

Abstract
Mice deficient in various mismatch repair (MMR) enzymes were examined to determine whether this repair pathway is involved in antibody class switch recombination. Splenic B cells from mice deficient in Msh2, Mlh1, Pms2, or Mlh1 and Pms2 were stimulated in culture with lipopolysaccharide (LPS) to induce immunoglobulin (Ig)G2b and IgG3, LPS and interleukin (IL)-4 to induce IgG1, or LPS, anti–δ-dextran, IL-4, IL-5, and transforming growth factor (TGF)-β1 to induce IgA. After 4 d in culture, cells were surface stained for IgM and non-IgM isotypes and analyzed by FACS®. B cells from MMR-deficient mice show a 35–75% reduction in isotype switching, depending on the isotype and on the particular MMR enzyme missing. IgG2b is the most affected, reduced by 75% in Mlh1-deficient animals. The switching defect is not due to a lack of maturation of the B cells, as purified IgM+IgD+ B cells show the same reduction. MMR deficiency had no effect on cell proliferation, viability, or apoptosis, as detected by [3H]thymidine incorporation and by propidium iodide staining. The reduction in isotype switching was demonstrated to be at the level of DNA recombination by digestion-circularization polymerase chain reaction (DC-PCR). A model of the potential role for MMR enzymes in class switch recombination is presented.
Keywords: class switch recombination, msh2, mlh1, pms2
Activation of mature B lymphocytes which express IgM and IgD on their surface leads to the expression of downstream Ig heavy chain genes that confer different effector functions upon the antibody molecule while maintaining the same antigen-binding V domain. This process occurs by means of an intrachromosomal, deletional DNA recombination event between switch (S)1 regions located 5′ of each heavy chain C region gene except Cδ. The S regions consist of 1–10 kb stretches of tandemly repeated G-rich sequences. The mechanism of class switch recombination (CSR) and the enzymes involved in cutting, aligning, and ligating the DNA during CSR are all unknown. Since S regions do not share significant stretches of identity, CSR is not thought to involve homologous recombination, as this requires ∼200 nucleotides (nt) of identity between recombining sequences 1 2. Sequence identities between the donor S and acceptor S region are found at the sites of recombination, but they are very short 3 and are typical of illegitimate recombination or end-joining reactions 4. Induction of double-strand breaks (DSBs) is thought to initiate switch recombination 5, and components of the Ku complex, which are required for DSB repair, are also required for switch recombination 6 7 8. The nt excision repair protein, XP-C, is not required for CSR 9.
We have examined whether mismatch repair (MMR) enzymes may play a role in CSR. Aside from the repair of mispaired bases due to DNA polymerase errors, these enzymes are also involved in recombination. MMR enzymes recognize DNA distortions—they bind to Holliday junctions 10 and heteroduplex DNA 11. MMR enzymes can suppress recombination between divergent sequences 12 13 and are involved in the removal of heterologous DNA at the ends of homologous recombining segments 14. In addition, several groups have reported that mice deficient in the MMR enzymes Mlh1, Pms2, or Msh2 have reduced levels and/or an altered pattern of somatic mutation in the V region genes 15 16 17 18 19. V gene somatic hypermutation results in small insertions, deletions, and point mutations similar to those found surrounding the sites of switch recombination 3 20 21. Both somatic mutation and CSR occur with similar kinetics after B cell activation 22 and occur in germinal centers (GCs), although CSR also occurs in the T cell–rich areas of the spleen and lymph nodes 22 23. MMR enzymes are known to be expressed and are functional in human tonsil GCs 24. In addition to altered somatic hypermutation, it has been observed that Msh2-deficient mice have reduced antigen-specific T-dependent serum IgG responses 18 25, T-independent IgG3 responses 25, and also reduced numbers of IgG antibody-forming cells in the periarteriolar lymphatic sheath (PALS [25]). Increased apoptosis occurs in GCs of Msh2−/− mice and has been proposed to account for the reduced secondary responses in these mice 25. Alternatively, we propose that impaired CSR could directly account for the reduced IgG expression in Msh2−/− mice.
To test this alternative hypothesis, we asked whether deficiencies in MMR enzymes would alter the ability of B cells to undergo isotype switching in vitro. We measured class switching by surface staining for expression of IgM and non-IgM isotypes, and also at the DNA level by digestion-circularization PCR (DC-PCR). We found no impairment in the ability of MMR-deficient cells to divide and survive in culture; however, CSR by these cells is seriously compromised.
Materials and Methods
Mice.
Mice made deficient in Pms2 and Mlh1 by gene targeting were obtained from R.M. Liskay, Oregon Health Sciences University, Portland, OR 26 27. Mice heterozygous for mlh1 and pms2 were mated to generate mice heterozygous at both the mlh1 and pms2 loci. These double heterozygotes were then mated to generate mice homozygous for the null mutation at both loci. msh2 mutant mice were generated by replacing exon 7 with a neomycin cassette (Hofland, N., R. Smits, W. Edelmann, R. Kucherlapati, and R. Fodde, manuscript in preparation). The phenotype of these mice resembles that of previously described msh2 mutant mouse lines 28 29. All mouse strains were carried as heterozygotes, and wild-type (wt) littermates were used as controls.
Cells and Cell Culture.
B cells were isolated from spleens by depletion of RBCs by lysis in Gey's solution for 5 min on ice and by depletion of T cells with a cocktail of anti-T cell reagents, anti-CD4 (GK1.5), anti-CD8 (3.168), and anti-Thy1 (HO13.4 and J1J10), followed by anti–rat κ chain mAb (MAR18.5) and guinea pig complement (Pelfreeze Biochem). Viable cells were isolated by flotation on Ficoll/Hypaque gradients (δ = 1.09). 106 B cells were cultured at 2 × 105/ml in 6-well plates for 4 d in RPMI 1640 (BioWhittaker), with 10% FCS (Hyclone), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from GIBCO BRL), and 5 × 10−5 M 2-ME (Sigma Chemical Co.). LPS (50 μg/ml; Sigma Chemical Co.), recombinant murine (rm)IL-4 (800 U/ml; gift of W. Paul, National Institutes of Health, Bethesda, MD), rmIL-5 (150 U/ml; PharMingen), human TGF-β1 (2 ng/ml; R&D Systems), and anti–δ-dextran (0.3 ng/ml; gift of C. Snapper, Uniformed Services University of the Health Sciences, Bethesda, MD) were added at the initiation of culture. In one experiment (see Table ), a combination of LPS plus dextran sulfate (30 μg/ml; Amersham Pharmacia Biotech) was used to induce IgG2b.
Table 2.
Isotype Switching Is Decreased in MMR-deficient IgM+IgD+ Mature B Cells
| Percentage of B cells | Percentage of wt switching | |||||
|---|---|---|---|---|---|---|
| IgM+IgD+ | IgG1 (LPS + IL-4) | IgG3 (LPS) | IgG2b (LPS + DS) | IgG2b (LPS + TGF-β) | ||
| Msh2− | T-depleted | 62 | – | 48 | 58 | |
| IgM+IgD+ | 79 | 88 | 55 | 44 | 66 | |
| Mlh1− | T-depleted | – | – | – | – | |
| IgM+IgD+ | 80 | 61 | 88 | − | − | |
| Pms2− | T-depleted | – | – | – | – | |
| IgM+IgD+ | 71 | 69 | 55 | – | – | |
| Mlh1−/Pms2− | T-depleted | 47 | – | 48 | 28 | |
| IgM+IgD+ | 85 | 50 | – | 31 | 44 | |
| Wt | T-depleted | 100 | 100 | 100 | 100 | |
| IgM+IgD+ | 88 | 100 | 100 | 100 | 100 | |
FACS® Analysis.
Before staining, cells were given a brief acid treatment to remove Fc receptor–bound Ig 30. Pelleted cells were drained and resuspended in 500 μl of 50 mM NaOAc, pH 5.2, 85 mM NaCl, 5 mM KCl, 1% FCS. After 2 min on ice, cells were washed twice in FACS buffer (PBS, 1% FCS, 0.2% NaN3) and stained for FACS® analysis. FITC-goat anti–mouse IgM, PE-goat F(ab′)2 anti–mouse IgG1, IgG2b, and IgG3, and PE-goat anti–mouse IgA were all purchased from Southern Biotechnology Associates. PE-anti–mouse IgDb was purchased from PharMingen. Cells were analyzed on a FACScan™ (Becton Dickinson) and gated on live lymphocytes based on forward and side scatter. IgM+IgD+ cells were sorted by FACS® and were 90–95% pure; contaminating cells were mostly IgM negative.
Analysis of DNA Synthesis, Cell Viability, and Cell Cycle.
To measure cell division, cells were cultured at 105/ml for 3 d. During the final 4 h, each well was pulsed with 1 μCi [3H]thymidine (2 Ci/mmol; ICN). Plates were harvested onto filter-mats (Wallac) and read on a 1205 Betaplate (LKB/Wallac). Data shown are the mean cpm of triplicate wells. For apoptosis and cell cycle analysis, cultured cells were pelleted, fixed in 70% ethanol for >24 h, resuspended in a buffer to facilitate extraction of low molecular weight DNA (nine parts 0.05 M Na2HPO4, and one part 25 mM citric acid, containing 1% Triton X-100), and stained with 20 μg/ml propidium iodide for FACS® analysis according to Hotz et al. 31. Modfit cell cycle analysis was used for quantitation.
DC-PCR.
Genomic DNA was isolated from cells cultured for 4 d under conditions used for switching analysis. DC-PCR was performed as described 32. In brief, DNA was digested with EcoRI overnight (2 μg/100 μl) and then ligated overnight (180 ng/100 μl) with T4 ligase (400 U; New England Biolabs). Circularized DNA was then dialyzed against deionized dH2O (0.05-μm VMWP filters; Millipore) before PCR analysis. PCR primers and conditions were as described for acetylcholine receptor (AchR) and IgG1 32 and IgG2b 33, except that HotStar Taq DNA polymerase (QIAGEN) was used. Plasmid standards P2A0 and P4AP 32 were used as templates with the primers for AchR and IgG1, respectively, to determine the PCR conditions under which the amount of product depends on the amount of input template in a linear fashion. Radiolabeled PCR products were analyzed on 8% polyacrylamide gels, dried, and exposed to X-ray film overnight at room temperature. Densitometry was performed on films with a Personal Densitometer SI (Molecular Dynamics). For quantitation, the amounts of Sμ-γ1 and Sμ-γ2b PCR products were normalized to the amount of AchR product.
Results
To evaluate whether MMR enzymes are involved in CSR, T-depleted spleen cells were isolated from mice deficient in one or more of the MMR enzymes and assessed for ability to undergo isotype switching in vitro. Our analysis included mice made deficient by targeted gene replacement in the MutS homologue Msh2, and the MutL homologues Mlh1 and Pms2. Splenic B cells from MMR-deficient mice or their wt littermates were stimulated in culture with either LPS to induce IgG2b and IgG3, LPS and IL-4 to induce IgG1, or LPS, anti–δ-dextran, IL-4, IL-5, and TGF-β1 to induce IgA.
Analysis of Proliferation, Cell Cycle, and Cell Viability.
As isotype switching is dependent on cell division 34, we first determined whether the MMR-deficient cells could synthesize DNA and cycle at the same rate as wt cells, and whether or not they were more prone to cell death. We used two methods to examine these questions. First, [3H] thymidine incorporation was used to measure DNA synthesis. MMR-deficient cells cultured under all three conditions used to induce isotype switching showed no difference in [3H]thymidine incorporation compared with wt cells (Fig. 1). Second, cells cultured for 2 or 4 d were stained with propidium iodide to quantify the percentage of cells in cycle and the amount of apoptosis during the culture period. MMR-deficient cells were indistinguishable from wt cells cultured under all three conditions with regard to both the percentage of cells in cycle and the percentage of cells undergoing apoptosis on day 2 (Table ) and on day 4 (not shown). In addition, recovery of viable cells on day 4 was similar between wt and MMR-deficient cultures (Table ). We conclude that MMR-deficient B cells divide and survive in culture as well as wt cells.
Figure 1.
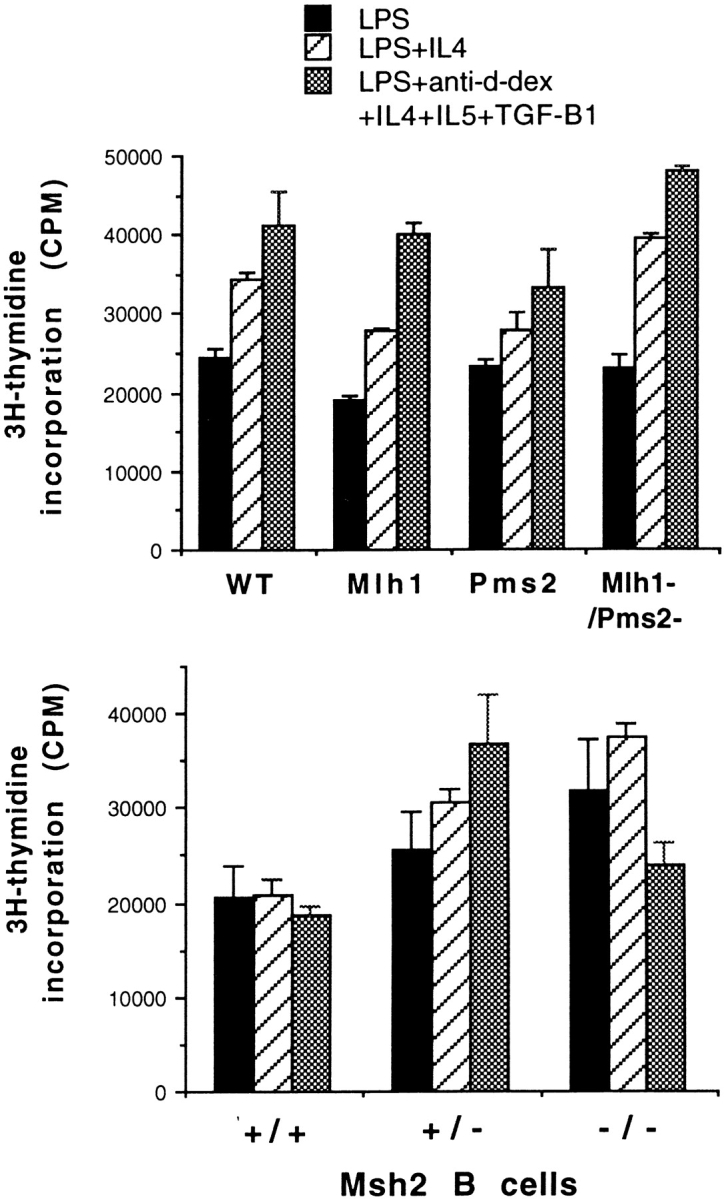
Splenic B cells from MMR-deficient mice incorporate [3H]thymidine as well as wt littermates. B cells were cultured under conditions used to induce isotype switching for 3 d and pulsed for the final 4 h with [3H]thymidine before harvesting. A representative of three separate experiments is shown.
Table 1.
Cell Cycle Analysis of Wt and MMR-deficient B Cells on Day 2
| B cells | Treatment | Percentage of cycling cells in each phase on day 2 | Percentage of apoptotic cells on day 2 (<2n) | Viable cell recovery on day 4 | ||
|---|---|---|---|---|---|---|
| G0/G1 | S | G2/M | ||||
| ×10−6 | ||||||
| Wt-msh | LPS | 33.6 | 50.2 | 16.2 | 3.4 | 8.4 |
| Pms2−/− | 29.5 | 48.3 | 22.2 | 1.6 | 7.7 | |
| Mlh1−/− | 32.4 | 48.6 | 19.1 | 1.4 | 9.5 | |
| Wt-pms2 | 30.5 | 48.4 | 21.1 | 1.9 | 14.6 | |
| Wt-msh | LPS + IL-4 | 38.3 | 45.4 | 16.3 | 3.3 | 9.6 |
| Msh2−/− | 34.6 | 47.8 | 17.6 | 2.6 | 7.7 | |
| Pms2−/− | 35.2 | 45.0 | 19.8 | 1.9 | 7.2 | |
| Mlh1−/− | 36.9 | 45.0 | 18.1 | 1.7 | 8.6 | |
| Wt-pms2 | 33.9 | 50.4 | 15.7 | 2.7 | 6.8 | |
| Wt-msh | LPS + α–δ dextran | 50.8 | 37.8 | 11.4 | 1.8 | 7.2 |
| Msh2−/− | 48.6 | 41.2 | 10.2 | 0.8 | 9.0 | |
| Pms2−/− | 49.7 | 39.7 | 10.6 | 0.9 | 7.1 | |
| Mlh1−/− | 52.8 | 35.3 | 11.9 | 1.1 | 8.7 | |
| Wt-pms2 | 52.1 | 37.5 | 10.4 | 1.2 | 7.4 | |
FACS® Analysis Demonstrates Reduction of Class Switching in B Cells from MMR-deficient Mice.
Isotype switching was measured after 4 d in culture with the stimulators described above. Cells were surface stained with FITC–anti-IgM and PE–anti-isotype reagents for FACS® analysis. A representative experiment using B cells from a mouse deficient in both Mlh1 and Pms2 is shown in Fig. 2. The specificity of the staining reagents and the induction of switching are demonstrated in the first two columns with wt B cells. LPS and IL-4, but not LPS alone, induce switching to IgG1. LPS induces switching to IgG3 and IgG2b, but addition of IL-4 downregulates switching to these isotypes. Comparison of the second and third columns shows that switching to all isotypes is reduced in B cells from Mlh1−/Pms2− mice. 44% of wt B cells expressed IgG1, compared with only 23% of Mlh1−/Pms2− B cells. IgG3 and IgA were also reduced by ∼50%. IgG2b was reduced even more, as 12% of wt B cells expressed IgG2b, compared with only 3% of Mlh1−/Pms2− B cells.
Figure 2.
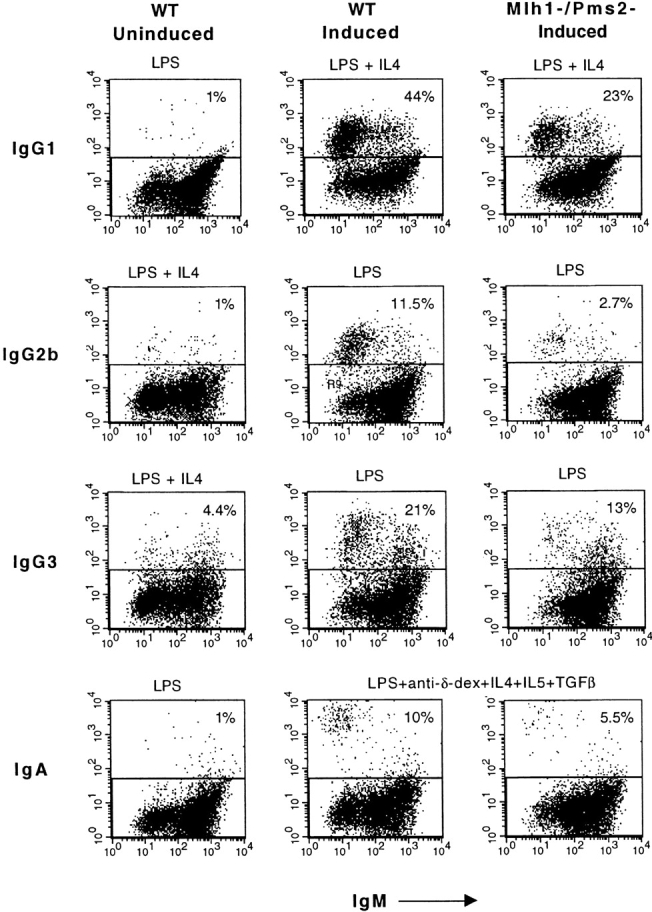
Representative FACS® analysis of splenic B cells from wt and Mlh1-/Pms2-deficient mice induced to switch in vitro. The first column (negative control) shows cells stimulated under conditions that do not induce the indicated isotype. Induced switching by wt cells (second column) is compared with switching by Mlh1−/Pms2− B cells (third column).
The results of many such experiments with Mlh1-, Pms2-, Mlh1-/Pms2-, and Msh2-deficient B cells are shown in Fig. 3. Compared with wt B cells, switching to all isotypes is reduced in B cells from all MMR-deficient mice. The extent of the reduction varies between isotypes, as well as between the different enzyme deficiencies. IgG1 and IgG3 are reduced by 1.6–2-fold, while IgG2b is the most affected isotype, reduced by 2–4-fold. IgA is reduced by two- to threefold. IgG1 and IgG2b are reduced more in Mlh1-deficient B cells than in Pms2-deficient cells (P < 0.05 and P < 0.0005, respectively). Switching by Mlh1−/Pms2− B cells is similar to that by B cells deficient only in Mlh1. These data are consistent with the fact that Mlh1 functions as part of an Mlh1/Pms2 heterodimer, and also in another heterodimer with Mlh3 35.
Figure 3.

MMR deficiency reduces isotype switching by 35–75%. Data are compiled from several experiments with mutant mice and their wt littermates. Mlh1− (n = 4) and Pms2− (n = 5), error bars show 1 SD; Mlh1−/Pms2− and Msh2− (n = 2), error bars show range.
It has been reported that Msh2-deficient mice have reduced numbers of mature IgM+IgD+ B cells 18 25, and it was possible that this was the cause of the reduced ability of these cells to undergo isotype switching. We analyzed the percentage of spleen cells that are IgM+IgD+ from the MMR-deficient mice by FACS® and found a small but reproducible decrease compared with wt mice (Table ). To determine if the decrease in isotype switching might be due to this decrease in mature B cell number, we sorted IgM+IgD+ cells from MMR-deficient and from wt mice by FACS® and determined the ability of these mature B cells to switch compared with unsorted T-depleted spleen cells. Isotype switching by the IgM+IgD+ sorted population was comparable to switching by T-depleted spleen cells, and switching was reduced in MMR-deficient mature IgM+IgD+ cells compared with wt. Again, IgG2b was reduced more than IgG1 (Table ).
MMR Deficiency Reduces Class Switching at the Level of DNA Recombination.
To verify that the decrease in isotype switching occurs at the level of DNA recombination, we used a quantitative DC-PCR assay to measure switch recombination 32. After EcoRI digestion of genomic DNA, Sμ and Sγ2b or Sμ and Sγ1 reside on the same DNA fragment if switching to that isotype has occurred. To detect these fragments, EcoRI-digested genomic DNA was ligated under dilute conditions to form circles, and then primers directing PCR amplification across the ligated EcoRI junctions were used to generate PCR products of uniform size, independent of where recombination took place within the switch regions. Primers specific for a control EcoRI fragment that does not undergo rearrangement (AchR) were used to control for the efficiency of the digestion and circularization. Plasmid standards for AchR and Sγ1 32 were used to establish PCR conditions in the linear range. As shown by serial dilution of standards and samples in Fig. 4, the amount of the PCR products depended on the amount of template added. For quantitation (Table ), the Sμ-Sγ2b and Sμ-Sγ1 DC-PCR results were normalized to the amount of AchR product.
Figure 4.

DC-PCR analysis demonstrates that switch recombination is reduced at the DNA level in MMR-deficient mice. Standard shown is a titration (threefold dilution) of plasmids P2A0 and P4AP with the same primers used to detect AchR and Sμ-Sγ1, respectively. Titration of sample templates was by twofold dilutions. DC-PCR for Sμ-Sγ1 and Sμ-γ2b was repeated on these same samples, as well as on samples from Msh2− cells (not shown), with similar results.
Table 3.
Comparison of Results from DC-PCR and FACS® Analyses from the Same Experiment
| IgG2b | IgG1 | |||
|---|---|---|---|---|
| DC-PCR | FACS® | DC-PCR | FACS® | |
| % | ||||
| Pms2− | 24 | 41 | 72 | 59 |
| Mlh1−/Pms2− | 12 | 34 | 69 | 52 |
The DC-PCR analysis indicated that the frequency of Sμ-Sγ2b switch recombination in Pms2− and Mlh1−/Pms2− B cells was three- to eightfold lower than in wt B cells (Fig. 4 and Table ). The reduction in Sμ-Sγ1 recombination was less dramatic, but still detectable in Pms2− and Mlh1−/Pms2− cells. Quantitation of the FACS® and DC-PCR data (Table ) shows that the two methods compare reasonably well, and that IgG2b is more severely affected than IgG1 by both assays. We conclude that the reduction in isotype switching observed in MMR-deficient B cells occurs at the level of DNA recombination.
Discussion
We have found that deficiency in any of three MMR enzymes, Mlh1, Pms2, or Msh2, results in a decrease in isotype switching in vitro. By FACS® analysis of surface Ig, the effect ranged from a 35% decrease in IgG1 in Pms2−/− and Msh2−/− B cells to a 75% decrease in IgG2b in Mlh1−/− B cells, the average reduction being 50%. This decrease was shown to occur at the level of DNA recombination by DC-PCR analysis. Although MMR-deficient B cells are capable of isotype switching, the process is clearly less efficient.
We found no evidence that MMR deficiency has any effect on the ability of B cells to grow and to enter cell cycle in response to the stimulation conditions used to induce isotype switching. It has been reported that Msh2-deficient mice have decreased numbers of mature IgM+IgD+ B cells 18. However, this decrease is observed predominantly in the bone marrow, and only marginally in the spleen 25. We found there to be an average of 10% fewer IgM+IgD+ B cells in spleens from MMR-deficient mice, and when sorted by FACS®, these mature B cells also have the phenotype of reduced isotype switching in vitro.
It has been observed at day 8 after immunization that GCs from msh2 −/− mice were smaller than wt and showed increased apoptosis relative to wt mice 25. The authors suggest that the reduction in IgG antibody-forming cells in the PALS and IgG production in these mice may be explained by the death of cells that undergo high rate proliferation. However, increased apoptosis was only observed in vivo; no increase in apoptosis or decrease in proliferation was observed in msh2 −/− B cells in vitro by these authors. Thus, it is possible that the apoptosis observed in vivo is an indirect effect of the msh2 mutation. Msh2-deficient mice were found to have normal T cell populations and normal architecture of PALS and spleen, including GC formation and follicular dendritic cell (FDC) network, suggesting the effect is on the B cells themselves 25. However, it is possible that the apoptosis detected in GCs may be related to a lack of positive selection due to the altered somatic mutation of V region genes observed in msh2 −/− mice 16 17 18. Decreased somatic hypermutation should lead to a decrease in antibody affinity maturation, consistent with the finding that msh2 −/− mice show no memory response upon secondary immunization 25. We conclude that the decrease in IgG responses in msh2−/− mice is a direct result of impaired CSR.
We have developed a model demonstrating a possible role for MMR enzymes in switch recombination (Fig. 5). The model is based on an illegitimate priming mechanism for CSR 3 21. In our model, CSR is initiated by DSBs, and occurs by an end-joining type of recombination in which a single-strand end from one S region uses bits of homology to prime DNA synthesis on the other, and vice versa. Small stretches of identity at switch region junctions range from only 1 to 5 nt, yet microhomologies on this order have been proposed to prime DNA synthesis for gap filling as a mechanism for end-joining 36. As the 3′ ends must have perfectly paired nt in order to prime DNA synthesis, heterologous DNA surrounding the DSB must be removed.
Figure 5.

Model for a possible role for MMR enzymes in isotype switch recombination. This model for CSR (references 3 and 21) is similar to a model for end-joining in Xenopus laevis oocytes proposed by Lehman et al. (reference 36), involving gap repair DNA synthesis primed by microhomologies. MMR enzymes may be involved in 3′ end-processing to remove nonhomologous DNA surrounding a DSB in order to reach bits of identity that can be used to prime DNA synthesis in an end-joining reaction. In S. cerevisiae, Msh2 is required for the removal of heterologous 3′ ends >30 nt in length during DSB-induced recombination between homologous sequences (reference 37).
We hypothesize that MMR enzymes may be involved in processing the 3′ ends produced by DSBs based on experiments performed in Saccharomyces cerevisiae. Sugawara et al. 14 showed that the MMR genes msh2 and msh3 are required for removal of nonhomologous DNA surrounding a DSB in order to reach the region homologous to the template for the repair of the DSB. Msh2 and 3 are essential for DSB repair when the heterologous DNA segment is 30 nt or greater in length 37. However, if the segment is only 20 nt long, repair is inhibited by 50–70% and if 10 nt long, by 15% in the msh2-deficient yeast. DNA polymerase δ was found to contribute to removal of shorter stretches of nonhomology 37. The mechanism of the removal of nonhomologous ends in yeast requires the Rad1–Rad10 complex, an endonuclease physically associated with Msh2. Thus, we propose that the role of MMR enzymes in CSR may be to produce 3′ single-strand ends capable of priming DNA synthesis by removing heterologous DNA to expose a few nt complementary to the other S region. The microhomology used for priming DNA synthesis may result in the short bits of identity observed at the S–S junctions 3.
This model provides several possible explanations for why CSR is not completely inhibited in the MMR-deficient B cells. It is possible that DNA polymerase δ or an exonuclease, such as the B cell–specific exonuclease BCAN 38, could partially substitute in the absence of MMR enzymes. In addition, 3′ end-processing may not be required for switch events when an adequate microhomology between the two combining S regions exists very near the sites of the DSB. The finding that some isotypes are more dependent on MMR enzymes than others may be due to the fact that the sequences of different S regions differ, and thus their dependence on 3′ end-processing may differ. Finally, the redundancy among different MMR enzymes is not well understood. This redundancy might vary depending on the particular function of the MMR enzyme and might help to account for the differential dependence of CSR on the three MMR enzymes examined here.
In yeast, Msh2, but not Mlh1 or the homologue of Pms2 (yeast Pms1), is required for the removal of heterologous sequences surrounding a DSB in order to repair it by homologous recombination 14. This result differs from our finding that deletion of any of the three genes for MMR enzymes that we tested results in a reduction in CSR. We propose two possible explanations for this difference. CSR differs from homologous recombination, as it does not involve recombination between homologous sequences. Whether the end-joining type of recombination in yeast requires MMR enzymes has not been reported. In addition, although yeast and mammalian MMR enzymes have been shown to be similar, they are not identical, and thus some of their functions might differ.
Acknowledgments
We are grateful to Dr. R. Michael Liskay for the generous gift of the Mlh1- and Pms2-deficient mice, and to Michael Twarog for assistance in typing mice. We thank Dr. Martin Marinus and Dr. Rachel Gerstein for helpful discussions, and Dr. Sean Bradley for review of this manuscript.
This work was supported by a grant to J. Stavnezer from the National Institutes of Health (AI-23283). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Allergy and Immunology.
Footnotes
1used in this paper: AchR, acetylcholine receptor; CSR, class switch recombination; DC-PCR, digestion-circularization PCR; DSB, double strand break; GC, germinal center; MMR, mismatch repair; nt, nucleotide(s); PALS, periarteriolar lymphatic sheath; S, switch; wt, wild-type
Note added in proof. Another group has recently reported that B cells from Msh2-deficient mice show reduced IgG switching in vitro and also in vivo (Ehrenstein, M.R., and M.S. Neuberger. 1999. EMBO (Eur. Mol. Biol. Organ.) J. 18:3484–3490).
References
- Bollag R.J., Waldman A.S., Liskay R.M. Homologous recombination in mammalian cells. Annu. Rev. Genet. 1989;23:199–225. doi: 10.1146/annurev.ge.23.120189.001215. [DOI] [PubMed] [Google Scholar]
- Liskay R.M., Letsou A., Stachelek J.L. Homology requirement for efficient gene conversion between duplicated chromosomal sequences in mammalian cells. Genetics. 1987;115:161–167. doi: 10.1093/genetics/115.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W., Hertz G.Z., Scappino L., Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 1993;21:365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth D.B., Wilson J.H. Nonhomologous recombination in mammalian cellsrole for short sequence homologies in the joining reaction. Mol. Cell. Biol. 1986;6:4295–4304. doi: 10.1128/mcb.6.12.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuerffel R.A., Du J., Thompson R.J., Kenter A.L. Ig Sγ3 DNA-specific double strand breaks are induced in mitogen-activated B cells and are implicated in switch recombination. J. Immunol. 1997;159:4139–4144. [PubMed] [Google Scholar]
- Rolink A., Melchers F., Andersson J. The SCID but not the RAG-2 gene product is required for Sμ-S∈ heavy chain class switching. Immunity. 1996;5:319–330. doi: 10.1016/s1074-7613(00)80258-7. [DOI] [PubMed] [Google Scholar]
- Casellas R., Nussenzweig A., Wuerffel R., Pelanda R., Reichlin A., Suh H., Qin X.-F., Besmer E., Kenter A., Rajewsky K., Nussenzweig M.C. Ku80 is required for immunoglobulin isotype switching. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis J.P., Gu Y., Lansford R., Sonoda E., Ferrini R., Davidson L., Rajewsky K., Alt F.W. Ku70 is required for late B cell development and immunoglobulin heavy chain switching. J. Exp. Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H.M., Cheo D.L., Friedberg E., Storb U. The inactivation of the XP-C gene does not affect somatic hypermutation or class switch recombination of immunoglobulin genes. Mol. Immunol. 1997;34:527–533. doi: 10.1016/s0161-5890(97)00064-3. [DOI] [PubMed] [Google Scholar]
- Alani E., Lee S., Kane M.F., Griffith J., Kolodner R.D. Saccharomyces cerevisiae Msh2, a mispaired base recognition protein, also recognizes Holliday junctions in DNA. J. Mol. Biol. 1997;265:289–301. doi: 10.1006/jmbi.1996.0743. [DOI] [PubMed] [Google Scholar]
- Alani E., Chi N.W., Kolodner R.D. The Saccharomyces cerevisiae protein Msh2 binds to duplex oligonucleotides containing mismatched DNA basepairs and insertions. Genes Dev. 1995;9:234–247. doi: 10.1101/gad.9.2.234. [DOI] [PubMed] [Google Scholar]
- Rayssiguier C., Thaler D.S., Radman M. The barrier to recombination between E. coli and S. typhimurium is disrupted in mismatch-repair mutants. Nature. 1989;342:396–401. doi: 10.1038/342396a0. [DOI] [PubMed] [Google Scholar]
- Selva E.M., New L., Crouse G.F., Lahue R.S. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae . Genetics. 1995;139:1175–1188. doi: 10.1093/genetics/139.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara N., Paques F., Calaiacovo M., Haber J.E. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proc. Natl. Acad. Sci. USA. 1998;94:9214–9219. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascalho M., Wong J., Steinberg C., Wabl M. Mismatch repair co-opted by hypermutation. Science. 1998;279:1207–1210. doi: 10.1126/science.279.5354.1207. [DOI] [PubMed] [Google Scholar]
- Frey S., Bertocci B., Delbos F., Quint L., Weill J.C., Reynaud C.A. Mismatch repair deficiency interferes with the accumulation of mutations in chronically stimulated B cells and not with the hypermutation process. Immunity. 1998;9:127–134. doi: 10.1016/s1074-7613(00)80594-4. [DOI] [PubMed] [Google Scholar]
- Phung Q.H., Winter D.B., Cranston A., Tarone R.E., Bohr V.A., Fishel R., Gearhart P.J. Increased hypermutation at G and C nucleotides in immunoglobulin variable genes from mice deficient in the MSH2 mismatch repair protein. J. Exp. Med. 1998;187:1745–1751. doi: 10.1084/jem.187.11.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C., Ehrenstein M.R., Neuberger M.S., Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- Winter D.B., Phung Q.H., Umar A., Baker S.M., Tarone R.E., Tanaka K., Liskay R.M., Kunkel T.A., Bohr V.A., Gearhart P.J. Altered spectra of hypermutation in antibodies from mice deficient for the DNA mismatch repair protein PMS2. Proc. Natl. Acad. Sci. USA. 1998;95:6953–6958. doi: 10.1073/pnas.95.12.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W., Wilson M., Stavnezer J. Mutations, duplication, and deletion of recombined switch regions suggest a role for DNA replication in the immunoglobulin heavy-chain switch. Mol. Cell. Biol. 1989;9:1850–1856. doi: 10.1128/mcb.9.5.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W., Stavnezer J. Copy choice mechanism of immunoglobulin heavy chain switch recombination. Mol. Cell. Biol. 1990;10:397–400. doi: 10.1128/mcb.10.1.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob J., Kassir R., Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl) acetyl. I. The architecture and dynamics of the responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toellner K.-M., Gulbranson-Judge A., Taylor D.R., Sze D.M.-Y., MacLennan I.C.M. Immunoglobulin switch transcript production in vivo related to the site and time of antigen-specific B cell activation. J. Exp. Med. 1996;183:2303–2312. doi: 10.1084/jem.183.5.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K., Kim J., Kim H.-S., Shin H.S. Isolated human germinal center centroblasts have an intact mismatch repair system. J. Immunol. 1998;161:6128–6132. [PubMed] [Google Scholar]
- Vora K.A., Tumas-Brundage K.M., Lentz V.M., Cranston A., Fishel R., Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J. Exp. Med. 1999;189:471–481. doi: 10.1084/jem.189.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S.M., Bronner C.E., Zhang L., Plug A.W., Robatzek M., Warren G., Elliott E.A., Yu J., Ashley T., Arnheim N. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- Baker S.M., Plug A.W., Prolla R.A., Bronner C.E., Harris A.C., Yao X., Christie D.-M., Monell C., Arnheim N., Bradley A. Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat. Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- DeWind N., Dekker M., Berns A., Radman M., TeRiele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- Reitmair A.H., Schmits R., Ewel A., Bapat B., Redston M., Mitri A., Waterhouse P., Mittrucker H.W., Wakeham A., Liu B. MSH2 deficient mice are viable and susceptible to lymphoid tumours. Nat. Genet. 1995;11:64–70. doi: 10.1038/ng0995-64. [DOI] [PubMed] [Google Scholar]
- Kumagai K., Abo T., Sekizawa T., Sasaki M. Studies of surface immunoglobulins on human B lymphocytesdissociation of cell-bound immunoglobulins with acid pH or at 37°C. J. Immunol. 1975;115:982–987. [PubMed] [Google Scholar]
- Hotz M., Gong J., Traganos F., Darzynkiewicz Z. Flow cytometric detection of apoptosiscomparison of the assays of in situ DNA degradation and chromatin changes. Cytometry. 1994;15:237–244. doi: 10.1002/cyto.990150309. [DOI] [PubMed] [Google Scholar]
- Chu C.C., Paul W.E., Max E.E. Quantitation of immunoglobulin μ-γ1 heavy chain switch region recombination by a digestion-circularization polymerase chain reaction method. Proc. Natl. Acad. Sci. USA. 1992;89:6978–6982. doi: 10.1073/pnas.89.15.6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballantyne J., Henry D.L., Muller J.R., Briere F., Snapper C.M., Kehry M., Marcu K.B. Efficient recombination of a switch substrate retrovector in CD40-activated B lymphocytesimplications for the control of CH gene switch recombination. J. Immunol. 1998;161:1336–1347. [PubMed] [Google Scholar]
- Hodgkin P.D., Lee J.-H., Lyons A.B. B cell differentiation and isotype switching is related to division cycle number. J. Exp. Med. 1996;184:277–281. doi: 10.1084/jem.184.1.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolodner R.D., Marsischky G.T. Eukaryotic DNA mismatch repair. Curr. Opin. Genet. Dev. 1999;9:89–96. doi: 10.1016/s0959-437x(99)80013-6. [DOI] [PubMed] [Google Scholar]
- Lehman C.W., Trautman J.K., Carroll D. Illegitimate recombination in Xenopuscharacterization of end-joined junctions. Nucleic Acids Res. 1994;22:434–442. doi: 10.1093/nar/22.3.434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paques F., Haber J.E. Two pathways for removal of nonhomologous DNA ends during double-strand break repair in Saccharomyces cerevisiae . Mol. Cell. Biol. 1997;17:6765–6771. doi: 10.1128/mcb.17.11.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter A.L., Tredup J. High expression of a 3′ to 5′ exonuclease activity is specific to B lymphocytes. Mol. Cell. Biol. 1991;11:4398–4404. doi: 10.1128/mcb.11.9.4398. [DOI] [PMC free article] [PubMed] [Google Scholar]