acad. Sci. USA. 1993;90:1696–1700. doi: 10.1073/pnas.90.5.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- August A., Gibson S., Kawakami Y., Kawakami T., Mills G.B., Dupont B. CD28 is associated with and induces the immediate tyrosine phosphorylation and activation of the Tec family kinase ITK/EMT in the human Jurkat leukemic T-cell line. Proc. Natl. Acad. Sci. USA. 1994;91:9347–9351. doi: 10.1073/pnas.91.20.9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasen S., Pages F., Peyron J.F., Cantrell D.A., Olive D. Two distinct regions of the CD28 intracytoplasmic domain are involved in the tyrosine phosphorylation of Vav and GTPase activating protein-associated p62 protein. Int. Immunol. 1998;10:481–489. doi: 10.1093/intimm/10.4.481. [DOI] [PubMed] [Google Scholar]
- Nunes J.A., Truneh A., Olive D., Cantrell D.A. Signal transduction by CD28 costimulatory receptor on T cells. B7-1 and B7-2 regulation of tyrosine kinase adaptor molecules. J. Biol. Chem. 1996;271:1591–1598. doi: 10.1074/jbc.271.3.1591. [DOI] [PubMed] [Google Scholar]
- Atluru S., Atluru D. Evidence that genistein, a protein-tyrosine kinase inhibitor, inhibits CD28 monoclonal-antibody-stimulated human T cell proliferation. Transplantation. 1991;51:448–450. doi: 10.1097/00007890-199102000-00035. [DOI] [PubMed] [Google Scholar]
- June C.H., Ledbetter J.A., Gillespie M.M., Lindsten T., Thompson C.B. T-cell proliferation involving the CD28 pathway is associated with cyclosporine-resistant interleukin 2 gene expression. Mol. Cell. Biology. 1987;7:4472–4481. doi: 10.1128/mcb.7.12.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulfing C., Davis M.M. A receptor/cytoskeletal movement triggered by costimulation during T cell activation. Science. 1998;282:2266–2269. doi: 10.1126/science.282.5397.2266. [DOI] [PubMed] [Google Scholar]
- Viola A., Schroeder S., Sakakibara Y., Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283:680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- Dustin M.L., Shaw A.S. Costimulationbuilding an immunological synapse. Science. 1999;283:649–650. doi: 10.1126/science.283.5402.649. [DOI] [PubMed] [Google Scholar]