

Abstract
Galectin-1 (GAL-1), a member of a family of conserved β-galactoside–binding proteins, has been shown to induce in vitro apoptosis of activated T cells and immature thymocytes. We assessed the therapeutic effects and mechanisms of action of delivery of GAL-1 in a collagen-induced arthritis model. A single injection of syngeneic DBA/1 fibroblasts engineered to secrete GAL-1 at the day of disease onset was able to abrogate clinical and histopathological manifestations of arthritis. This effect was reproduced by daily administration of recombinant GAL-1. GAL-1 treatment resulted in reduction in anticollagen immunoglobulin (Ig)G levels. The cytokine profile in draining lymph node cells and the anticollagen IgG isotypes in mice sera at the end of the treatment clearly showed inhibition of the proinflammatory response and skewing towards a type 2–polarized immune reaction. Lymph node cells from mice engaged in the gene therapy protocol increased their susceptibility to antigen-induced apoptosis. Moreover, GAL-1–expressing fibroblasts and recombinant GAL-1 revealed a specific dose-dependent inhibitory effect in vitro in antigen-dependent interleukin 2 production to an Aq-restricted, collagen type 2–specific T cell hybridoma clone. Thus, a correlation between the apoptotic properties of GAL-1 in vitro and its immunomodulatory properties in vivo supports its therapeutic potential in the treatment of T helper cell type 1–mediated autoimmune disorders.
Keywords: galectin-1, gene therapy, apoptosis, collagen-induced arthritis, T cells
Galectin (GAL)-11 is a member of a growing family of animal β-galactoside–binding proteins, which are highly conserved throughout animal evolution 1 2 3 and share significant sequence similarities in the carbohydrate recognition domain 1 4. The precise functions of individual members of this family have been difficult to assess in vivo by virtue of their widespread expression and overlapping specificities 3 4 5. Nevertheless, GAL-1 has been proposed to play key roles in a wide variety of biological events involving carbohydrate recognition, such as cell adhesion 6, cell growth regulation 7 8 9, metastasis 10, and immunomodulation 11 12.
This homodimeric protein, composed by subunits of ∼134 amino acids 1, recently has been shown to induce in vitro apoptosis of activated mature T cells 13 14 and particular subsets of non- or negatively selected CD4loCD8lo immature thymocytes 15. The apoptotic effect of GAL-1 depended upon the activation state of T cells and was mediated by engagement of CD43 or CD45, particularly the polylactosamine-enriched CD45RO splicing product 13 15. Expression of GAL-1 has been specially identified in lymphoid organs such as thymus 16 and lymph nodes 17, in activated macrophages 14 18 and T cells 8, and in immune privileged sites such as placenta 3 19 and cornea 20. All these observations suggest that GAL-1 could play an important role in generating and maintaining central and peripheral immune tolerance.
Rheumatoid arthritis (RA) is a common chronic autoimmune disease for which there is no effective therapy capable of preventing long-term progression and joint damage 21 22. To test in vivo the immunoregulatory properties of GAL-1, we used a collagen-induced arthritis (CIA) model, which is a widely accepted experimental model for human RA 23. Murine CIA is induced in genetically susceptible DBA/1 mice and resembles RA in that both cellular and humoral mechanisms are involved in the pathogenic process 21. Blood-derived cells that migrate into joints, in conjunction with activated synovial cells, produce inflammatory and type 1 cytokines together with degradative enzymes that progressively lead to the destruction of cartilage and bone 21 23.
Effective treatment of arthritis will require the elimination of arthritogenic lymphocytes that initiate and perpetuate joint inflammation, as well as the induction of tissue repair. Hence, GAL-1–induced apoptosis could provide for an ideal mechanism using an endogenous naturally occurring protein to terminate the autoimmune T cell attack, preventing the expansion of dominant autoaggressive clones, and assuring a minimum of detrimental bystander damage to the local parenchyma 24 25 26.
Our study deals with the concept of an in vivo role for GAL-1 in T cell–dependent immunoregulation and provides experimental data, using gene and protein therapy strategies, aimed at validating GAL-1's therapeutic potential.
Materials and Methods
Subcloning of Murine GAL-1 into pCDNA3 Expression Vector.
Murine GAL-1 cDNA (length, 495 bp) was obtained from the IMAGE Consortium (HGMP Resource Centre, MRC, Cambridge, UK; clone 330090) and subcloned immediately downstream of the CMV promoter of a HindIII/BamHI-cut pCDNA3 expression vector (Invitrogen) by using a PCR strategy. Oligonucleotide primers (5′) and (3′) were ordered from Oswell.
For cloning purposes, the sense deoxynucleotide primer 5′-CAAGCTTCCATGGCCTGTGGTCTGGTCGCCAGCA and the antisense primer 5′-GGGATCCTCACTCAAGGCCACGCACTT contained a HindIII and a BamHI restriction site, respectively, and 21 nucleotides that annealed to the coding sequence of mouse GAL-1 cDNA. The reaction mixture consisted of 100 ng/ml cDNA, 100 mM of each primer, 0.20 mM dNTPs, 1.5 mM MgCl2, 1× PCR buffer (10× buffer: 500 mM KCl, 100 mM Tris-HCl, pH 8.3, 0.01% wt/vol gelatin), and 25 U/ml Taq DNA polymerase (Appligen Oncor) to a final volume of 100 μl. The amplification procedure included a denaturation step at 94°C for 4 min, followed by 35 cycles of 1 min strand separation at 94°C, 1 min annealing at 56°C, and 3 min extension at 72°C, followed by an elongation step of 10 min at 72°C. The PCR reaction product was further purified by agarose gel electrophoresis, and then phenol-extracted, ethanol-precipitated, and finally its ends were blunted with Klenow fragment of DNA polymerase. After digestion, with HindIII and BamHI, the 495-bp product was ligated into the HindIII/BamHI sites of the eukaryotic CMV promoter-driven expression vector pCDNA3 by using T4 DNA ligase. The ligated DNA was then used for the transformation of competent Escherichia coli DH5α cells. Ampicillin-resistant clones were screened for the presence of the insert by HindIII/BamHI restriction. A recombinant clone that contained the 495-bp insert was named mGAL-1 and expanded to mass culture, and the plasmid DNA was purified by equilibrium centrifugation in CsCl-ethidium bromide gradients. DNA restriction enzymes were purchased from New England Biolabs or Boehringer Mannheim.
Antigen-dependent IL-2 Production Using a Collagen Type II–specific T Cell Hybridoma.
In vitro antigen presentation assays were performed trying to mimic the in vivo therapeutic protocols: for the gene therapy protocol, a collagen type II (CII)-specific and Aq-restricted T cell hybridoma clone (HCQ.6) was stimulated with CII (50 μg/ml) and cultured in 96-well plates at a density of 5 × 105 cells/ml in the presence of splenocytes from naive DBA/1 mice (5 × 106 cells/ml) as APCs, in DMEM supplemented with 10% FCS, 2-ME, streptomycin, penicillin, and glutamine as previously described 27. To analyze the influence of GAL-1 on antigen presentation, mGAL-1– or pCDNA3-transfected DBA/1 fibroblasts were added to some wells at increasing concentrations of 0.25, 0.5, and 1 × 106 cells/ml in a final volume of 200 μl. For the protein therapy protocol HCQ.6 cells were cultured in identical experimental conditions in the presence of splenocytes from naive DBA/1 mice and the specific antigen. Recombinant GAL-1 was added to some wells at concentrations ranging from 0.04 to 4 μg/ml at different time points of the assay. To test the specificity of the effect in both experimental protocols, some wells were supplemented with thiodigalactoside (TDG) at 100 mM or the rabbit polyclonal anti–GAL-1 Ab (1:50 or 1:100 dilutions).
Supernatants were collected after overnight culture and assessed for IL-2 production by a standard ELISA using an anti–mouse IL-2 capture Ab (18161D; PharMingen), a biotinylated anti–mouse IL-2 detecting Ab (18172D; PharMingen), and the streptavidin-biotinylated horseradish peroxidase complex. Controls included HCQ.6 cells cultured with APCs in the absence of the specific antigen; HCQ.6 cells cultured with CII in the absence of APCs; and APCs incubated with CII in the absence of HCQ.6 cells. Anti-CD3–stimulated HCQ.6 cells were used as positive controls of IL-2 secretion.
DNA Transfections.
GAL-1 expression was firstly assessed by transiently transfecting mGAL-1 into COS-7 cells. In brief, exponentially growing cells were harvested 24 h before transfection and replated at a density of 106 cells/plate in 90-mm tissue culture plates in DMEM (Bio-Whittaker) containing 10% FCS (GIBCO BRL). Cells were then transfected with 20 or 30 μg of vector DNA by the Ca2+-phosphate method as previously described 28.
Also, conditionally immortalized syngeneic DBA/1 fibroblasts were permanently cotransfected by the same method with 20 or 40 μg of mGAL-1 or pCDNA3 DNA and 2.5 μg of pSV2-Hygro for hygromycin B selection, previously linearized with PvuI. Transfected DBA/1 cells were selected in DMEM medium containing 10% FCS, and 200 μg/ml hygromycin B (Boehringer Mannheim). Hygromycin-resistant clones, transfected with mGAL-1, were pooled and assessed by Western blot for expression of mGAL-1. Cells transfected with pCDNA3 alone were maintained as a population and were used as controls.
Western Blot Analysis.
Serum-free supernatants were collected from transiently and permanently transfected cells, centrifuged at 1,000 g for 5 min to discard cell debris, and stored frozen at −70°C. Then, 5 ml of these supernatants were made to 0.5% SDS final concentration and boiled for 5 min. Proteins were precipitated with 9 vol of methanol overnight at −20°C. This solution was then centrifuged at 4°C at 21,000 g for 30 min in a SS34 Sorvall rotor (Sorvall Instruments) and the precipitated proteins were dissolved in 200 μl SDS-PAGE loading buffer with 2-ME. Cells were also collected in PBS by scrapping with a rubber policeman and centrifuged at 1,000 g for 10 min. The cell pellet was resuspended in 1 ml of ice-cold lysis buffer containing 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1% NP-40, 10 mM EDTA, and a protease inhibitor cocktail (1 mM PMSF, 1 mg/ml leupeptin, 1 mg/ml pepstatin A, 10 mM iodoacetamide, and 1 mM sodium vanadate) and left on ice for 30 min. The solution was centrifuged at 4°C for 10 min at 10,000 g and the resultant cell lysate was mixed 1:1 with 2× SDS-PAGE loading buffer.
Samples corresponding to supernatants and cell lysates were boiled for 5 min, cooled on ice, and resolved on a 14% PAGE. After electrophoresis, the separated proteins were electroblotted onto nitrocellulose membranes (Schleicher and Schüll) and probed with a 1:500 dilution of a rabbit polyclonal anti–human GAL-1 antibody obtained as previously described 29. Blots were incubated with a 1:2,000 dilution of a horseradish peroxidase–conjugated donkey anti–rabbit F(ab)2 IgG (Amersham International), developed by using the ECL system and finally exposed to Amersham Hyperfilm for 1 min. Recombinant GAL-1 was used as a positive control of immunodetection and quantitation. Rainbow protein molecular weight markers were from Amersham International.
Production of Recombinant GAL-1.
Human recombinant GAL-1 was obtained as described by Hirabayashi et al. 29. In brief, the expression plasmid pH14GAL was constructed from the plasmid pUC540 (KanR) and a cDNA for GAL-1 derived from a human lung cDNA library. E. coli strains of SCS1 and Y1090 were then transformed with pH14GAL and GAL-1 expression was assessed by Western blot analysis. Finally, the recombinant protein was purified by affinity chromatography on an asialofetuin-agarose column. The hemagglutinating activity was measured as previously described 14 29 and the NH2-terminal amino acid sequence was determined with an ABI 477A pulsed-liquid sequencer (Applied Biosystems, Inc.). Lipopolysaccharide content of the purified sample was 60 ng/mg protein, determined with a colorimetric endotoxin determination reagent (Pyrodick).
Induction of Arthritis.
Bovine CII was purified from hyaline cartilage as previously described 30. Male DBA/1 mice (8–12 wk old) were immunized with 100 μg of CII emulsified in CFA (Difco) by intradermal injection at the base of the tail. The day of the disease onset in our study oscillated between days 20 and 23 after immunization with CII with a 95% incidence by day 24. Mice were maintained according to approved Home Office protocols and following Institute guidelines. The number of mice used in these studies was the minimum required to achieve statistical significance.
Experimental Design.
Two therapeutic protocols were used at onset of arthritis in our study. One was a gene therapy protocol in which DBA/1 fibroblasts expressing mGAL-1 were intraperitoneally injected into DBA/1 arthritic mice (n = 10) at the day of disease onset (4 × 106 cells/mouse). Cells permanently transfected with pCDNA3 expression vector alone were injected in the control group (n = 10). This number of cells was chosen from previous experience as being sufficient to obtain a therapeutic benefit when expressing IFN-β and a TNF antagonist. The other was a protein therapy protocol, which used recombinant human GAL-1 (100 μg diluted in 100 μl PBS) administered daily to DBA/1 mice (n = 10) by intraperitoneal injections over an 11-d period starting on the day of disease onset. Mice receiving daily intraperitoneal injections of 100 μl PBS (n = 8) were used as controls for this protocol. Due to inter-individual differences between immunized mice, recombinant GAL-1 protein treatment began on the corresponding day of arthritis onset of each individual animal. For mice receiving gene therapy it was technically difficult to proceed on this manner so they all were injected with cells on day 21.
Clinical Monitoring of Arthritis.
Starting on day 15 after immunization, mice were inspected daily for onset of the disease and macroscopic signs of arthritis according to two clinical parameters: paw swelling and clinical score. Paw swelling was assessed by measuring the thickness of the affected hind paw with calipers. The clinical severity of arthritis was monitored and scored on a daily basis using a scoring system as follows: 0, normal; 1, slight swelling and/or erythema; 2, pronounced edematous swelling; 3, ankylosis. Each limb was graded, resulting in a maximal clinical score of 12 per animal and expressed as the mean score on a given day. Severity and limb recruitment was also assessed by counting the number of affected paws. Arthritis was monitored over a 12-d treatment period by a blinded observer, after which the mice were killed. Inguinal lymph nodes and spleens were removed and disaggregated, and cells were cultured in in vitro assays.
Histopathologic Assessment.
Arthritic hindpaws (one or two per mouse) were removed post mortem on day 12 of arthritis, fixed in 10% (wt/vol) phosphate-buffered formalin, and then decalcified in 5.5% EDTA in buffered formalin. Decalcified paws were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Microscopic evaluation of arthritic paws was performed in a blinded fashion. Arthritic changes in the ankle, distal interphalangeal, proximal interphalangeal, and metacarpophalangeal joints were classified as normal, moderate, or severe based on the following criteria: normal, control nonarthritic joint; moderate, pannus formation, cartilage loss, synovitis, and erosions present but intact joint architecture; severe, marked synovitis, with extensive erosions and disrupted joint architecture.
Anti-collagen Ab ELISA.
At day 12 after onset, serum levels of anti-CII total IgG and the IgG1 and IgG2a isotypes were measured by modification of an ELISA as described previously 30. In brief, microtiter plates (Nunc) were coated with 2 μg/ml native bovine CII, blocked, and incubated with serially diluted test sera. Bound IgG was detected by incubation with alkaline phosphatase–conjugated goat anti–mouse IgG (Jackson ImmunoResearch Labs.) or sheep anti–mouse IgG1 or IgG2a (The Binding Site), followed by substrate (dinitrophenyl phosphate). Plates were washed three times between steps with 0.01% Tween 20/PBS (vol/vol). Optical densities were measured at 405 nm in a Wallac 1420 spectrophotometer. To obtain anti-CII antibody concentrations, serum samples were titered in parallel to a standard of affinity-purified anti-CII IgG 30.
Cell Culture for Cytokine Determination.
As mice were killed, spleens and inguinal lymph nodes were excised, teased apart to make a single cell suspension, washed, and cultured in 96-well plates at a density of 5 × 106 cells/ml (200 μl/well) in DMEM containing 10% heat-inactivated FCS, 2 mM glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 × 10-5 M 2-ME. Cells were cultured in medium alone, or in the presence of 5 μg/ml Con A (Sigma Chemical Co.) or bovine CII (100 μg/ml) in Tris-buffered saline. Supernatants were collected after 72 h, which was found to be the optimal incubation time for cytokine determination, and stored at −20°C until analyzed. Levels of IFN-γ and IL-5 were detected by a capture ELISA as previously described 31. In brief, 96-well flat-bottomed plates (Corning) were coated with the appropriate primary anticytokine capture Ab (5 μg/ml) in PBS, incubated overnight at 4°C, and blocked with 2% BSA in PBS. Samples and standards were then incubated overnight at 4°C, and, after washing, biotinylated anticytokine detecting Ab was added at a concentration of 2 μg/ml for 2 h. Streptavidin-biotinylated horseradish peroxidase complex (Amersham International) was finally added for 1 h at a 1:1,000 dilution and the color was developed with 3,3′, 5,5′-tetramethylbenzidine (Kirkegaard and Perry Labs.). The reaction was finally stopped by adding 100 μl 4.5 N H2SO4 and the optical density was read at 450 nm in a Wallac 1420 spectrophotometer. Each sample was assayed in triplicate and the results were presented as the mean ± SEM of three independent experiments. The antibody pairs used were as follows, listed by capture/biotinylated detection: IFN-γ, R4-GA2/XMG1.2; and IL-5, TRFK5/TRFK4. All antibodies were supplied by the American Type Culture Collection, courtesy of Dr. Abrams, DNAX (Palo Alto, CA). Standard curves were generated using mouse recombinant IFN-γ and IL-5 at concentrations ranging from 4.5 to 10,000 pg/ml.
Analysis of Hypodiploid DNA Content.
After treatment was accomplished, inguinal lymph nodes and spleens were removed and lymph node cells and splenocytes were subsequently analyzed for susceptibility to antigen-induced apoptosis by measuring the nuclear DNA content by flow cytometry as described by Nicoletti et al. 32. Lymph nodes or spleens from mice with compatible disease evolution were pooled together and analyzed. In brief, cells corresponding to mGAL-1– and pCDNA3-treated animals were cultured in 24-well plates (Corning) in complete medium at a density of 2 × 106 cells/well in the absence or in the presence of CII (100 μg/ml). Cells were recovered after 24 h, washed with ice-cold PBS, and processed for apoptotic cell detection. In brief, cell pellets were gently resuspended in 1 ml hypotonic fluorochrome solution: 50 μg/ml propidium iodide (PI; Sigma Chemical Co.), diluted in 0.1% sodium citrate plus 0.1% Triton X-100 in 12 × 75 polystyrene tubes and kept at 4°C for 3 h in the dark. The PI fluorescence emission of individual nuclei was filtered through a 585/42 nm band pass filter and measured on a logarithmic scale by a FACScan® cytometer (Becton Dickinson). Cell debris was excluded from analysis by appropriately gating on physical parameters. The number of apoptotic cells was assessed by evaluating the percentage of hypodiploid nuclei in the <2 N DNA peak (M1), and distinguished from necrotic cells by analyzing the light scatter profile. Positive controls of apoptosis included cells cultured in the presence of recombinant GAL-1 (4 μg/ml).
DNA Fragmentation Assay.
Lymph node and spleen cells of mGAL-1–treated or control mice were also processed for DNA fragmentation as previously described 14. In brief, cells cultured for 24 h in the absence or presence of CII (100 μg/ml) were harvested, washed with TNE buffer (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 2 mM EDTA, pH 8), and lysed by the addition of 0.5% SDS. Cell lysates were incubated at 56ºC for 3 h in the presence of 100 μg/ml proteinase K. DNA was further purified by successive phenol–chloroform extractions and mixed with 3 M sodium acetate, pH 5.2, and absolute ethanol. The mixture was incubated overnight at −20°C and the purified DNA was washed, resuspended in TE buffer (10 mM Tris-HCl and 1 mM EDTA, pH 7.5) and treated with 5 ml of 1 mg/ml DNase free-RNase A for 1 h. Samples were finally resuspended in loading dye and resolved on a 1.8% agarose gel in Tris-Borate-EDTA buffer, containing 0.5 μg/ml ethidium bromide.
Statistical Analysis.
The Mann-Whitney U test was used to compare nonparametric data for statistical significance using the Minitab computer package. The χ2 test was used for analysis of histological data.
Results
Permanently Transfected Syngeneic DBA/1 Fibroblasts Express High Levels of mGAL-1.
Syngeneic DBA/1 fibroblasts 44 engineered to express mGAL-1 were used in the in vivo gene therapy protocol. The CMV promoter-driven pCDNA3 expression vector containing the 495-bp mGAL-1 cDNA insert was used to establish stable transfectants by long-term hygromycin B selection. As it is clearly shown in Fig. 1, cells constitutively expressed this β-galactoside–binding protein in their whole cell lysates (lane I) not only in its predominant monomeric form of apparent molecular weight of 14.5 kD but also in its dimeric form of 29 kD. Moreover, these cells were able to secrete GAL-1 to the culture medium, as shown in lane J. Similarly, transiently transfected COS-7 cells expressed high levels of GAL-1 (lanes D and E, 20 and 30 μg of transfected DNA, respectively), although it could not be immunodetected in concentrated COS-7 culture supernatants (lanes F and G). Neither COS-7 cells nor DBA/1 fibroblasts showed any immunoreactivity with the anti–GAL-1 Ab when transfected with control pCDNA3 expression vector alone, either in their whole cell lysates (lanes C and K, respectively) or in their cell culture supernatants (lane H for COS-7 cells). Affinity purified recombinant GAL-1, which was used in the in vivo protein therapy protocol, gave rise to a single protein band of the predicted molecular weight, which immunoreacted strongly with the anti–GAL-1 Ab (lanes A and B). To semiquantitatively assess the concentration of mGAL-1 in the medium of DBA/1-transfected cells, we used different concentrations of recombinant GAL-1 and scanned a Western blot. Assuming similar epitopes are recognized by the polyclonal antibodies in the human and murine proteins, 106 DBA/1 cells produced 0.32 μg/ml per 24 h mGAL-1. This assessment may be an underestimation of the real mGAL-1 concentration.
Figure 1.

Western blot analysis of COS-7 cells and DBA/1 fibroblasts engineered to express murine GAL-1. Cell extracts and supernatants of transiently transfected COS-7 cells (lanes C–H) and permanently transfected DBA/1 fibroblasts (lanes I–K) were resolved on a 14% polyacrylamide slab gel, transferred to nitrocellulose, and probed with a 1:500 dilution of an anti–GAL-1 Ab. The immunoreactive profile of rGAL-1 is indicated in lanes A and B (0.75 and 1 μg, respectively). Extracts from COS-7 cells transfected with mGAL-1 (lanes D and E; 20 and 30 μg of transfected DNA, respectively) or control pCDNA3 (lane C) were analyzed for GAL-1 detection in parallel with their supernatants (lane F, pCDNA3; lanes G and H, GAL-1). GAL-1 was highly expressed in cell extracts (lane I) and supernatants (lane J) of transfected syngeneic DBA/1 fibroblasts but not in control transfectants (lane K). Molecular mass standards are shown on the left.
GAL-1 Suppresses Clinical Manifestations of CIA.
The potential of GAL-1 to ameliorate joint disease was explored by two different therapeutical approaches: gene therapy and protein therapy. A single cell injection (4 × 106 cells/mouse) at the day of disease onset of syngeneic DBA/1 fibroblasts engineered to express mGAL-1 was able to abrogate clinical manifestations of established CIA. In broad agreement, overall disease progression was markedly attenuated when GAL-1 protein (100 μg/mouse per day) was administered daily from the day of the arthritis onset.
Severity of arthritis was monitored by paw swelling (Fig. 2, a and d), clinical score (Fig. 2b and Fig. e), and by the number of affected paws (Fig. 2c and Fig. f). Pooled data from separate experiments showed that on day 12 after disease onset, gene therapy using mGAL-1–transfected fibroblasts significantly reduced hind paw swelling (Fig. 2 a; P = 0.0002) and arthritis progression as determined by the clinical score (Fig. 2 b; P = 0.0002), compared with control constructs of pCDNA3-transfected fibroblasts. Disease amelioration was clearly manifested and reached statistical significance (P = 0.01) as early as 3 d after the start of the treatment. As to the protein therapy protocol, daily administration of GAL-1 protein from the day of the disease onset, resulted in a significant reduction on day 12 after onset in hind paw swelling (Fig. 2 d; P = 0.0008) and clinical score (Fig. 2 e P = 0.0009) to similar inhibitory levels raised by gene therapy, in comparison to saline-treated controls. Marked amelioration of CIA by GAL-1 was clearly reflected by a reduction in the number of arthritic paws as indicated in Fig. 2 c (gene therapy protocol) and f (protein therapy protocol).
Figure 2.

Therapeutic effect of GAL-1 in murine CIA. Arthritis was induced in male DBA/1 mice by intradermal injection of 100 μg native bovine CII in CFA. Hind paw swelling (a and d) was monitored with calipers from the day of disease onset onwards and expressed in mm (mean ± SEM). Clinical score (b and e), was assessed as described in Materials and Methods (mean ± SEM). Number of arthritic paws was assessed visually (c and f) (mean ± SEM). A single intraperitoneal injection at the day of disease onset of mGAL-1 expressing DBA/1 fibroblasts (4 × 106 cells/mouse; panels a, b, and c, □; n = 10), or daily administration for 11 d of recombinant GAL-1 (100 μg) from the day of established arthritis (d, e, and f, □; n = 10), was able to significantly reduce paw swelling in comparison to pCDNA3 control transfectants (a, b, and c, ♦; n = 10) or saline-treated mice (d, e, and f, ♦; n = 8). SEM bars are not shown where they are smaller than the symbol. Two sets of independent in vivo experiments were carried out with very similar results. The results shown are representative of one such experiment.
In brief, arthritic mice experienced a clear and remarkable decline in the development of the ongoing inflammatory disease when treated with GAL-1 by both therapeutic strategies. The specificity of this therapeutic effect was highlighted by the rapid progression and disease evolution in virtually all mice treated with control pCDNA3-transfected fibroblasts (Fig. 2, a–c) or PBS (Fig. 2d–f).
GAL-1 Inhibits Histopathological Manifestations of CIA.
Histopathological findings tightly paralleled clinical data of individual mice (Fig. 3). 12 d after disease onset, microscopic analysis of hematoxylin and eosin–stained hind paw sections showed that most of joints of GAL-1–treated mice of both therapeutic protocols (Fig. 3A and Fig. B) were only mildly affected (P < 0.05; χ2) in comparison to control groups (Fig. 3 C). Synovitis, mononuclear cell infiltration, and cartilage erosion were dramatically diminished in joints corresponding to GAL-1–treated mice, particularly those engaged in the gene therapy protocol (Fig. 3 D). 75% of these mice showed normal joints and 20% mild joint lesions, consisting of small erosions limited to the cartilage–pannus junction. In contrast, 65% of control mice exhibited severe arthritis (Fig. 3 D) accompanied by massive leukocyte infiltration, cartilage destruction, bone erosion, and loss of joint integrity. Overall, protein therapy appeared to be less effective at protecting joint structure than was gene therapy (data not shown).
Figure 3.
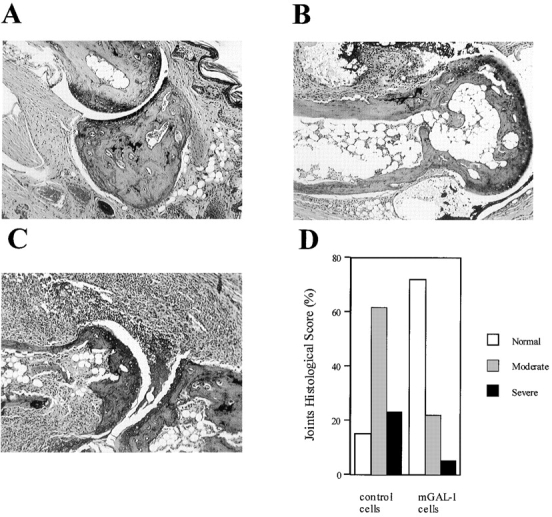
Histopathological assessment of arthritic joints. Mice treated with mGAL-1–transfected fibroblasts (4 × 106 cells/mouse) showed mainly normal joints (A) and some moderately arthritic joints (B). Control mice treated with pCDNA3-transfected cells (4 × 106 cells/mouse) showed moderate (B) and severe arthritis (C) accompanied by synovitis, erosions, and loss of joint integrity. A–C, hematoxylin and eosin staining; original magnification: ×100. Percentage of histological score per group is shown in D.
GAL-1 Reduces Anti-CII IgG Levels and Induces a Bias from a Type 1– to a Type 2–mediated Immune Response.
To determine whether GAL-1 could affect humoral responses to CII over the treatment period, an analysis of anti-CII IgG levels was performed on sera of treated and control mice subjected to both therapeutic protocols. As shown in Fig. 4 a, arthritic mice engaged in the gene therapy protocol with mGAL-1 experienced a biologically and statistically significant reduction in total anti-CII IgG levels (P = 0.0048) 12 d after the start of the treatment, compared with control mice treated with pCDNA3-transfected fibroblasts. This dramatic decline in anti-CII IgG levels was clearly manifested in mice receiving a daily dose of recombinant GAL-1 (Fig. 4 d; P = 0.0018) in comparison to saline-treated controls. It should be emphasized that overall reduction in anti-CII IgG levels by GAL-1 was strictly correlated in each individual mouse with marked amelioration of joint disease.
Figure 4.

Reduction of total anti-CII IgG levels in sera from GAL-1–treated mice and changes in anti-CII IgG2a and IgG1 isotypes. Arthritic mice engaged in the gene therapy (a, b, and c) or protein therapy (d, e, and f) protocols were killed on day 12 after onset and terminally bled, and sera were analyzed for total anti-CII IgG levels by standard ELISA as described in Materials and Methods. Mice receiving mGAL-1–expressing fibroblasts or recombinant GAL-1 (white bars) had a significant reduction in anti-CII Ab levels in comparison to control mice receiving pCDNA3 control fibroblasts or saline (black bars). A significant increase in anti-CII IgG1 levels was seen in all the GAL-1–treated groups as compared with their controls. Mean values of different groups (n = 10) are shown (mean ± SEM).
It has been hypothesized that the balance of cytokines produced by Th1/Th2 subsets of T helper cells plays an important role in the development of the autoimmune response 21 and that a type 2 cytokine pattern is involved in the remission of CIA 31. This prompted us to investigate whether GAL-1 treatment could modify the Th1/Th2 balance in the arthritogenic process. Thus, anti-CII IgG2a (Th1) and IgG1 (Th2) subclasses were further determined in sera of treated and control mice, after gene and protein therapy with GAL-1. A definitive reduction in anti-CII IgG2a isotype (Fig. 4 b; P = 0.00147), accompanied by a slight but significant increase in absolute levels of anti-CII IgG1 (Fig. 4 c; P = 0.0048) were observed at the end of the treatment in sera of DBA/1 mice engaged in the gene therapy protocol in comparison to control mice. In broad agreement, treatment with recombinant GAL-1 resulted in a pronounced shift from an IgG2a (Fig. 4 e; P = 0.0018) to an IgG1 isotype profile (Fig. 4 f; P = 0.0055) in comparison to saline-treated controls.
To elucidate whether class switching to IgG1 was correlated with changes in the cytokine secretion pattern, inguinal lymph nodes and spleens were excised at the end of the treatment from GAL-1–treated or untreated mice. Cells were cultured in the presence of CII and supernatants were analyzed after 72 h for IFN-γ (Fig. 5 a) and IL-5 (Fig. 5 b) production. The main differences were found at the level of draining lymph nodes of mice engaged in the gene therapy protocol with GAL-1, where IL-5 raised to mean high levels of 1,800 pg/ml (P < 0.005), in comparison to the lower levels exhibited by arthritic mice, treated with control vector alone (<250 pg/ml). Consistently, gene therapy with GAL-1 resulted in a severe decline in IFN-γ to background levels of <50 pg/ml (P < 0.005), whereas the control group secreted large amounts of this proinflammatory cytokine (>500 pg/ml). No significant differences in cytokine secretion could be detected at the level of spleens between experimental and control groups (data not shown).
Figure 5.


Cytokine profile in draining lymph node cells after gene therapy with mGAL-1. Arthritic DBA/1 mice treated with mGAL-1 transfected cells (white bars) or control transfectants (black bars) were killed on day 12 after onset. Inguinal lymph nodes from two mice with compatible clinical scores were excised and pooled together. Cells (5 × 106 cells/ml) were cultured in triplicate in 96-well plates in the presence of CII (100 μg/ml). After 72 h, supernatants were collected and analyzed for IFN-γ (a) and IL-5 (b) detection by a capture ELISA. Controls included cells cultured in medium alone or with 5 μg/ml Con A (data not shown). Mean values of different groups are indicated (mean ± SEM) as a combination of two independent experiments.
In brief, GAL-1 treatment promoted a clear shift of the arthritogenic process, mainly at the level of draining lymph node cells, skewing the balance towards a type 2–polarized immune response and inducing a remission state in the evolution of the ongoing inflammatory autoimmune disease.
Susceptibility to Antigen-induced Apoptosis Is Increased in GAL-1–treated Mice.
It is well known that T cells cycling in response to antigenic stimuli are driven into apoptosis by potent TCR restimulation 33. Since GAL-1 has been shown to trigger apoptosis of activated T cells in vitro 13 14, we investigated whether this protein was able to increase the susceptibility of T cells in vivo to antigen-induced apoptosis, thus providing a potential explanatory mechanism for the suppression of the arthritogenic process.
Mice engaged in the gene therapy protocol were killed at the end of the treatment, draining lymph nodes and spleens were excised, and cells were cultured for 24 h in the presence or absence of CII for apoptotic cell detection. Lymph node cells from all GAL-1–treated mice experienced higher susceptibility to antigen-induced apoptosis (Fig. 6 B), as shown by FACS® analysis of hypodiploid DNA content (38 ± 3%) and the increased ladder pattern of DNA cleavage into oligonucleosomal-sized fragments of ∼180–200 bp (inset, lane 2). Moreover, microscopic examination of lymph node cells revealed the typical features of apoptosis, including chromatin condensation and reduction of the cytoplasmic volume (data not shown). In contrast, pCDNA3-treated mice (Fig. 6 A) showed background levels of hypodiploid DNA content (9 ± 2%) and lower levels of fragmentation (inset, lane 1). In the absence of CII, the levels of apoptosis were between 2 and 6% in both experimental groups.
Figure 6.

Increased susceptibility of lymph node cells to antigen-induced apoptosis after gene therapy with GAL-1. Arthritic mice receiving mGAL-1–transfected fibroblasts (B; n = 10) or control pCDNA3 transfectants (A; n = 10) were killed on day 12 after onset. Inguinal lymph nodes from two mice with compatible clinical scores were excised and pooled together. Cells (5 × 106 cells/ml) were cultured in 24-well plates in the presence of CII (100 μg/ml). After 24 h, cells were collected and processed for apoptotic cell detection. FACS® analysis revealed increased hypodiploid DNA content after PI staining in mGAL-1–treated mice (B), in a representative one out of five independent histograms with compatible results. In histograms: x axis, PI uptake (FL2-H); y axis, relative cell number (counts). Lymph nodes cells from galectin-treated mice showed higher levels of DNA fragmentation (inset, lane 2) compared with controls (inset, lane 1). Positive controls of apoptosis (up to 85%) included cells cultured in the presence of 4 μg/ml recombinant GAL-1 for 18 h (data not shown).
On the other hand, no significant differences could be detected between splenocytes from experimental and control groups in their susceptibility to undergo apoptosis in response to TCR restimulation (data not shown). The results presented herein suggest a correlation between the molecular properties reported for GAL-1 in vitro 14 18 and its therapeutic potential in vivo.
GAL-1 Impairs T Cell Function during Antigen Presentation to a CII-specific T Cell Hybridoma.
The experimental conditions used by gene and protein therapy in vivo were reproduced in vitro to study the influence of GAL-1 on IL-2 production and apoptosis during antigen presentation. The capacity of splenocytes from naive DBA/1 mice to present CII to an Aq-restricted, CII-specific T cell hybridoma clone (HCQ.6) was evaluated in the presence of mGAL-1–transfected syngeneic fibroblasts. After 24-h cultures, cell supernatants were collected and analyzed for IL-2 production.
The presence of mGAL-1–transfected fibroblasts decreased the level of IL-2 production in one order of magnitude, when added at concentrations of 5 × 105 to 1 × 106 cells/ml (Table ). No changes in IL-2 production could be detected when fibroblasts were added at 2.5 × 105 cells/ml, indicating a critical inhibitory concentration. As clearly shown, TDG, a β-galactoside–specific sugar, was able to partially prevent this effect when added at a concentration of 100 mM, while the anti–GAL-1 Ab did not neutralize GAL-1 activity at any of the dilutions tested (data not shown).
Table 1.
mGAL-1–expressing Cells Induce Dose-dependent Inhibition of IL-2 Production during Antigen Stimulation of an Aq-restricted, CII-specific T Cell Hybridoma
| HCQ.6 cells (5 × 105/ml) | Spleen APC (5 × 106/ml) | CII (50 μg/ml) | DBA/1 mGAL-1 | DBA/1 PCDNA-3 | TDG (100 mM) | IL-2 |
|---|---|---|---|---|---|---|
| cells/ml | cells/ml | pg/ml | ||||
| + | + | + | – | 878.0 ± 20.09 | ||
| + | + | + | 106 | – | 51.13 ± 9.37 | |
| + | + | + | 5 × 105 | – | 51.64 ± 7.25 | |
| + | + | + | 2.5 × 105 | – | 581.83 ± 89.83 | |
| + | + | + | 106 | – | 653.66 ± 50.01 | |
| + | + | + | 5 × 105 | – | 595.10 ± 84.74 | |
| + | + | + | 2.5 × 105 | – | 581.01 ± 90.96 | |
| + | + | + | 106 | + | 244.04 ± 39.45 | |
| + | + | + | 5 × 105 | + | 252.39 ± 67.53 | |
| + | – | + | – | 33.06 ± 3.25 | ||
| + | + | – | – | 35.38 ± 1.37 |
HCQ.6 (5 × 105 cells/ml), an Aq-restricted, CII-specific T cell hybridoma, was stimulated with bovine CII (50 μg/ml) presented by splenocytes from naive DBA/1 mice (5 × 106 cells/ml) in the presence of syngeneic fibroblasts transfected with mGAL-1 construct or control pCDNA3 expression vector. Specificity was assayed by adding TDG (100 mM) to the culture. Cells were cocultured for 24 h and IL-2 was measured in the supernatants by a capture ELISA. Anti-CD3–stimulated HCQ.6 cells reached maximal levels of IL-2 secretion (1,475 ± 204 pg/ml). Anti–GAL-1 rabbit antibody used at a dilution of 1:100 did not reverse the inhibition by GAL-1, indicating that it had no neutralizing activity (data not shown).
For extrapolation to the protein therapy protocol, the capacity of rGAL-1 to inhibit T cell function during CII-specific activation of HCQ.6 cells was also studied. As deduced from Table , a dose- and time-dependent inhibitory effect on IL-2 production was observed when GAL-1 was added at concentrations ranging from 0.04 to 4 μg/ml at different time points of the assay. Interestingly, 2 h of incubation with rGAL-1 at 4 μg/ml were sufficient to totally abrogate IL-2 secretion, whereas 4 h were required to achieve maximal apoptotic effect as assessed by DNA fragmentation assay (data not shown). This indicates different mechanisms leading to IL-2 inhibition and apoptosis. As mentioned for transfected fibroblasts, rGAL-1 inhibitory activity was partially counteracted by TDG but not by the specific antigalectin Ab, confirming that the carbohydrate recognition domain was clearly involved in this function and that this polyclonal Ab had no neutralizing capacity. The peak in IL-2 production was confirmed by incubating HCQ.6 cells with anti-CD3 mAb, whereas background levels of this cytokine were obtained when APCs or CII were respectively omitted from the assay.
Table 2.
Concentration and Time-dependent Inhibition of IL-2 Production by rGAL-1 upon Antigen Cell Stimulation of an Aq-restricted, CII-specific T Cell Hybridoma
| HCQ.6 cells (5 × 105/ml) | Spleen APC (5 × 106/ml) | CII (50 μg/ml) | rGAL-1 | TDG (100 mM) | IL-2 |
|---|---|---|---|---|---|
| μg/ml | pg/ml | ||||
| + | + | + | 611.00 ± 32.15 | ||
| + | + | + | 4 (18 h) | – | 25.90 ± 3.2 |
| + | + | + | 4 (4 h) | – | 29.95 ± 2.21 |
| + | + | + | 4 (2 h) | – | 56.52 ± 4.53 |
| + | + | + | 0.4 (18 h) | – | 48.77 ± 6.25 |
| + | + | + | 0.4 (4 h) | – | 97.13 ± 9.37 |
| + | + | + | 0.4 (2 h) | – | 382.59 ± 12.91 |
| + | + | + | 0.04 (18 h) | – | 362.10 ± 21.89 |
| + | + | + | 0.04 (4 h) | – | 399.60 ± 20.82 |
| + | + | + | 0.04 (2 h) | – | 400.02 ± 12.93 |
| + | + | + | 4 (18 h) | + | 119.22 ± 7.93 |
| + | + | + | 4 (4 h) | + | 203.57 ± 9.64 |
| + | − | + | – | 33.57 ± 1.96 | |
| + | + | – | – | 29.45 ± 2.53 |
HCQ.6 (5 × 105 cells/ml), an Aq-restricted, CII-specific T cell hybridoma, was stimulated with bovine CII (50 μg/ml) presented by splenocytes from naive DBA/1 mice (5 × 106 cells/ml) in the presence of rGAL-1 (0.04, 0.4, or 4 μg/ml), added for different time periods (2, 4, or 18 h) as indicated. Specificity was assayed by adding TDG (100 mM) to the culture. The anti–GAL-1 Ab (1:100) had no neutralizing activity (data not shown). Cells were cocultured for 24 h and IL-2 was measured in the supernatants by a capture ELISA. Anti-CD3–stimulated HCQ.6 cells reached maximal levels of IL-2 secretion (1,147 ± 165 pg/ml).
Taken together, these findings unequivocally suggest an inhibitory effect of GAL-1 on antigen-specific T cell function. Finally, to address the possibility that this effect could be related to galectin's apoptotic properties, HCQ.6 cells alone were incubated in the presence of GAL-1, revealing a time-dependent increase on DNA fragmentation and hypodiploid DNA content, when GAL-1 was added at its highest concentration of 4 μg/ml, which has been previously shown to be the critical apoptotic threshold 14 34 (data not shown). This high concentration of GAL-1 is probably necessary because it exists as a monomer at lower concentrations. It has been shown that the dimeric form of GAL-1 is required for its biological effect 13 15, which is probably needed for cross-linking of cell surface receptors.
Discussion
Attempts to dissect the functional roles for GAL-1 in vivo have been unsuccessful in comparison to the overwhelming information reached at the biochemical and molecular level. Targeted disruption of GAL-1 gene in knockout mice resulted in the absence of major phenotypic abnormalities, suggesting that other proteins could potentially compensate for the absence of GAL-1, as suggested for null mutations in ostensibly important genes 5. Nevertheless, their conservation throughout animal evolution and their widespread distribution, strongly suggest that they could be implicated in critical biological functions such as cell adhesion 6, cell growth regulation 7 8 9 and immunomodulation 11 12.
In this study we provide definitive experimental data supporting the concept of an in vivo therapeutic role for GAL-1 in a murine experimental model of RA, by using gene and protein therapy strategies. A single injection at the day of the disease onset of genetically modified DBA/1 fibroblasts engineered to secrete GAL-1 was sufficient to achieve a dramatic arrest in overall disease progression, as judged by clinical, histopathological, and immunological manifestations of arthritis. This effect was reproduced by continuous daily administration of recombinant GAL-1.
Research over the past decade identified immunomodulatory properties for β-galactoside–binding proteins in two experimental models of autoimmune myasthenia gravis 11 and autoimmune encephalomyelitis 12. However, only in the last few years has evidence been raised concerning the molecular mechanism involved in these properties. GAL-1 has been shown to induce in vitro apoptosis of activated T cells 13 14 34 through the recognition of selectively glycosylated receptors such as CD43 and CD45, particularly the CD45RO splicing product 35. Furthermore, GAL-1 has been implicated in T cell receptor–mediated apoptosis, as a gear of the complex machinery involved in the elimination of nonselected or negatively selected cells during thymocyte maturation 15 52.
Results presented here establish for the first time a correlation between in vitro apoptotic properties of GAL-1 and its therapeutic potential in vivo, providing an ideal noninflammatory mechanism to terminate the autoimmune T cell attack. Susceptibility to apoptosis was increased in lymph node cells from mice engaged in the gene therapy protocol. Hence, GAL-1–induced apoptosis might eliminate the first wave of arthritogenic T cells, which are responsible for clinical disease and thus prevent the expansion of dominant autoaggressive clones and concomitant epitope spreading 26. The importance of dysregulated apoptosis in the etiology of autoimmune diseases has been highlighted by the occurrence of autoimmune disorders in MRL-lpr/lpr or C3H-gld/gld mice strains that carry spontaneous mutations in Fas or Fas ligand genes 36, and in particular by the dominant Fas gene mutation associated to Canale-Smith syndrome, a human autoimmune lymphoproliferative disorder 37.
Two major pathogenic processes have been clearly identified in the development of RA, the first involving abnormal synoviocyte proliferation, and the second dependent on T cell and macrophage activation 38. Hence, attempts to induce apoptosis either in rheumatoid synovium or activated immune cells will be clearly beneficial for the treatment of joint disease. In this context, Okamoto et al. 39 investigated the apoptotic effects of Fas ligand–transfected cells on proliferating human rheumatoid synovium engrafted in severe combined immunodeficient mice. Moreover, Zhang et al. 40 reported the amelioration of CIA after adenoviral-mediated gene transfer of Fas ligand to arthritic joints.
GAL-1 treatment resulted in an overall reduction of anti-CII Ab levels, skewing the balance towards a type 2–mediated immune response, a novel function for a β-galactoside–binding protein, which remains to be further investigated. Nevertheless, one might speculate that the ongoing autoimmune response, mainly driven by Th1-proinflammatory cells, could be inhibited by apoptosis of memory and activated T cells 13. Once the inflammatory action of type 1 cytokines has been removed, it is feasible that a previously repressed Th2 response will become apparent. This observation may provide a potential association between GAL-1–induced apoptosis and immune deviation. Supporting our finding, Varadhachary et al. 41 and Zhang et al. 42 have recently reported that only Th1 effector cells were susceptible to TCR- and Fas ligand–mediated apoptosis, leading to selective Th2 survival. Moreover, the results we obtained by GAL-1 treatment regarding a Th1/Th2 switch are quantitatively stronger than those we previously reported expressing a soluble TNF receptor 43 or IFN-β 44. The change in Ig isotypes, i.e., reduction in IgG2a and increase in IgG1, reflects the change in T cell help and has important therapeutic implications. IgG2a is a complement fixing antibody, contributing to tissue damage by triggering the production of anaphylatoxins that lead to extravasation and infiltration of neutrophils which in turn secrete pathogenic mediators of inflammation. This effect on the humoral response is relevant also in multiple sclerosis and myasthenia gravis where antibodies and complement have been implicated, as in RA, in the pathology of the disease.
It seems that a novel paradigm is providing a breakthrough in galectin research. Overall opposite functions from GAL-1 have been assigned to galectin-3, a 29-kD member of this protein family with similar carbohydrate specificity. GAL-1 has been shown to induce T cell apoptosis 13 14, whereas galectin-3 has been conversely shown to prevent cell death 45. Thus, galectin-1 and -3 may represent an additional family of proteins similar to the Bcl-2 family, where different members exhibit sequence similarity, yet have the opposite effects on cell survival 46. In view of the results presented in this paper, the limits of the paradigm could be further extended to the regulation of the Th1/Th2 balance. Recent findings suggest that galectin-3 inhibited the transcription and release of IL-5 protein from antigen-specific T cell lines and human eosinophils 47. On the other hand, our results demonstrate the ability of GAL-1 to skew the immune response in vivo towards a Th2 profile, inducing a decrease in IFN-γ and a clear increase in IL-5 production.
Finally, we have also demonstrated that GAL-1–transfected fibroblasts as well as recombinant GAL-1 induced a specific and dose-dependent inhibitory effect in vitro using a CII-specific T cell hybridoma clone. Increasing concentrations of GAL-1 augmented apoptosis and inhibited IL-2 secretion. These two effects appear to be regulated by different signaling pathways 52. It is important to note that the dose-dependent effect was greatly enhanced during antigen presentation, i.e., triggering through the TCR. Without specific antigen, GAL-1 had to be added to the cell culture at the high concentration of 4 μg/ml to get significant apoptosis (data not shown). Our results strengthen the hypothesis put forward by Perillo et al. 15 and Vespa et al. 52 that thymocytes and T cells of the CD4+CD8+, CD4−CD8−, CD4+CD8−, and CD4−CD8+ are more sensitive to GAL-1–induced apoptosis after triggering of their TCR with anti-CD3 Ab. Thus, it appears that GAL-1 induces a second signal that together with TCR signaling sensitizes dividing T cells to apoptose 15 52. The fact that in our experiments lymph node cells and not spleen-derived cells showed clear effects after GAL-1 treatment indicates that alternative signaling mechanisms could function in different organs that regulate GAL-1–induced functions.
Localization of GAL-1 in lymphoid organs such as thymus 16 and lymph nodes 17 support the idea that GAL-1 may play a key role in the context of the immune system. In this regard, using an mRNA differential display PCR, GAL-1 gene was found to be induced in activated but not resting T cells and then secreted to the extracellular milieu to act as an autocrine negative growth factor 8. Consequently, we recently purified a proapoptotic GAL-1–like protein from peritoneal rat macrophages 14, and found that its expression was differentially regulated according to the activation state of the cells 18.
In our study, gene therapy using GAL-1–secreting fibroblasts reached similar therapeutic benefits to those found by daily administration of GAL-1. However, gene therapy offers unique advantages such as a single injection and therapeutic effects at lower concentrations and in a local environment, overcoming the adverse effects of protein therapy 22 48 49. Gene transfer strategies for RA 38 are currently designed for inhibiting proinflammatory cytokines 43 50 51, matrix-degrading enzymes 38, and survival of activated synovial cells 38 39. Despite the advantages of gene therapy, the majority of the work to date refers to the constitutive expression of therapeutic genes. As with GAL-1, long-term expression of biological agents can have secondary effects such as altering the immune response to infectious pathogens such as viruses 49. To prevent such an outcome, transcriptionally regulated gene expression is necessary. The use of the tetracycline-inducible operon, which is activated by tetracycline derivatives already used in the clinic, is a possible solution to this problem 54.
To our knowledge, this study is the first approach aimed at using the survival of activated arthritogenic T lymphocytes as a therapeutic target and that uses a naturally occurring protein and not a synthetic compound such as bisindolylmaleimide VIII 53.
In recent years it has been postulated that bone marrow transplantation is the way to treat certain autoimmune diseases such as RA and multiple sclerosis 55 56. The hypothesis behind this proposal is that the old “memory” of autoimmune disease will be deleted by radiation therapy while newly transplanted bone marrow will be tolerized to the current autoantigens driving the immune response. This resetting of the immune system may also be induced by GAL-1 treatment. Further experiments will be required to evaluate these interesting clinical applications.
Acknowledgments
We are grateful to all the staff of the Biological Services Unit at The Kennedy Institute of Rheumatology, specially to Mr. P. Warden. We would also like to thank to Drs. R.N. Maini and M. Londei for critically reviewing the manuscript; Dr. M. Kahan for assistance with FACS® analysis; Mr. P. Conolly and Dr. L. Marinova-Mutafchieva for help in histological studies; and Dr. C. Landa and Dr. C. Sotomayor for encouragement and support.
These studies were supported in part by a joint grant from the British Council and CONICOR (16/96), a personal award from Fundación Antorchas and the British Council for Advanced Studies in the United Kingdom to G.A. Rabinovich, a grant from CONICET (4921/96), and by the Arthritis Research Campaign, United Kingdom.
Footnotes
1used in this paper: CIA, collagen-induced arthritis; CII, collagen type II; GAL-1, galectin-1; mGAL-1, mouse GAL-1; PI, propidium iodide; RA, rheumatoid arthritis; rGAL-1, human recombinant GAL-1; TDG, thiodigalactoside
Gordon Daly and Hanna Dreja contributed equally to this work.
References
- Barondes S.H., Cooper D.N.W., Gitt M.A., Leffler H. Galectinsstructure and function of a large family of animal lectins. J. Biol. Chem. 1994;269:20807–20810. [PubMed] [Google Scholar]
- Barondes S.H., Castronovo V., Cooper D.N.W., Cummings R.D., Drickamer K., Feizi T., Gitt M.A., Hirabayashi J., Hughes R.C., Kasai K. Galectinsa family of animal β-galactoside-binding lectins. Cell. 1994;76:597–598. doi: 10.1016/0092-8674(94)90498-7. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J., Kasai K. The family of metazoan metal-independent β-galactoside-binding lectinsstructure, function and molecular evolution. Glycobiology. 1993;3:297–304. doi: 10.1093/glycob/3.4.297. [DOI] [PubMed] [Google Scholar]
- Leffler H. Introduction to galectins. Trends Glycosci. Glycotech. 1997;45:9–19. [Google Scholar]
- Poirrier F., Robertson E.J. Normal development of mice carrying a null mutation in the gene encoding the L-14 S-type lectin. Development. 1993;119:1229–1236. doi: 10.1242/dev.119.4.1229. [DOI] [PubMed] [Google Scholar]
- Van den Brüle F.A., Buicu C., Baldet M., Sobel M.E., Cooper D.N.W., Marschal P., Castronovo V. GAL-1 modulates human melanoma cell adhesion to laminin. Biochem. Biophys. Res. Commun. 1995;209:760–767. doi: 10.1006/bbrc.1995.1564. [DOI] [PubMed] [Google Scholar]
- Wells V., Mallucci L. Identification of an autocrine negative growth factormouse β-galactoside-binding protein is a cytostatic factor and cell growth regulator. Cell. 1991;64:91–97. doi: 10.1016/0092-8674(91)90211-g. [DOI] [PubMed] [Google Scholar]
- Blaser C., Kaufmann M., Muller C., Zimmerman C., Wells V., Mallucci L., Pircher H. Beta-galactoside-binding protein secreted by activated T cells inhibits antigen-induced proliferation of T cells. Eur. J. Immunol. 1998;28:2311–2319. doi: 10.1002/(SICI)1521-4141(199808)28:08<2311::AID-IMMU2311>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Allione A., Wells V., Forni G., Mallucci L., Novelli F. Beta-galactoside-binding protein (β-GBP) alters the cell cycle, up-regulates expression of the α- and β-chains of the IFN-γ receptor, and triggers IFN-γ-mediated apoptosis of activated human T lymphocytes. J. Immunol. 1998;161:2114–2119. [PubMed] [Google Scholar]
- Bresalier R.S., Mazurek N., Sternberg L.R., Bird J.C., Yunker C.K., Makker P.N., Raz A. Metastasis of human colon cancer is altered by modifying expression of the β-galactoside binding protein galectin-3. Gastroenterology. 1998;115:287–296. doi: 10.1016/s0016-5085(98)70195-7. [DOI] [PubMed] [Google Scholar]
- Levy G., Tarrab-Hazdai R., Teichberg V.I. Prevention and therapy with electrolectin of experimental autoimmune myasthenia gravis in rabbits. Eur. J. Immunol. 1983;13:500–507. doi: 10.1002/eji.1830130613. [DOI] [PubMed] [Google Scholar]
- Offner H., Celnik B., Bringman T., Casentini-Borocz D., Nedwin G.E., Vanderbark A. Recombinant human β-galactoside-binding lectin suppresses clinical and histological signs of experimental autoimmune encephalomyelitis. J. Neuroimmunol. 1990;28:177–184. doi: 10.1016/0165-5728(90)90032-i. [DOI] [PubMed] [Google Scholar]
- Perillo N.L., Pace K.E., Seilhamer J.J., Baum L.G. Apoptosis of T-cells mediated by GAL-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- Rabinovich G.A., Iglesias M.M., Modesti N.M., Castagna L.F., Todel C.W., Riera C.M., Sotomayor C.E. Activated rat macrophages produce a GAL-1-like protein that induces apoptosis of T cellsbiochemical and functional characterization. J. Immunol. 1998;160:4831–4840. [PubMed] [Google Scholar]
- Perillo N.L., Uittenbogaart C.H., Nguyen J.T., Baum L.G. GAL-1, an endogenous lectin produced by thymic epithelial cells, induces apoptosis of human thymocytes. J. Exp. Med. 1997;185:1851–1858. doi: 10.1084/jem.185.10.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L.G., Pang M., Perillo N.L., Wu T., Delegaene A., Uittenbogaart C.H., Fukuda M., Seilhamer J.J. Human thymic epithelial cells express an endogenous lectin, GAL–1, which binds to core 2 O-glycans on thymocytes and T lymphoblastoid cells. J. Exp. Med. 1995;181:877–887. doi: 10.1084/jem.181.3.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum L.G., Seilhamer J.J., Pang M., Levine W.B., Beynon D., Berliner J.A. Synthesis of an endogenous lectin, GAL-1 by human endothelial cells is up-regulated by endothelial cell activation. Glycoconjugate J. 1995;12:63–68. doi: 10.1007/BF00731870. [DOI] [PubMed] [Google Scholar]
- Rabinovich G.A., Castagna L.F., Landa C.A., Riera C.M., Sotomayor C.E. Regulated expression of a 16-kd galectin-like protein in activated rat macrophages. J. Leukocyte Biol. 1996;59:363–370. doi: 10.1002/jlb.59.3.363. [DOI] [PubMed] [Google Scholar]
- Iglesias M.M., Rabinovich G.A., Ivanovic V., Sotomayor C.E., Wolfenstein-Todel C. GAL-1 from ovine placentaamino-acid sequence, physiochemical properties and implications in T-cell death. Eur. J. Biochem. 1998;252:400–407. doi: 10.1046/j.1432-1327.1998.2520400.x. [DOI] [PubMed] [Google Scholar]
- Allen H.J., Sucato D., Woynarowska B., Gottstine S., Sharma A., Bernacki R.J. Role of galaptin in ovarian carcinoma adhesion to extracellular matrix in vitro. J. Cell. Biochem. 1990;43:43–57. doi: 10.1002/jcb.240430105. [DOI] [PubMed] [Google Scholar]
- Feldmann M., Brennan F.M., Maini R.N. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Chernajovsky Y., Feldmann M., Maini R.N. Gene therapy in rheumatoid arthritis via cytokine regulationfuture perspectives. Br. Med. Bull. 1995;51:503–516. doi: 10.1093/oxfordjournals.bmb.a072976. [DOI] [PubMed] [Google Scholar]
- Durie F.H., Fava R.A., Noelle R.J. Collagen-induced arthritis as a model of rheumatoid arthritis. Clin. Immunol. Immunopathol. 1994;73:11–18. doi: 10.1006/clin.1994.1164. [DOI] [PubMed] [Google Scholar]
- Vaishnaw A.K., McNally J.D., Elkon K.B. Apoptosis in the rheumatic diseases. Arthritis Rheum. 1997;40:1917–1927. doi: 10.1002/art.1780401102. [DOI] [PubMed] [Google Scholar]
- Revillard J.P., Adorini L., Goldman M., Kabelitz D., Waldmann H. Apoptosispotential for disease therapies. Immunol. Today. 1998;19:291–293. doi: 10.1016/s0167-5699(98)01279-1. [DOI] [PubMed] [Google Scholar]
- Gold R., Hartung H.P., Lassman H. T-cell apoptosis in autoimmune diseasestermination of inflammation in the nervous system and other sites with specialized immune-defense mechanism. Trends Neurosci. 1997;20:399–404. doi: 10.1016/S0166-2236(97)01079-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunsberg U., Gustafsson K., Jansson L., Michaëlsson E., Ährlund-Richter L., Petterson S., Mattsson R., Holmdahl R. Expression of a transgenic class II Ab gene confers susceptibility to collagen-induced arthritis. Eur. J. Immunol. 1994;24:1698–1702. doi: 10.1002/eji.1830240736. [DOI] [PubMed] [Google Scholar]
- Triantaphyllopoulos K.A., Croxford J.L., Baker D., Chernajovsky Y. Cloning and expression of murine IFN β and a TNF antagonist for gene therapy of experimental allergic encephalomyelitis. Gene Therapy. 1998;5:253–263. doi: 10.1038/sj.gt.3300570. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J., Ayaki H., Soma G., Kasai K. Production and purification of a recombinant human 14 kDa β-galactoside-binding lectin. FEBS Lett. 1989;250:161–165. doi: 10.1016/0014-5793(89)80711-2. [DOI] [PubMed] [Google Scholar]
- Williams R.O., Feldmann M., Maini R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri C., Williams R.O., Walmsley M., Feldmann M. Relationship between Th1/Th2 cytokine patterns and the arthritogenic response in collagen-induced arthritis. Eur. J. Immunol. 1996;26:1511–1518. doi: 10.1002/eji.1830260716. [DOI] [PubMed] [Google Scholar]
- Nicoletti I., Migliorati G., Pagliacci M.C., Grignani F., Riccardi C.A. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-o. [DOI] [PubMed] [Google Scholar]
- McFarland H.L., Critchfield J.M., Racke M.K., Mueller J.P., Nye S.H., Boehme S.A., Lenardo M.J. Amelioration of autoimmune reactions by antigen-induced apoptosis of T cells. Adv. Exp. Med. Biol. 1995;383:157–166. doi: 10.1007/978-1-4615-1891-4_18. [DOI] [PubMed] [Google Scholar]
- Rabinovich G.A., Modesti N.M., Castagna L.F., Landa C.A., Riera C.M., Sotomayor C.E. Specific inhibition of lymphocyte proliferation and induction of apoptosis by CLL-I, a β-galactoside-binding lectin. J. Biochem. 1997;122:365–373. doi: 10.1093/oxfordjournals.jbchem.a021762. [DOI] [PubMed] [Google Scholar]
- Cyster J.G., Fowell D., Barclay A.N. Antigen determinants encoded by alternatively spliced exons of CD45 are determined by the polypeptide but influenced by glycosylation. Int. Immunol. 1994;6:1875–1881. doi: 10.1093/intimm/6.12.1875. [DOI] [PubMed] [Google Scholar]
- Singer G.G., Carrera A.C., Marshak-Rothstein A., Martínez A.C., Abbas A.K. Apoptosis, Fas and systemic autoimmunitythe MRL-lpr/lpr model. Curr. Opin. Immunol. 1994;6:913–920. doi: 10.1016/0952-7915(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Fisher G.H., Rosenberg F.J., Straus S.E., Dale J.K., Middelton L.A., Yin A.Y., Strober W., Lenardo M.J., Puck J.M. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–946. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- Jorgensen C., Gay S. Gene therapy in osteoarticular diseaseswhere are we? Immunol. Today. 1998;19:387–391. doi: 10.1016/s0167-5699(98)01296-1. [DOI] [PubMed] [Google Scholar]
- Okamoto K., Asahara H., Kobayashi T., Matsuno H., Hasunuma T., Kobata T., Sumida T., Nishioka K. Induction of apoptosis in rheumatoid synovium by Fas ligand gene transfer. Gene Therapy. 1998;5:331–338. doi: 10.1038/sj.gt.3300597. [DOI] [PubMed] [Google Scholar]
- Zhang H., Yang Y., Horton J.L., Samoilova E.B., Judge T.A., Turka L.A., Wilson J.M., Chen Y. Amelioration of collagen-induced arthritis by CD95 (Apo-1/Fas)-ligand gene transfer. J. Clin. Invest. 1997;100:1951–1957. doi: 10.1172/JCI119726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadhachary A.S., Perdow S.N., Hu C., Ramanarayanan M., Salgame P. Differential ability of T cell subsets to undergo activation-induced cell death. Proc. Natl. Acad. Sci. USA. 1997;94:5778–5783. doi: 10.1073/pnas.94.11.5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Brunner T., Carter L., Dutton R.W., Rogers P., Bradley L., Sato T., Reed J.C., Green D., Swain S.L. Unequal death in T helper cell (Th) 1 and Th2 effectorsTh1, but not Th2 effectors undergo rapid Fas/FasL-mediated apoptosis. J. Exp. Med. 1997;185:1837–1849. doi: 10.1084/jem.185.10.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mageed R.A., Adams G., Woodrow D., Podhajcer O.L., Chernajovsky Y. Prevention of collagen-induced arthritis by gene delivery of soluble p75 TNF receptor. Gene Therapy. 1998;5:1584–1592. doi: 10.1038/sj.gt.3300785. [DOI] [PubMed] [Google Scholar]
- Triantaphyllopoulos K.A., Williams R.O., Tailor H., Chernajovsky Y. Amelioration of collagen-induced arthritis and suppression of IFN γ, IL-12 and TNF α production by IFN β gene therapy. Arthritis Rheum. 1999;42:90–99. doi: 10.1002/1529-0131(199901)42:1<90::AID-ANR12>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Yang R.Y., Hsu D.K., Liu F.T. Expression of galectin-3 modulates T cell growth and apoptosis. Proc. Natl. Acad. Sci. USA. 1996;93:6737–6742. doi: 10.1073/pnas.93.13.6737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroemer G. The proto-oncogene Bcl-2 and its role in regulating apoptosis. Nat. Med. 1997;3:614–620. doi: 10.1038/nm0697-614. [DOI] [PubMed] [Google Scholar]
- Cortegano I., del Pozo V., Cárdaba B., de Andrés B., Gallardo S., del Amo A., Arrieta I., Jurado A., Palomino P., Liu F.-T., Lahoz C. Galectin-3 down-regulates IL-5 gene expression on different cell types. J. Immunol. 1998;161:385–389. [PubMed] [Google Scholar]
- Evans C.H., Robbins P.D. Pathways to gene therapy in rheumatoid arthritis. Curr. Opin. Rheumatol. 1996;8:230–234. doi: 10.1097/00002281-199605000-00011. [DOI] [PubMed] [Google Scholar]
- Chernajovsky Y., Annenkov A., Herman C., Triantaphyllopoulos K., Gould D., Dreja H., Moyes S.P., Croxford J.L., Mageed R.A., Podhajcer O.L., Baker D. Gene therapy for rheumatoid arthritistheoretical considerations. Drugs Aging. 1998;12:29–41. doi: 10.2165/00002512-199812010-00004. [DOI] [PubMed] [Google Scholar]
- Otani K., Nita I., Macaulay W., Georgescu H.I., Robbins P.D., Evans C.H. Suppression of antigen-induced arthritis in rabbits by ex vivo gene therapy. J. Immunol. 1996;156:3558–3562. [PubMed] [Google Scholar]
- Chernajovsky Y., Adams G., Podhajcer O.L., Mueller G.M., Robbins P.D., Feldmann M. Inhibition of transfer of collagen-induced arthritis into SCID mice by ex vivo infection of spleen cells with retroviruses expressing soluble tumor necrosis factor receptor. Gene Therapy. 1995;2:731–735. [PubMed] [Google Scholar]
- Vespa G.N., Lewis L.A., Kozak K.R., Moran M., Nguyen J.T., Baum L.G., Miceli M.C. Galectin-1 specifically modulates TCR signals to enhance TCR apoptosis but inhibit IL-2 production and proliferation. J. Immunol. 1999;162:799–806. [PubMed] [Google Scholar]
- Zhou T., Song L., Yang P., Wang Z., Lui D., Jope R.S. Bisindolylmaleimide VIII facilitates Fas-mediated apoptosis and inhibits T cell mediated autoimmune diseases. Nat. Med. 1999;5:42–48. doi: 10.1038/4723. [DOI] [PubMed] [Google Scholar]
- Bohl D., Naffakh N., Heard J.-M. Long-term control of erythropoietin secretion by doxycycline in mice transplanted with engineered primary myoblasts. Nat. Med. 1997;3:299–305. doi: 10.1038/nm0397-299. [DOI] [PubMed] [Google Scholar]
- Burt R.K., Burns W.H., Miller S.D. Bone marrow transplantation for multiple sclerosisreturning to Pandora's box. Immunol. Today. 1997;12:559–561. doi: 10.1016/s0167-5699(97)01168-7. [DOI] [PubMed] [Google Scholar]
- Wicks I., Cooley H., Szer J. Autologous hemopoietic stem cell transplantationA possible cure for rheumatoid arthritis? Arthritis Rheum. 1997;40:1005–1011. doi: 10.1002/art.1780400603. [DOI] [PubMed] [Google Scholar]