

Abstract
C-reactive protein (CRP) is an acute phase serum protein that shares several functions with immunoglobulin (Ig)G including complement activation and binding to receptors on monocytes and neutrophils. The identity of the receptor for CRP has been the target of extensive research. We previously determined that CRP binds to the high affinity receptor for IgG, FcγRI (CD64). However, this interaction could not account for the majority of binding of CRP to neutrophils or monocytic cells. We now determine that CRP also interacts with FcγRIIa (CD32), the low affinity receptor for IgG on monocytes and neutrophils. COS-7 cells were transfected with a construct containing the human FcγRIIA cDNA. CRP binding and the presence of CD32 were detected by mAb and analyzed by two-color flow cytometry. Cells expressing CD32 bound CRP in a dose-dependent and saturable manner consistent with receptor binding. CRP bound to transfectants and K-562 cells with similar kinetics, and in both cases binding was completely inhibited by aggregated IgG. On monocytic cell lines, treatment with Bt2cAMP increased FcγRII expression and enhanced CRP binding. CRP also specifically precipitated FcγRI and FcγRII from the monocytic cell line, THP-1. It is suggested that the major receptor for CRP on phagocytic cells is FcγRII.
Keywords: C-reactive protein, Fc receptors, FcγRII, receptors, immunoglobulin G
C-reactive protein (CRP) is an acute phase serum protein in humans and a member of the pentraxin family of proteins 1. Serum concentrations of CRP increase from less than one to hundreds of micrograms per milliliter as part of the inflammatory response to infection or acute injury. Functionally, CRP shares many properties with IgG. These properties include specific ligand binding, activation of the complement cascade through the binding of C1q 2, and opsonic activity 3 4 5.
The presence of a receptor for CRP was first suggested by reports of opsonic activity of CRP 3 4 5. Several investigators have reported the presence of a unique receptor for CRP on mononuclear cells and neutrophils. Tebo et al. 6 and Zahedi et al. 7 concluded that two receptors for CRP were present on mononuclear cells in humans and mice. It was noted that there was some possible association with Fc receptors (FcRs) as IgG could inhibit binding. Other authors also noticed a functional interaction with FcRs but maintained that a specific receptor for CRP was also present 8 9. Our laboratory reported that CRP interacted specifically with the high affinity receptor for IgG, FcγRI 10 11. However, it was apparent that this receptor could not account for all the binding to mononuclear cells and neutrophils as cells lacking FcγRI were also capable of binding CRP, and upregulation of FcγRI did not significantly enhance CRP binding. Examination of the binding pattern of CRP with various cell lines and blood cell populations suggested to us that CRP might bind to FcγRII. In the study presented here, we demonstrate binding of CRP to FcγRIIa expressed in COS-7 cells by transient transfection. Studies of monocytic cell lines done in parallel indicate that FcγRIIa is responsible for the majority of CRP binding to these cells. It is suggested that FcγRIIa is the previously described “unique” receptor for CRP.
Materials and Methods
Preparation of CRP.
Human CRP was purified from human pleural fluids exactly as previously described 12. The purity of the preparation was determined by 12% SDS-PAGE. No contaminating proteins were detected on overloaded gels.
Reagents and Antibodies.
The monoclonal anti-CRP antibody, 2C10, was the gift of Dr. Larry Potempa (ImmTech, Evanston, IL) and was used as a hybridoma tissue culture supernatant. FITC-conjugated F(ab′)2 goat anti–mouse IgG (FITC-GAM) was obtained from Caltag Labs. or from ICN. Affinity-purified F(ab′)2 PE-GAM was obtained from Caltag Labs. Aggregated IgG (aggIgG) was prepared from human IgG (Sigma Chemical Co.) by incubation at 63°C for 30 min at 10–12 mg/ml. AT10 (an anti-CD32 mAb) and FITC-AT10 were purchased from Serotec. FITC-FLI8.26 (2003) (an anti-CD32 mAb) was purchased from PharMingen. This antibody was absorbed with mock-transfected COS-7 cells for 30 min on ice to remove nonspecific binding. PE-C1KM5 (an anti-CD32 mAb) was purchased from Caltag Labs. Anti-CD64 mAb 197 and anti-CD32 mAb IV.3 were purchased from Medarex. Protein A–Sepharose CL6B and Affigel 15 were purchased from Sigma Chemical Co. and Bio-Rad, respectively. Bt2cAMP was purchased from Sigma Chemical Co.
Cell Culture.
The human erythroleukemia cell line K-562, the human monocytic cell lines THP-1 and U-937, and COS-7 cells were obtained from the American Type Culture Collection. All cell lines were maintained in RPMI with 10% FCS (Hyclone) and 50 μg/ml gentamycin, except for COS-7 cells, which were maintained in DMEM with 10% FCS and 50 μg/ml gentamycin.
Treatment of U-937 Cells with Bt2cAMP.
Cells were counted and resuspended at 0.3 × 106 cells/ml in medium containing 1 mM Bt2cAMP, and incubated for 60 h before analysis.
Cell Transfections.
The human FcγRIIA cDNA clone in the pcDSRα296 transient transfection vector 13 was obtained from Dr. Kevin Moore (DNAX Research Institute, Palo Alto, CA). Cells were transfected using the GenePORTER™ transfection reagent from Gene Therapy Systems Inc. Control cells received GenePORTER™ reagent only. 6-well tissue culture plates were seeded with 2.5 × 105 cells/well and cultured for 18–24 h until the cells were 70–80% confluent. Transfection was performed according to the manufacturer's protocol, except cells were centrifuged to increase transfection efficiency. Plates were centrifuged at 300 g for 10 min at 21°C and then incubated at 37°C for 5 h. Cells were cultured overnight in DMEM with 10% FCS. 18 h after transfection, cells were detached using PBS containing 5 mM EDTA and replated on 100-mm tissue culture plates (two 100-mm tissue culture plates for cells from each 6-well plate) and cultured in DMEM. Flow cytometric assays were performed 66–78 h after transfection. The percentage of transfected cells was 75–85% as determined using either FITC-FLI8.26 or PE-C1KM5.
CRP Binding Assay.
Cells were washed twice in ice-cold PBS containing 0.05% azide and 0.1% globulin-free BSA (PAB). Between 5 × 105 and 106 cells per tube were washed with 1 ml of ice-cold PAB. Cells were pelleted at 180 g for 5 min at 4°C and resuspended in PAB with CRP at the concentrations indicated in a total volume of 80 μl. Cells were incubated for 1 h in the presence of CRP, then washed twice with 1 ml of PAB. Cells were then incubated for 30 min at 4°C with mAb 2C10 culture supernatant. Cells were washed twice with 1 ml PAB and then incubated with PE-GAM (1 μg/106 cells) or FITC-GAM (a 1:100 final dilution) for 30 min. After this last incubation, cells were washed twice with 1 ml PAB as above and then resuspended in 0.4–0.5 ml PAB for analysis by flow cytometry. FcγRI levels were determined using mAb 32.2 and PE-GAM. Levels of FcγRII were determined by binding of FITC-AT10. The percentage of dead cells was determined using 7-Aminoactinomycin D (Molecular Probes). To test inhibition by aggIgG, cells were incubated with aggIgG at an increasing concentration for 30 min before the addition of CRP. There was no significant change in the fluorescence of the secondary antibodies in the presence of aggIgG.
Flow Cytometry.
Cells were analyzed using a Becton Dickinson FACSCalibur® flow cytometer equipped with CellQuest software (Becton Dickinson). The population analyzed was gated by forward and side scatter to exclude dead cells. A minimum of 30,000 cells was collected. For all measurements of CRP binding, the binding of 2C10 and the anti–mouse secondary antibody were subtracted. Results are presented as the geometric mean channel fluorescence (gMCF).
Precipitation of Cell Surface Receptors.
THP-1 cells were treated with 50 μg/ml pronase (Calbiochem) for 30 min at 37°C, washed, and radiolabeled with 125I using lactoperoxidase 14. Membrane extracts were prepared at 5 × 107 cells/ml using 1% NP-40 in PBS with 1 mM AEBSF, 1 μM pepstatin A, 10 μM E-64, 10 μM bestatin, 100 μM leupeptin, and 2 μg/ml aprotinin (lysis buffer). CRP beads were prepared by coupling 18 mg CRP/ml Affigel 15 according to the manufacturer's protocol. Antibody beads were prepared by incubation of 5 μg anti-CD32 mAb (IV.3 or AT10) or anti-CD64 mAb 197 with 15 μl protein A–Sepharose. 2.5 × 106 cell equivalents were incubated with CRP beads, anti-CD32 Sepharose, or anti-CD64 Sepharose in lysis buffer for 4 h at 4°C. The beads were then washed four times with lysis buffer and once with 0.1% NP-40 in the same buffer. The samples were boiled for 3 min in Laemmli's sample buffer and run on 4–20% gradient SDS-PAGE gels (Novex). The gels were dried and autoradiography was performed using a Storm imaging system (Molecular Dynamics).
Data Analysis.
The binding kinetics of CRP to cells were analyzed using GraphPad PRISM™ software (GraphPad Software, Inc.). ImageQuant software (Molecular Dynamics) was used for quantitation of radioactive bands. Experiments were repeated at least twice.
Results
Binding of CRP to Transfected COS-7 Cells and to K-562 Cells.
Transfection of COS-7 cells with pcDSRα296 containing the cDNA for CD32 resulted in 70–85% of cells expressing the CD32 marker. When these cells were incubated with CRP, a dose-dependent and saturable binding of CRP was seen (Fig. 1 A). The binding of CRP was fitted to a single site model using GraphPad Prism™ software. Saturation was predicted at 100 μg/ml with an apparent equilibrium binding constant (K D ) of 6.6 × 10−8 M (7.6 μg/ml). This binding curve is similar to the binding of CRP to K-562 cells, which express FcγRII as the only FcR (Fig. 1 B). The apparent K D is 9.7 × 10−8 M (11.2 μg/ml) with saturation at ∼100 μg/ml. Background binding to mock-transfected cells was not above baseline binding of secondary antibody. The apparent affinity of CRP for transfected and K-562 cells was significantly higher than the reported values for IgG binding to FcγRII (K D > 10−7 M) 15 and similar to the previously reported figure for CRP binding to K-562 cells (3.8 × 10−8 M) 6. In separate experiments using 125I-labeled aggIgG, an apparent K D of 23 μg/ml was measured (data not shown).
Figure 1.
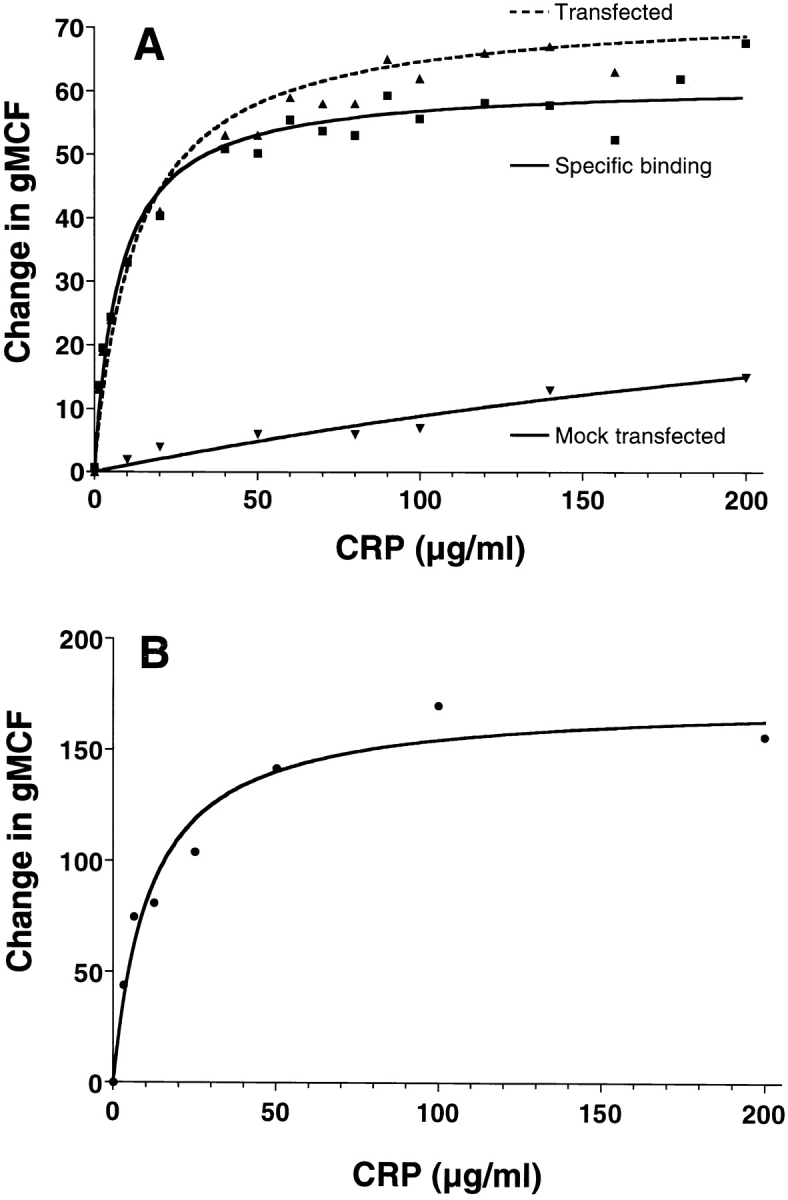
CRP binds to COS–7 cells transfected with the FcγRIIA cDNA, and to K-562 cells with similar kinetics. Cells were incubated with increasing doses of CRP and binding was detected with 2C10 and PE-GAM, as described in Materials and Methods. Results are expressed as the increase in gMCF compared with the secondary antibody controls. (A) COS-7 cells transfected with the FcγRIIA plasmid or pcDSRα296, or mock-transfected. Specific binding was determined by subtracting gMCF of mock-transfected cells from gMCF of transfected cells. (B) K-562 cells.
Binding of CRP to transfected cells was also examined by two-color analysis to confirm that CRP bound to cells expressing CD32 (Fig. 2). Single color staining with anti-CD32 (Fig. 2 B) or CRP (Fig. 2 C) showed staining of 72% (with a background binding of 6% to mock-transfected) and 63% (with a background binding of <1% of mock-transfected) of transfected cells, respectively. In the two-color analysis, CRP binding to CD32-transfected COS cells directly correlated with anti-CD32 binding with 52% of the cells stained for both markers (Fig. 2 D).
Figure 2.

Analysis of CRP binding to transfected COS-7 cells by two-color flow cytometry. COS-7 cells were transfected with the FcγRIIA plasmid, pcDSRα296. Mock-transfected cells received no DNA. Cells were incubated with 200 μg/ml of CRP and binding was detected with 2C10 and PE-GAM as described in Materials and Methods. Levels of FcγRII were determined by binding of FITC-AT10. A, B, C, and D, transfected; E, F, G, and H, mock-transfected. A and E, staining with 2C10 and PE-GAM; B and F, staining for CD-32 only; C and G, staining for CRP only; D and H, staining for CRP and CD32.
Binding of CRP to CD32-Transfected COS-7 Cells and to K-562 Cells Is Blocked by IgG.
To determine if CRP was binding to the same site on FcγRII as IgG, the effect of aggIgG on CRP binding was examined. As shown in Fig. 3, aggIgG inhibited CRP binding to K-562 cells in a dose-dependent manner with 50% inhibition at ∼80 μg/ml of aggIgG. This finding is consistent with previous reports by us 10 11 and others 5 6 7 that aggIgG effectively blocks CRP binding to its receptor. A nearly identical inhibition profile was obtained using aggIgG to inhibit CRP binding to CD32-transfected COS cells (Fig. 3). For both cell types, aggIgG did not affect the background binding of the secondary antibodies measured in the absence of CRP.
Figure 3.

CRP binding to FcγRIIa is inhibited by aggIgG. K-562 cells or COS-7 cells transfected with the FcγRIIA plasmid, pcDSRα296, were incubated with 25 μg/ml of CRP in the presence of increasing concentrations of aggIgG. CRP binding was detected with 2C10 and PE-GAM as described in Materials and Methods. Results are expressed as the increase in gMCF compared with the secondary antibody controls. The solid line represents binding of CRP to K-562 cells and the dotted line represents the binding of CRP to transfected COS cells in the absence of aggIgG.
Enhanced Binding of CRP to Bt2cAMP-treated U-937 Cells.
Agents that affect the differentiation state may alter the expression of FcR on monocytic cell lines. The U-937 cell line expresses FcγRI and FcγRII. Exposure of U-937 cells to Bt2cAMP has been reported to decrease the expression of FcγRI while increasing the expression of FcγRII 17. We tested the effect of exposure of U-937 cells to Bt2cAMP on the amount of binding of CRP. Since both FcγRI and FcγRII bind CRP, a change in the level of CRP binding might suggest that one or the other receptor is more important in CRP binding. As shown in Fig. 4, Bt2cAMP decreased the expression of FcγRI and increased the expression of FcγRII as previously reported. Associated with this increase in FcγRII, a marked increase in CRP binding was seen (Fig. 4). Thus, the binding of CRP to FcγRII may be quantitatively more important than the binding to FcγRI.
Figure 4.

Treatment of U-937 cells with Bt2cAMP increases CRP binding. U-937 cells were grown for 60 h in medium containing 1 mM Bt2cAMP before analysis of CRP binding. CRP binding was detected with 2C10 and PE-GAM as described in Materials and Methods. Results are expressed as the increase in gMCF compared with the secondary antibody controls. (Inset) FcγRI and FcγRII levels were measured using mAb 32.2 and PE-F(ab′)2 GAM IgG and PE-C1KM5, respectively.
Precipitation of FcR by CRP.
In an attempt to identify proteins reacting with CRP on FcR-bearing cells, receptor precipitation studies were performed. THP-1 cells express both FcγRI and FcγRII. Attempts to immunoprecipitate surface-labeled proteins with immobilized CRP showed weak and nonspecific bands. However, it had previously been determined that the interaction of IgG with FcγRII could be enhanced by pronase treatment of cells 18. We recently determined that pronase treatment of FcγRII-bearing cells also markedly increases the binding of CRP (Stein, M.-P., J.C. Edberg, R.P. Kimberly, E.K. Mangan, D. Bharadwaj, C. Mold, and T.W. Du Clos, manuscript submitted for publication). When pronase-treated cells were radiolabeled and precipitated with CRP, distinct binding of two major bands was seen. These bands with Mr of 70 and 42 kD corresponded to the bands precipitated by mAb to CD64 and CD32 (Fig. 5).
Figure 5.
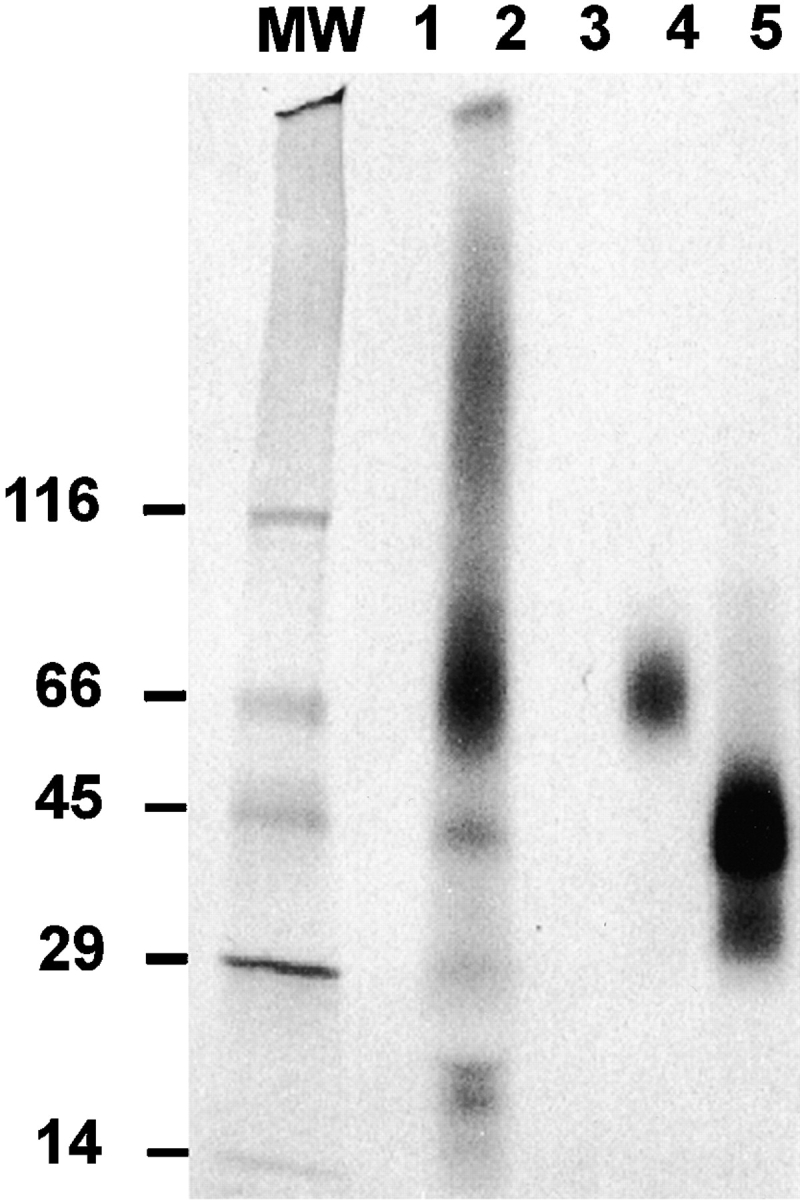
Immobilized CRP precipitates FcγRI and FcγRII from radiolabeled, pronase-treated THP-1 cells. THP-1 cells were treated with 50 μg/ml pronase, radiolabeled with 125I, and lysed with 1% NP-40. (A) Membrane extracts (2.5 × 106 cell equivalents) were incubated with CRP beads, anti-CD32 (mAb IV.3) protein A–Sepharose, or anti-CD64 (mAb 32.2) protein A–Sepharose, and then washed. The samples were boiled in Laemmli's sample buffer and analyzed by SDS-PAGE. Autoradiography was performed using a Storm imaging system. MW, mol wt markers: lane 1, Affigel; lane 2, CRP-Affigel; lane 3, protein A–Sepharose; lane 4, anti-CD64–protein A–Sepharose; lane 5, anti-CD32–protein A–Sepharose.
Discussion
The binding of CRP to FcγRII was demonstrated in cells transfected with the gene for human FcγRIIA. Binding was dose dependent and resembled the binding of CRP to the erythroleukemia cell line, K-562, which expresses FcγRII as the only FcR. Because accessory molecules are not necessary for the expression of FcγRII, it is probable that CRP binds directly to FcγRII and not to associated molecules. This finding was also confirmed by the complete inhibition of CRP binding to K-562 and CD32-transfected COS cells by aggIgG. The binding and inhibition curves for the two cell types were similar. These studies suggest that not only is CRP capable of binding to FcγRII, there is reason to believe it is the only receptor on K-562 cells, which express only the low affinity IgG receptor.
Precipitation of surface-labeled proteins from the monocytic cell line THP-1 with immobilized CRP demonstrated bands with the Mr of FcγRI (70 kD) and FcγRII (42 kD). The 42 kD band could be precleared from the lysate by adsorption with immobilized anti-CD32 mAb, confirming its identity as FcγRII (data not shown).
Our previous studies demonstrated that CRP also binds to the high affinity receptor for IgG, FcγRI 10 11. These studies revealed that mononuclear cells were capable of binding CRP through FcγRI, but FcγRI could not account for all of the binding. Mononuclear cells and monocytic cell lines express FcγRII as well as FcγRI. Our current findings demonstrate the precipitation of both FcγRI and FcγRII from the THP-1 cell line, after pronase activation of FcγRII. However, treatment of U-937 cells with Bt2-cAMP, which increased FcγRII expression, also increased CRP binding, whereas treatment of U-937 cells with rIFN-γ to increase FcγRI did not increase CRP binding (our unpublished results). In our previous studies, binding to U-937 cells was completely inhibited by aggIgG but only 20% inhibited by monomeric IgG 10. These findings indicate that CRP binding to FcγRII accounts for most of the binding to these monocytic cell lines.
Taken together, our results support the hypothesis that CRP binding to phagocytic cells is mediated by FcRs. This is further supported by studies in a mouse model in which knockout mice lacking all three FcRs fail to bind CRP (Stein, M.-P., C. Mold, and T.W. Du Clos, manuscript in preparation). These findings thus suggest that there is no unique receptor for CRP, as was postulated previously, and that the major receptor for CRP on monocytic cells is FcγRIIa.
In conclusion, the acute phase protein CRP, an ancient mediator of innate immunity, binds to FcγRII, considered a receptor for the acquired immune response, with an affinity comparable to that of IgG. It is attractive to speculate that during the acute phase response, high levels of circulating CRP may influence signaling by IgG complexes through FcγRII. It remains to be determined whether CRP will have a positive or negative influence. Future studies will examine the direct effect of CRP on receptor activation and the effect of CRP on IgG-mediated signaling.
Acknowledgments
We thank Dr. Kevin Moore for the donation of the FcγRIIA construct, and Dr. Larry Potempa for the mAb 2C10. We thank Dr. Robert P. Kimberly and Dr. Jeffrey C. Edberg (University of Alabama at Birmingham) for helpful discussions.
This work was supported in part by the Veteran's Administration and by National Institutes of Health grant AI28358.
Footnotes
The first two authors contributed equally to the work.
References
- Osmand A.P., Friedenson B., Gewurz H., Painter R.H., Hofmann T., Shelton E. Characterization of C-reactive protein and the complement subcomponent C1t as homologous proteins displaying cyclic pentameric symmetry (pentraxins) Proc. Natl. Acad. Sci. USA. 1977;74:739–743. doi: 10.1073/pnas.74.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan M.H., Volanakis J.E. Interactions of C-reactive protein with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J. Immunol. 1974;112:2135–2147. [PubMed] [Google Scholar]
- Hokama Y., Coleman M.K., Riley R.F. In vitro effects of C-reactive protein on phagocytosis. J. Bacteriol. 1962;83:1017–1024. doi: 10.1128/jb.83.5.1017-1024.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kindmark C.-O. Stimulating effect of C-reactive protein on phagocytosis of various species of pathogenic bacteria. Clin. Exp. Immunol. 1971;8:941–948. [PMC free article] [PubMed] [Google Scholar]
- Mortensen R.F., Osmand A.P., Lint T.F., Gewurz H. Interaction of C-reactive protein with lymphocytes and monocytescomplement-dependent adherence and phagocytosis. J. Immunol. 1976;117:774–781. [PubMed] [Google Scholar]
- Tebo J.M., Mortensen R.F. Characterization and isolation of a C-reactive protein receptor from the human monocytic cell line U-937. J. Immunol. 1990;144:231–238. [PubMed] [Google Scholar]
- Zahedi K., Tebo J.M., Siripont J., Klimo G.F., Mortensen R.F. Binding of human C-reactive protein to mouse macrophages is mediated by distinct receptors. J. Immunol. 1989;142:2384–2392. [PubMed] [Google Scholar]
- Zeller J.M., Kubak B.M., Gewurz H. Binding sites for C-reactive protein on human monocytes are distinct from IgG Fc receptors. Immunology. 1989;67:51–55. [PMC free article] [PubMed] [Google Scholar]
- Müller H., Fehr J. Binding of C-reactive protein to human polymorphonuclear leukocytesevidence for association of binding sites with Fc receptors. J. Immunol. 1986;136:2202–2207. [PubMed] [Google Scholar]
- Crowell R.E., Du Clos T.W., Montoya G., Heaphy E., Mold C. C-reactive protein receptors on the human monocytic cell line U-937. Evidence for additional binding to FcγRI. J. Immunol. 1991;147:3445–3451. [PubMed] [Google Scholar]
- Marnell L.L., Mold C., Volzer M.A., Burlingame R.W., Du Clos T.W. C-reactive protein binds to FcγRI in transfected COS cells. J. Immunol. 1995;155:2185–2193. [PubMed] [Google Scholar]
- Du Clos T.W., Zlock L., Marnell L.L. Definition of a C-reactive protein binding determinant on histones. J. Biol. Chem. 1991;266:2167–2171. [PubMed] [Google Scholar]
- Stuart S.G., Trounstine M.L., Vaughn D.J.T., Koch T., Martins C.L., Mellman I., Moore K.W. Isolation and expression of cDNA clones encoding a human receptor for IgG (FcγRII) J. Exp. Med. 1987;166:1668–1684. doi: 10.1084/jem.166.6.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G.S., Reisfeld R.A. Protein iodination with solid state lactoperoxidase. Biochemistry. 1974;13:1014–1021. doi: 10.1021/bi00702a028. [DOI] [PubMed] [Google Scholar]
- Ravetch J.V., Kinet J.-P. Fc receptors. Annu. Rev. Immunol. 1991;9:457–492. doi: 10.1146/annurev.iy.09.040191.002325. [DOI] [PubMed] [Google Scholar]
- Ballou S.P., Kushner I. C-reactive protein and the acute phase response. Adv. Int. Med. 1992;37:313–336. [PubMed] [Google Scholar]
- Sheth B., Dransfield I., Partridge L.J., Barker M.D., Burton D.R. Dibutyryl cyclic AMP stimulation of a monocyte-like cell line U937a model for monocyte chemotaxis and Fc receptor-related functions. Immunology. 1988;63:483–490. [PMC free article] [PubMed] [Google Scholar]
- van de Winkel J.G., van Ommen R., Huizinga T.W., de Raad M.A., Tuijnman W.B., Groenen P.J., Capel P.J., Koene R.A., Tax W.J. Proteolysis induces increased binding affinity of the monocyte type II FcR for human IgG. J. Immunol. 1989;143:571–578. [PubMed] [Google Scholar]