

Abstract
Activated vascular endothelial cells (ECs) express major histocompatibility complex (MHC) class II molecules in vitro and in vivo in acute and chronic allograft rejection. However, human ECs may be limited in their ability to effectively activate CD4+ T cells, because they do not express members of the B7 family (CD80 and CD86) of costimulatory molecules. In this study, we show that ECs promote the full activation of CD4+ T cells via trans-costimulatory interactions. By reverse transcriptase polymerase chain reaction, Western blot, and FACS® analysis, we could not detect the expression of CD80 and CD86 on activated ECs and found minimal expression on purified CD4+ T cells. In contrast, both CD80 and CD86 were expressed in allogeneic CD4+ T cell–EC cocultures. Expression of CD86 peaked at early times between 12 and 24 h after coculture, whereas CD80 was not expressed until 72 h. Addition of anti-CD86 but not anti-CD80 monoclonal antibodies to cocultures inhibited IL-2 production and the proliferation of CD4+ T cells to allogeneic donor human umbilical vein ECs (HUVECs), as well as to skin and lung microvascular ECs. Furthermore, we found that interferon γ–activated ECs but not untreated ECs induced mRNA and cell surface expression of CD80 and CD86 on CD4+ T cells, and these T cells were functional to provide a trans-costimulatory signal to autologous CD4+ T cells. Blockade of MHC class II and lymphocyte function–associated antigen 3 but not other EC cell surface molecules on IFN-γ–activated ECs inhibited the induction of CD86 on CD4+ T cells. Transmigration of purified populations of monocytes across EC monolayers similarly resulted in the induction of functional CD86, but also induced the de novo expression of the cytokines interleukin (IL)-1α and IL-12. In addition, EC-modified monocytes supported enhanced proliferation of allogeneic and autologous CD4+ T cells. Taken together, these data define the ability of the endothelium to modify CD4+ T cells and monocytes for trans-costimulatory events. This unique function of the endothelium in alloimmune T cell activation has functional consequences for the direct and the indirect pathways of allorecognition.
Keywords: endothelium, T lymphocyte, monocyte, allorecognition, transplantation
Recognition of alloantigen (donor major or minor histocompatibility antigens) by CD4+ T cells is the initiating event that ultimately leads to graft rejection. Recipient CD4+ T cells may recognize either intact allo-MHC class II molecules on donor cells (known as direct allorecognition) or peptides derived from allo-MHC molecules, shed from the allograft and subsequently processed and presented bound to MHC class II on recipient APCs (known as indirect allorecognition) 1. Irrespective of the pathway of allorecognition, full activation of naive or previously activated CD4+ T cells requires a second signal that may be provided by soluble factors such as cytokines, but more often is provided by cell surface costimulatory molecules such as B7-1 (CD80) and B7-2 (CD86) 2 3. These molecules are expressed by professional APCs and bind the counterreceptors CD28 and cytolytic T lymphocyte–associated antigen (CTLA)41 expressed by CD4+ T cells. Ligation of CD28 is necessary for maximal CD4+ T cell cytokine production, proliferation, and prevention of activation-induced apoptosis 4 5. In contrast, ligation of CTLA4 delivers a negative signal to CD4+ T cells 6.
It is well established that activated endothelial cells (ECs) express class II MHC molecules in vitro and in vivo in rejecting allografts, and may therefore provide antigen-dependent signals to CD4+ T cells for direct activation 7 8 9 10 11. An unresolved issue is whether ECs can provide the costimulatory signals required to fully activate CD4+ T cells 12 13 14 15 16. Current evidence suggesting a role for ECs in CD4+ T cell activation in vivo is based heavily on in vitro studies, and only a few studies have provided indirect evidence in vivo 17 18. The majority of in vitro studies show that class II–expressing ECs can induce IL-2 production and alloproliferation of bulk populations of CD4+ T cells, an effect largely dependent on costimulation by endothelial lymphocyte function–associated antigen (LFA)-3 12 13 14 15. However, inhibition of CD2–LFA-3 interactions with anti–LFA-3 mAbs only partially inhibits EC-induced CD4+ T cell cytokine production and alloproliferation, suggesting a role for other costimulatory molecules in this process. Although B7-dependent costimulation has been demonstrated in allogeneic mouse EC-induced T cell activation 19 and xenogenic pig EC-induced human T cell activation 20 21, firm conclusions have not been made as to whether CD28–B7 interactions are functional in the activation of human CD4+ T cells by human ECs. This issue is relevant for a proactive role of ECs in rejection, because activation of naive CD4+ T cells (induction of primary immune responses) and the activation of previously activated CD4+ T cells is dependent on CD28-mediated costimulation 22 23. Indeed, in the absence of CD28-mediated signals, naive CD4+ T cells may be rendered refractory to further stimulation by antigen (clonal anergy) 24, or reactivation responses may be qualitatively different 25. Furthermore, recent studies have demonstrated a requirement for B7 costimulation at the local site of inflammation 26. In these studies we investigate the role of CD28–B7 interactions in human EC-induced CD4+ T cell alloactivation. Our data provide new insight into the ability of ECs to modify leukocytes to promote direct (and perhaps indirect) allorecognition and define novel mechanisms by which allograft ECs may promote transplant rejection.
Materials and Methods
Reagents and Antibodies.
Antibodies used in these studies include seven anti-CD86 mAbs; IT2.2 and FUN-1 clone (PharMingen), anti–human CD86 (Serotec), anti–human CD86 (B7-F3, gift from P. Linsley, Bristol Myers Squibb, Princeton, NJ), HF2 3D1, HA5 2B7, HA3 1F9 (gifts from V.J. Kuchroo, Brigham and Women's Hospital, Boston, MA), anti–human CD80 (PharMingen), anti–human CD25 (PharMingen, San Diego, CA); anti–human LFA-3 (IE6, gift from P. Hochman, Biogen Inc., Cambridge, MA), anti–human CD40 (220; a gift from D. Hollenbaugh, Bristol Myers Squibb), anti–human OX40 (Ancell Corp.), anti–human intercellular adhesion molecule (ICAM)-1 (RR1/1, a gift from T.A. Springer, Center for Blood Research, Harvard Medical School, Boston, MA), anti–HLA-DR (LB3.1, a gift from A.H. Lichtman, Brigham and Women's Hospital), and negative control mouse IgG (K16/16, a gift of M. Gimbrone, Brigham and Women's Hospital). Human CTLA4 Ig and control fusion protein were gifts from Dr. Peter Linsley (Bristol Myers Squibb). Cytokines used were recombinant human IFN-γ (Genzyme) and TNF-α (a gift from Biogen Inc.). Other reagents used included recombinant soluble CD154 (a gift from D. Hollenbaugh), LPS (Sigma Chemical Co.), and PHA (Sigma Chemical Co).
Cell Isolation and Culture.
Endothelial cells were isolated from human umbilical cords as previously described 27 and were grown in M199 medium (BioWhittaker) containing 10% FCS (GIBCO BRL), EC growth supplement, 1% penicillin/streptomycin, l-glutamine, and heparin. Cultured cells were harvested in trypsin/ethylenediaminetetraacetic acid (Sigma Chemical Co.) and subcultured for use at passages 2–4. Saphenous vein ECs were a gift from Dr. P. Libby (Brigham and Women's Hospital) 28. Single donor, dermal, and lung microvascular ECs were purchased from Clonetics.
Cell membrane fractions of ECs or CD4+ T cells were prepared as previously described 29. In brief, untreated or IFN-γ–treated ECs (2–4 × 106) were harvested by gentle scraping, washed, and resuspended in lysis buffer containing 0.25 M sucrose, 10 mM Tris (pH 7.4), 10 mM NaCl, 0.1 M MgCl2, and 1 mM PMSF. The cells were lysed by homogenization and were centrifuged at 1,000 g for 15 min. Supernatants were centrifuged at 100,000 g for a further 30 min. All manipulations were performed at 4°C. Pellets were resuspended in RPMI and added directly to CD4+ T cells (106) in 96-well plates. For select experiments, cell membranes were prepared from unactivated or mitogen-activated CD4+ T cells (107 cells/condition).
PBMCs were isolated by Ficoll-Hypaque gradient centrifugation from blood obtained from healthy volunteers. CD4+ T cells were isolated from PBMCs by positive selection using anti-CD4–coated magnetic beads (Dynal Inc.) according to the manufacturer's instructions. Magnetic beads were subsequently removed using Detachabead (Dynal Inc.). In some experiments CD4+ T cells were further purified by negative depletion of CD14 and HLA-DR expressing cells using a CD14-coated microbead column (MiniMACS separation column; Miltenyi Biotec) and panning on anti–HLA-DR (LB3.1) coated plastic culture dish respectively. The purity of the CD4+ T cells using these methods was 98 and 99.7%, respectively. Purity was assessed by double stain FACS® analysis for CD3 and CD4 cell surface markers. Purified cells were unactivated as assessed by the lack of spontaneous proliferation, IL-2 and IFN-γ production, and CD25 cell surface expression, as previously described 30. Human monocytes were isolated from platelet pheresis residues by centrifugation on density gradients (LSM; Organon Teknika), followed by counterflow centrifugation elutriation 31. Monocytes isolated by this technique are >90% pure and are relatively unactivated as determined by minimal alterations in cell surface activation markers. In some experiments, monocytes were isolated from PBMCs by positive selection using CD14-coated microbeads (MiniMACS separation column; Miltenyi Biotec).
CD80-, CD86-, and neomycin-transfected Chinese hamster ovary (CHO) cells (a gift from Dr. G. Freeman, Dana Farber Cancer Institute, Boston, MA) were cultured in collagen-coated tissue culture flasks in complete RPMI with 10% FCS. Cells were harvested by trypsinization and fixed in 0.4% PFA before addition to EC–CD4+ T cell cocultures.
CD4+ T cell–EC Coculture.
Primary cultures of ECs (passages 3–4) were treated with IFN-γ (1,000 U/ml) for 72 h to upregulate class II MHC. IFN-γ–treated ECs (5 × 104/well) were then irradiated (1,750 rads) and cocultured with resting CD4+ T cells (5 × 105/well) in 96-well cell culture plates in a final volume of 200 μl. Additional cells or reagents were added as indicated. Coculture supernatants were taken at days 3 and 5 for cytokine analysis by specific ELISA. Proliferation was assessed after 6 d by [3H]thymidine incorporation for the last 18 h of coculture. Cells were harvested by an automated cell harvester and incorporated radioactivity was assessed by a Beckman Betamax counter.
In separate experiments, we examined whether ECs modify CD4+ T cells to express functional CD86. CD4+ T cells were cultured on IFN-γ–treated human umbilical vein EC (HUVEC) monolayers for 24–72 h and then reisolated by positive selection using CD4-coated magnetic beads (Dynal Inc.). These cells were termed “EC-modified” CD4+ T cells. EC-modified CD4+ T cells (105) were irradiated (1,750 rads) and cocultured with resting autologous CD4+ T cells (5 × 105) in the presence of submitogenic doses of PHA (0.3 μg/ml). Coculture supernatants were taken at 24 h for specific ELISA. Proliferation was assessed after 3 d of coculture as described above.
Transmigration Assay.
Transmigration assays were performed using a protocol modified, as follows, from one described previously 32. HUVECs were seeded at 2 × 105 cells/cm2 on collagen-coated, 8-μm-pore size polycarbonate tissue culture Transwell inserts (Costar Corp.) and were cultured for 5 d to attain confluence. Confluency was assessed by exclusion of FITC-labeled dextran in control wells as previously described 32. HUVECs were pretreated with IFN-γ (1,000 U/ml) for 72 h to upregulate MHC class II. Purified, resting monocytes (5 × 106) were added onto the transwell and allowed to transmigrate across untreated or IFN-γ–treated ECs. HUVECs were washed thoroughly to remove IFN-γ before addition of monocytes. Transmigrated cells were counted and RNA was isolated from 2 × 106 cells 12 h after transmigration. Induction of monocyte CD86 and cytokine mRNA expression was determined by semiquantitative reverse transcriptase (RT)-PCR and RNase protection assay.
Isolation of RNA and RT-PCR Analysis.
Total RNA was prepared using the Ultraspec RNA isolation system (Biotecx) according to the manufacturer's instructions, and was quantified by spectrophotometry. cDNA was prepared by reverse transcription of 5 μg of RNA using random hexamer primers (100 ng/μl) and Moloney murine leukemia virus reverse transcriptase (50 μm/μl) (Stratagene) in a 50-μl reaction. 10 μl of cDNA was used for each PCR amplification reaction. PCR was performed with Taq DNA polymerase using the buffer supplied by the manufacturer (Boehringer Mannheim). The PCR primers were: human CD80, sense: 5′-CAT CAC GGA GGG TCT TCT AC-3′ and antisense: 5′-AGG ATC TTG GGA AAC TGT TGT-3′; and human anti-CD86, sense: 5′-AGG ACA AGG GCT TGT ATC AA-3′ and antisense: 5′-ATT GCT CGT AAC ATC AGG GA-3′. The PCR conditions were 94°C for 1 min, 60°C for 1 min, and 72°C for 2 min for 35 cycles. PCR products were analyzed by ethidium bromide staining in 1.5% agarose gels using standard techniques.
RNase Protection Assays.
RNase protection was performed using the Riboquant™ Multi-Probe RNase Protection assay system (PharMingen). RNA was isolated as described above. 32P-labeled probes were synthesized from the hCK-2 human cytokine multi-probe template set and were hybridized overnight with RNA samples in hybridization buffer according to the manufacturer's instructions. Samples were digested with RNase and T1 mix in RNase buffer and protected probes were purified and were run on a 5% acrylamide gel in 0.5% TBE buffer. Human control RNA and a dilution of the probe set (serving as size markers) were run in parallel. The gel was absorbed onto filter paper, dried, and exposed onto Kodak photographic paper at −70°C for 24 h.
Cytokine Assays.
IL-2 was assessed by specific ELISA. Primary and secondary antibodies were purchased from Genzyme and were used according to the recommended protocol. In brief, 96-well flat-bottomed ELISA plates (Falcon; Becton Dickinson Labware) were coated with primary antibody overnight at 4°C. Blocking was then performed with 4% BSA in PBS for 2 h at 37°C and neat coculture supernatants or standards were added to each well in duplicate for 1 h at 37°C. After the incubation, secondary biotinylated anti–IL-2 mAb was added and the ELISA was developed using avidin alkaline phosphatase (Sigma Chemical Co.) and phosphatase substrate (Sigma Chemical Co.). In between each step, the plates were washed in PBS with 0.01% Triton X-100. Plates were read at 405 nm in an E-Max ELISA plate reader (Molecular Devices).
Flow Cytometry.
Cell suspensions of CD4+ T cells, ECs, or monocytes were analyzed by direct immunofluorescence. In brief, 1–2 × 106 cells were incubated with FITC- and/or PE-conjugated mAbs at 4°C for 30 min and were fixed in 1% PFA. Stained cells were then analyzed by FACScan® (Becton Dickinson). Monocytes were preincubated with buffer containing 20% non-A non-B human serum before flow cytometry to block nonspecific Fc receptor binding and optimize specific binding.
Western Blotting.
Western blot was performed on ∼5 × 106 cells per condition. Cells were lysed in PBS containing 1% NP-40, and protease-inhibitor (Boehringer Mannheim) and lysates were separated by standard 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Blots were blocked overnight at 4°C in Tris-buffered saline containing 0.1% Tween 20 and 2% BSA before incubation with optimal concentrations of primary antibody diluted in TBS/0.1% Tween 20 for 12 h at 4°C. After four washes in TBS/0.1% Tween 20, blots were incubated in peroxide-conjugated goat anti–mouse secondary antibody (Jackson ImmunoResearch Labs.) at a 1:2,500 dilution in TBS for 4 h at 4°C. The blots were then washed and developed by chemiluminescence (Amersham Inc.).
Results
Expression of CD80 and CD86 in CD4+ T Cell–EC Cocultures.
To investigate a role for CD80 and CD86 in CD4+ T cell–EC interactions, we initially examined the expression of these molecules by resting or activated CD4+ T cells, and ECs alone or after coculture of both cell types (Fig. 1). Our findings were that TNF-α, IFN-γ, IL-1, or soluble CD40 ligand (CD40L)–stimulated HUVECs do not express CD80 or CD86 mRNA by RT-PCR; likewise resting CD4+ T cells do not express CD80 and express variable and low levels of CD86 mRNA. In contrast, we found a marked expression of both CD80 and CD86 mRNA in CD4+ T cell–HUVEC cocultures. CD86 mRNA was detected as early as 6 h and was maximal by 72 h of coculture, whereas CD80 mRNA was only expressed in cells harvested at 72 h of coculture (Fig. 1 A). CD86 protein was detected by Western blot in 72-h CD4+ T cell–HUVEC cocultures but not in resting CD4+ T cells or TNF-α, IFN-γ, or soluble CD40 ligand–activated HUVECs (Fig. 1 B). By FACS® analysis, anti-CD80 mAbs, seven anti-CD86 mAbs, and CTLA4 Ig did not bind to resting or cytokine-activated HUVECs, saphenous vein ECs, dermal microvascular ECs, or lung microvascular ECs; although positive control CD80- and CD86-transfected CHO cells consistently demonstrated high levels of binding (Fig. 1 C and data not shown). Thus, human ECs express neither CD80 nor CD86, but both these molecules are induced in CD4+ T cell–EC cocultures.
Figure 1.

Expression of CD80 and CD86 in allogeneic CD4+ T cell–EC cocultures. (A) Semiquantitative RT-PCR was performed on RNA samples harvested from resting CD4+ T cells, IFN-γ–treated HUVECs, or cocultures of CD4+ T cell with IFN-γ–treated HUVECs at various time points. Illustrated is one experiment representative of four. Resting CD4+ T cells occasionally show low level expression of CD86. (B) Western blot analysis of 24-h TNF-α–, soluble CD40 ligand–, and IFN-γ–activated ECs, or CD4+ T cells or 72-h CD4+ T cell–IFN-γ–treated EC cocultures. Two different anti-CD86 antibodies were used for our studies: IT2.2 (PharMingen; lanes 1–3) and B7-F3 (Bristol Myers Squibb; lanes 4–7). Illustrated are two representative blots showing absent CD86 expression on ECs and CD4+ T cells, but enhanced expression in CD4+–EC cocultures. Positive controls were CD86-transfected CHO cells. Data is representative of four experiments. (C) FACS® analysis of untreated HUVECs or 72-h IFN-γ (1,000 U/ml)–treated HUVECs for CD80, CD86, and HLA-DR (positive control). Control isotype mouse IgG was used as a negative control. As positive controls, we used anti-CD80 mAb and anti-CD86 mAbs bound to CD80- and CD86-transfected CHO cells, respectively (not shown).
Function of CD86 and CD80 in CD4+ T Cell–Allogeneic EC Cocultures.
We next wished to determine the function of CD86 and CD80 in CD4+ T cell–EC interactions. Purified CD4+ T cells were rested for 48 h and were cocultured with either HUVECs, saphenous vein ECs, dermal microvascular ECs, or lung microvascular ECs, untreated or treated with IFN-γ for 72 h to upregulate class II MHC. Anti-CD86 mAbs, anti-CD80 mAbs, or control isotype antibodies were added to cocultures as indicated by each experiment (Fig. 2). We found that anti-CD86 mAbs consistently inhibited IFN-γ–treated HUVEC-induced CD4+ T cell alloproliferation (Fig. 2 A). Maximal inhibition with anti-CD86 mAbs was variable (20–50%) and was less than that observed by blocking CD2–LFA-3 interactions with anti–LFA-3 mAbs. In addition, we found that the combination of anti-CD86 and anti–LFA-3 mAbs provided additive inhibition of CD4+ T cell alloproliferation (Fig. 2 A) and IL-2 production (data not shown). The inhibitory effects of anti-CD86 mAbs was dose dependent (Fig. 2 B) and was observed when other types of human ECs were cocultured with CD4+ T cells (Fig. 2 C). In contrast, interruption of CD28–CD80 interactions using anti-CD80 mAbs did not inhibit EC-induced CD4+ T cell alloproliferation, most likely because CD80 is not expressed at early time points in the CD4+ T cell–EC coculture (Fig. 1 A). Thus, CD86 but not CD80 is functional in allogeneic EC-induced CD4+ T cell activation.
Figure 2.

Function of CD80 and CD86 in allogeneic CD4+ T cell–EC cocultures. CD4+ T cells were cultured alone (white bars), with untreated HUVECs (gray bars), or with HUVECs that were treated with IFN-γ (1,000 U/ml) for 72 h (black bars). (A) Anti-CD86 antibody (IT2.2, clone, 10 μg/ml), LFA-3 (5 μg/ml), or combinations were added into cocultures of CD4+ T cells with IFN-γ–treated HUVECs as indicated. Data represents the mean of 10 experiments performed in triplicate cultures ± 1 SD. (B) Dose–response effect of anti-CD86 antibody (IT2.2 clone) on CD4+ T cell alloproliferation. We also find that anti-CD86 mAbs inhibit EC-induced CD4+ T cell IL-2 production (data not shown). Results are representative of four similar experiments performed in triplicate cultures ± 1 SD. (C) Coculture of CD4+ T cells with various IFN-γ–treated microvascular endothelial cells in the absence (black bars) or presence (gray bars) of anti-CD86 mAbs (10 μg/ml, IT2.2 clone). Results are representative of two similar experiments performed in triplicate cultures ± 1 SD (*P < 0.05). (A–C) In all experiments, proliferation was assessed after 6 d of coculture by [3H]thymidine uptake.
Human ECs Induce CD86 Expression by CD4+ T Cells.
Since we find that human ECs do not express CD86, we next wished to assess CD4+ T cell CD86 expression and function in our model. Previous studies have shown that activated CD4+ T cells may express both CD80 and CD86 33 34. CD4+ T cells were cultured with IFN-γ–treated HUVECs and CD86 expression determined by double stained FACS® analysis of cells. As illustrated in Fig. 3 A, we found low levels of CD86 on resting CD4+ T cells, but augmentation of CD4+ T cell CD86 expression after coculture with IFN-γ–treated ECs. Maximal expression of CD4+ T cell CD86 was observed after 72 h of coculture. We also confirmed that CD86 was induced de novo on CD4+ T cells by sorting and culturing CD86−CD4+ T cells with IFN-γ–treated ECs for 24 h. Consistently, we found that ECs induce de novo expression of CD86 on CD4+ T cells.
Figure 3.

EC induction of CD86 on CD4+ T cells. (A) Double stain FACS® analysis of resting CD4+ T cells, or CD4+ T cells that were cultured with IFN-γ–activated ECs for 72 h using FITC-conjugated CD3 and PE-conjugated CD86 antibodies (PE-conjugated mouse isotype antibody served as control). (B) The expression of CD86 by semiquantitative RT-PCR on RNA harvested from CD4+ T cells alone (lane 1), CD4+ T cells cultured with cell membrane preparations from unactivated ECs (lane 2), or CD4+ T cells cultured with cell membrane preparations from IFN-γ–treated ECs (lane 3). (C) CD86 and E-selectin expression by RT-PCR on RNA harvested from HUVECs (lane 1), HUVECs cultured with cell membrane preparations from resting CD4+ T cells (lane 2), or HUVECs cultured with cell membrane preparations from PHA-activated CD4+ T cells (lane 3). IFN-γ–treated monocytes were used as a positive control (lane 4). Membranes were prepared as described in Materials and Methods from 5 x106 cells. Data is representative of three similar experiments.
Furthermore, to determine whether EC-induced CD86 expression by CD4+ T cells was dependent on cell contact, we generated cell membranes from ECs and incubated them with CD4+ T cells. Membrane preparations from IFN-γ–activated ECs but not resting ECs induced CD4+ T cell CD86 mRNA expression by RT-PCR, consistent with the interpretation that induction of CD4+ T cell CD86 is mediated by a cell surface molecule(s) on activated ECs (Fig. 3 B). However, when membranes generated from unactivated or activated CD4+ T cells were incubated with ECs, they failed to induce EC expression of CD86. As a positive control, activated CD4+ T cell membranes enhanced EC E-selectin expression (Fig. 3 C).
Functionality of EC-induced CD4+ T Cell CD86.
Having established that ECs induce CD86 expression on CD4+ T cells, we next assessed function. CD4+ T cells were cultured with IFN-γ–treated ECs and were reisolated by positive selection after 24 h of coculture. These EC-modified CD4+ T cells were then irradiated and cocultured either alone or with resting autologous CD4+ T cells in the presence of submitogenic doses of PHA (0.3 μg/ml). Although resting CD4+ T cells alone and EC-modified CD4+ T cells alone failed to proliferate to low dose PHA, coculture of both cell types resulted in enhanced proliferation, which was inhibited by ∼50–80% by anti-CD86 mAbs (Fig. 4 A). This suggests that induced CD86 on EC-modified CD4+ T cells provides an effective costimulatory signal in trans to resting CD4+ T cells. However, the proliferative responses of CD4+ T cells in this trans-costimulation assay were less than those induced by ECs (Fig. 2 and data not shown). We note that this is consistent with the ability of ECs to provide additional costimulatory signals such as LFA-3–dependent signals as reported by others 13 35.
Figure 4.

EC-modified CD4+ T cells provide CD86-mediated trans-costimulation to autologous CD4+ T cells. (A) CD4+ T cells were cultured on a monolayer of IFN-γ–treated endothelial cells for 72 h and were reisolated from the coculture by positive selection using CD4-coated magnetic beads. These cells were then irradiated (1,750 rads) and 105 cells were cultured with autologous CD4+ T cells (5 × 105) in the presence of low dose PHA (0.3 μg/ml) for 4 d. Shown are the proliferative responses of CD4+ T cells. Anti-CD86 (10 μg/ml, IT2.2 clone), anti-CD80 (10 μg/ml, BB1-1 clone), or CTLA4 Ig (10 μg/ml) were added as indicated. Illustrated is one experiment representative of three similar experiments in triplicate cultures ± 1 SD. Anti–HLA-DR mAbs (LB3.1) do not inhibit CD4+ T cell proliferation (data not shown). (B and C) To determine the EC cell surface molecule(s) that mediates induction of CD4+ T cell CD86 expression, anti–HLA-DR (LB3.1), anti–LFA-3 (1E6), anti–ICAM-1 (RR1/1), anti-CD40(220), or anti-OX40 (Ancell) were added to the primary CD4+ T cell–EC coculture at optimal blocking concentrations. CD4+ T cells were reisolated, irradiated, and added to autologous CD4+ T cells in secondary cultures in the presence of low dose PHA as described above. Proliferation was assessed by [3H]thymidine incorporation for the final 18 h of a 6-d culture. Data is a representative of three experiments performed in triplicate cultures ± 1 SD. Irradiated stimulator cells alone failed to proliferate, as illustrated in A.
To determine the molecular basis for EC modification of CD4+ T cell costimulatory activity, we incubated CD4+ T cells with IFN-γ–treated ECs in the absence or presence of anti–ICAM-1, anti–LFA-3, anti–HLA-DR mAbs, anti-CD40 mAbs, anti-OX40 mAbs, or isotype control antibodies for 24 h. EC-modified CD4+ T cells were then reisolated and irradiated, and were added to resting CD4+ T cells in the presence of submitogenic doses of PHA as described above. We found that anti–LFA-3 and anti–HLA-DR, but not anti-CD40, anti-OX40L, anti–ICAM-1, or control antibody, inhibited the subsequent CD4+ T cell costimulatory effect (Fig. 4B and Fig. C, and data not shown). Anti-CD40 antibodies also failed to inhibit T cell–T cell trans-costimulation (Fig. 4 C). Thus, induction of functional CD86 on CD4+ T cells is in part dependent on interactions between CD4+ T cells and EC class II MHC and LFA-3.
Finally, to confirm functionality of CD86-dependent trans-costimulation for direct allorecognition (when signal one is provided by alloantigen on ECs rather than mitogen), CD4+ T cells were cocultured with IFN-γ–treated ECs in the presence of increasing numbers of CHO cells transfected with CD86. Mock-transfected CHO cells were used as a negative control. CD86-transfected, but not mock-transfected, CHO cells enhanced EC-induced CD4+ T cell alloproliferation (Fig. 5 A) and IL-2 production (data not shown) in a dose-dependent manner that is inhibited by anti-CD86 mAbs and CTLA4 Ig (Fig. 5 B). This data confirms that CD4+ T cells can receive CD86 costimulation in trans when signal one is provided by alloantigen on ECs.
Figure 5.


Effect of CD86 trans-costimulation on alloantigen-induced CD4+ T cell activation. (A) CD4+ T cells (5 × 105) were cocultured with IFN-γ–treated endothelial cells in the presence of increasing numbers of either CD86-transfected or mock-transfected CHO cells. CHO cells were fixed in 0.4% paraformaldehyde before coculture to inhibit spontaneous proliferation. (B) Anti-CD86 (10 μg/ml, IT2.2) or CTLA4 Ig (10 μg/ml) were added to cocultures as indicated. Proliferation was assessed by [3H]thymidine incorporation for the final 18 h of a 6-d culture. Shown is one experiment representative of four in triplicate cultures ± 1 SD. CD4+ T cells cocultured with CHO cells alone did not proliferate.
EC-induced CD86 Trans-costimulation Is Mediated by CD3+ CD4+ T Cells and Not by Contaminating CD4+ Dendritic Cells.
It has been reported that a subpopulation of peripheral blood CD4+ cells are HLA-DR+ and CD3− myeloid derived dendritic cells 36. We found that ∼2% of our CD4+ cells were CD3−, and therefore we wished to confirm that the CD86-dependent trans-costimulation described above is indeed due to CD86 expressed by CD4+ T cells and not to dendritic cell CD86. We further purified our CD4+ cells by negative selection for HLA-DR and CD14 expressing cells (as described in Materials and Methods). The resulting cells were CD4+CD3+HLA-DR− T cells (Fig. 6 A) and express low levels of CD86. However, consistently after coculture with IFN-γ–treated ECs, these T cells exhibit augmented CD86 expression (Fig. 6 B). Furthermore, these T cells provide effective trans-costimulation to autologous T cells in the presence of low dose mitogen (Fig. 6 C). Trans-costimulation is mediated in part by CD86 (with 25–75% inhibition observed with anti-CD86 mAbs) but also involves other cell surface molecules including LFA-3 (data not shown and Fig. 4). There is no antigen-dependent component to the T cell–T cell proliferative response, since anti–HLA-DR antibody (LB3.1) failed to inhibit proliferation with concentrations of mitogen (PHA > 0.3 μg/ml) used in our model. However, in the absence of mitogen or at low doses of mitogen (PHA < 0.1 μg/ml), anti–HLA-DR partially inhibits proliferative responses.
Figure 6.

EC-induced CD86 trans-costimulation is mediated by CD3+CD4+ T cells and not by contaminating CD4+ dendritic cells. (A) FACS® analysis of CD4+ T cells purified by positive selection and depletion of HLA-DR+ and CD14+ cells. Double staining of purified CD4+ T cells with FITC-conjugated CD3 and PE-conjugated CD4 antibodies. (B) CD3+CD4+ T cells were cocultured with IFN-γ–treated ECs for 72 h and were repurified by positive selection. FACS® analysis was then performed by double staining for CD3 and CD86. Histograms show shifts in PE-conjugated CD86 (shaded area) and control PE-isotype antibody staining by CD3+ cells gated within regions R1 (scattergram in A). Illustrated are CD86 expression in resting, 72-h EC-activated and PHA (1 μg/ml)-activated CD4+ T cells. (C) CD4+ T cells were modified by ECs to express CD86 (as above) and were irradiated and cocultured with resting autologous CD4+ T cells in the presence of low dose mitogen (PHA 0.3 μg/ml). Anti-CD86 (10 μg/ml) was added to cultures as described in Fig. 4. Shown is one experiment representative of four performed in triplicate cultures ± 1 SD. Irradiated stimulator cells alone failed to proliferate.
ECs Induce Functional Costimulatory Activity in Transmigrating Monocytes.
We next wished to determine whether ECs modify the costimulatory and antigen presenting capacity of monocytes. Recent studies suggest that transmigration of monocytes across ECs promotes their differentiation into dendritic cells 37 38. Indeed, a novel function of ECs may be to enhance the antigen presenting and costimulatory function of infiltrating APCs in the course of alloimmune inflammatory reactions and rejection. Monocytes were isolated by elutriation in order to obtain a relatively unactivated cell population. Cells were then allowed to transmigrate across resting or 72-h IFN-γ–treated confluent EC monolayers in transwells as described in Materials and Methods. After 12 h, cells were harvested from the lower chamber of the transwell and RNA was isolated for analysis by RT-PCR and RNase protection. We found that transmigration of monocytes across IFN-γ–treated ECs, and to a lesser extent resting ECs, resulted in the induction of monocyte CD86 mRNA expression by RT-PCR (data not shown) and enhanced CD86 protein expression by FACS® analysis (Fig. 7 A). Transmigration across transwell inserts not coated with ECs did not result in induction of CD86 (data not shown). We also found that transmigration of monocytes across IFN-γ–treated ECs resulted in the expression of the cytokine IL-1α as determined by RNase protection assay (Fig. 7 B); and coincubation of monocytes with IFN-γ–treated ECs resulted in induction of monocyte IL-12 expression (Fig. 7 C).
Figure 7.
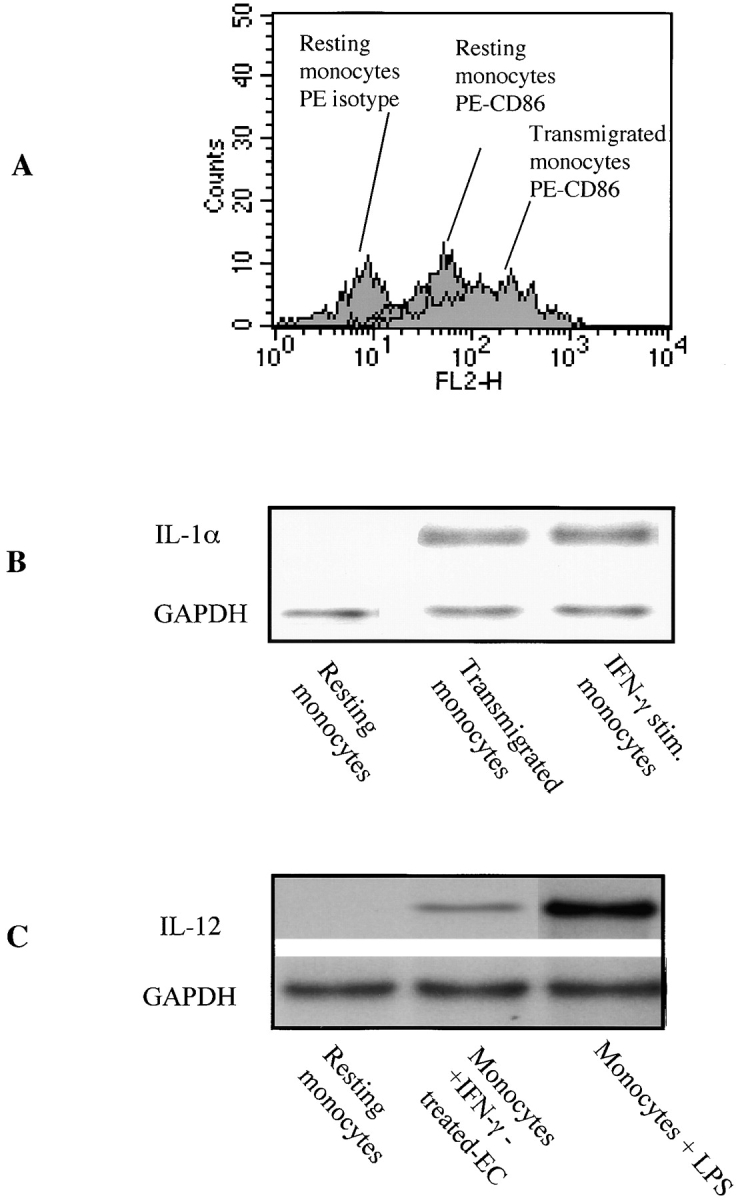
Transmigrated monocytes express enhanced costimulatory molecules. Purified populations of monocytes (107) were cultured with IFN-γ–treated HUVEC monolayers in the upper chamber of a 8-μm-pore polycarbonate transwell. (A) Monocyte CD86 protein expression was determined by FACS® 24 h after transmigration. Illustrated is the FACS® analysis of CD86 in resting monocytes and transmigrated monocytes. Isotype control PE antibody served as control. Representative analysis of three experiments. (B) Monocytes were harvested from the lower chamber after 12 h. Illustrated is the expression of IL-1α by RNase protection from resting, transmigrated monocytes and positive control IFN-γ–activated monocytes. (C) RNA was harvested from cocultures of monocytes and IFN-γ–activated endothelial cells. Illustrated is the expression of IL-12 by RNase protection in resting monocytes, monocytes cocultured with IFN-γ–activated ECs, and LPS (1 μg/ml)-stimulated monocytes. Unactivated or IFN-γ–activated ECs do not express IL-12, and IFN-γ alone only weakly induces IL-12 expression in resting monocytes (not shown).
Finally, we compared the ability of resting monocytes with that of EC-modified monocytes to activate allogeneic and autologous CD4+ T cells. After a 72 h coculture with IFN-γ–treated ECs, CD14+ monocytes were reisolated by positive selection and cultured with allogeneic or autologous CD4+ T cells at fixed responder/stimulator ratios. We found that EC-modified monocytes consistently induced greater proliferation of allogeneic CD4+ T cells than did resting monocytes (Fig. 8 A), an effect inhibited by CTLA4 Ig and anti–HLA-DR antibodies (Fig. 8 B). CTLA4 Ig caused a greater percentage of inhibition of CD4+ T cell proliferation induced by EC-modified monocytes, consistent with enhanced expression of CD86 by these cells. Thus, ECs augment the capacity of monocytes to provide costimulatory signals to CD4+ T cells activated by direct allorecognition. Furthermore, EC-modified monocytes induced proliferation of autologous CD4+ T cells. This suggests that ECs may donate alloantigen to monocytes for presentation to autologous CD4+ T cells via the indirect pathway of allorecognition.
Figure 8.

Functional effects of resting and EC-modified monocytes. (A) Monocytes were cultured on a monolayer of IFN-γ–treated ECs or a plastic culture dish as a control for 72 h and were reisolated by positive selection using CD14-coated magnetic beads. Monocytes were then irradiated (1,750 rads) and added at fixed ratios to either allogeneic or autologous CD4+ T cells (105) for 6 d. (B) Resting or EC-modified monocytes (as above) were cocultured with allogeneic CD4+ T cells in a mixed leukocyte reaction (stimulator/responder ratio 1:10). Anti–HLA-DR (10 μg/ml) or CTLA4 Ig (10 μg/ml) were added to cultures. Proliferation was assessed by [3H]thymidine incorporation after 6 d of coculture. Results shown are representative of three experiments in triplicate cultures ± 1 SD.
Discussion
Microvascular ECs express cell surface molecules that mediate both the recruitment into and the activation of leukocytes within vascularized solid organ transplants. Thus, it is proposed that interactions between CD4+ T cells and microvascular graft ECs are critical for rejection 12. Antigen-dependent activation of CD4+ T cells is initiated by interactions between the TCR and foreign peptide in association with MHC class II molecules. ECs express MHC class I and class II molecules and provide antigen-dependent signals to T cells in vitro 7 8 9 10 11 12 and in vivo 17 18. However, the ability of human endothelium to provide effective costimulatory signals for full CD4+ T cell activation is more controversial 12 13 14 15 16. Surprisingly, only endothelial LFA-3 has been identified to possess costimulatory function with little if any contribution of other human EC cell surface molecules. Endothelial LFA-3 interacts with T cell CD2 and initiates a series of activation responses in CD4+ T cells that result in IL-2, IL-4, and IFN-γ production 10 15 39. Since naive and previously activated CD4+ T cells are dependent on CD28 signaling for effective activation 22, we wished to examine in more detail the role of CD28–B7 interactions in EC-induced CD4+ T cell activation. Our results provide insight into how graft ECs may modify infiltrating leukocytes for provision of CD28-mediated costimulation in trans and promote CD4+ T cell activation via direct and indirect allorecognition.
In these studies, we confirm that human ECs do not express CD80 or CD86 mRNA, nor protein assessed by RT-PCR, Western blotting, and FACS® analysis, respectively. However, we do find that CD86 is induced and is functional in CD4+ T cell–EC interactions due to its expression on CD4+ T cells and the ability of these cells to deliver CD86 mediated costimulatory signals in trans. Indeed, we demonstrate induction of expression of CD86 on CD4+ T cells after coculture with ECs. Blockade of CD4+ T cell CD86 using several anti-CD86 mAbs caused a variable but consistent inhibition of CD4+ T cell proliferation and IL-2 production when alloantigen was presented to CD4+ T cells by several different human microvascular ECs. Furthermore, combined blockade of LFA-3 and CD86 results in additive inhibition of CD4+ T cell activation, suggesting that these molecules participate in parallel pathways of CD4+ T cell activation. We interpret these data to suggest that ECs, in addition to providing direct costimulatory signals to CD4+ T cells, predominantly via LFA-3, may promote CD28-dependent trans-costimulation by the induction of CD86 on CD4+ T cells. The ability of ECs to modify leukocytes for provision of trans-costimulatory signals probably provides an additional mechanism whereby ECs regulate inflammatory responses.
Although PHA-activated CD4+ T cells have been reported previously to express both CD80 and CD86, the functional importance of CD4+ T cell expression of these molecules is controversial. Azuma et al. have shown that CD4+ T cell clones expressing CD80 are able to stimulate T cell cytokine production and proliferation in a mixed lymphocyte reaction 33. Furthermore, Jeannin et al. reported that human effector T cells express CD86, and may costimulate naive T cell responses 34. In contrast, it has been reported that CD4+ T cell CD86 may be nonfunctional due to reduced posttranslational glycosylation 40. Our studies clearly demonstrate that CD4+ T cell CD86 is functional and may provide CD28-mediated costimulatory signals in trans to autologous T cells when signal one is provided by mitogen or alloantigen on ECs. The high purity of our CD4+ T cell preparations and the high expression of CD86 on CD4+ T cells after coculture with ECs suggests that CD86-mediated costimulation was provided by CD4+ T cells and not low numbers of contaminating CD86+ dendritic cells. To confirm this, we depleted our CD4+ T cell population of HLA-DR– and CD14-expressing cells to eliminate contaminating CD4+ APCs 36. The resulting highly purified CD4+ T cells proliferated to IFN-γ–treated ECs, expressed enhanced levels of CD86 after 72 h coculture with IFN-γ–treated ECs and provide functional T cell–T cell trans-costimulation. We note that EC stimulation of CD4+ T cells resulted in a discrete population of CD86 expressing CD4+ T cells; which may represent alloactivated CD4+ T cells. Although resting CD4+ T cells may express low levels of CD86, we suggest that this level is insufficient to provide effective costimulation. Indeed, anti-CD86 reagents fail to inhibit CD4+ T cell activation when signal one is provided by mitogen and costimulation is dependent on constitutive cell surface molecules expressed on ECs or resting CD4+ T cells 15 39.
EC induction of CD4+ T cell CD86 is mediated in part by stimulation of the TCR and T cell CD2. Anti–class II MHC mAbs and anti–LFA-3 mAbs inhibit the ability of ECs to modify CD4+ T cells to provide CD86-mediated costimulation. Consistent with these findings, we found by FACS® analysis that LFA-3 fusion protein and low doses of PHA additively promote CD86 protein expression in purified CD4+ T cells (data not shown). Therefore, EC LFA-3 may provide costimulation to CD4+ T cells via two distinct mechanisms. First, LFA-3 may directly costimulate cytokine production and CD4+ T cell proliferation via interactions in cis 35; and second, LFA-3 may promote trans-costimulation via the induction of CD86 on T cells. Although CD40 signals induce CD80 and CD86 expression on B cells 41, monocytes, and dendritic cells 42, we find that interruption of CD40L–CD40 interactions does not inhibit EC induction of functional CD4+ T cell CD86. This is consistent with the low levels of CD40 expression on resting and activated CD4+ T cells. Similarly, OX40L-OX40 interactions do not appear to be functional in EC induction of CD86 expression by CD4+ T cells. Consistent with these observations, neither anti-CD40 mAbs nor anti-CD40L mAbs inhibit alloactivation of CD4+ T cells by IFN-γ–treated ECs (data not shown).
Our findings that allogeneic ECs can fully activate bulk populations of CD4+ T cells are similar to those reported by other groups 13 15 35 43. However, they are different from those reported by Marelli-Berg et al., in which ECs were found to have limited costimulatory function 16. A common finding of all groups is that addition of B7-dependent costimulation in trans reconstitutes the ability of ECs to fully activate T cells, including naive CD4+ T cells 16 21. We now report that CD28–CD86 interactions are functional in EC-induced CD4+ T cell activation, via induction of functional CD86 on CD4+ T cells.
Although B7-mediated costimulation is required for induction of primary immune responses, our studies have not specifically addressed whether ECs can activate naive CD4+ T cell populations. Previous reports have shown that human ECs fail to activate naive (CD45RA) CD4+ T cells 15 16, which would seem contradictory to our demonstration of functional CD86 on EC-modified CD4+ T cells. It is possible that the levels of CD86 expressed by EC-modified CD4+ T cells is insufficient to activate naive CD4+ T cells. Alternatively, ECs may be unable to induce CD86 expression and thus to promote trans-costimulation in pure populations of naive CD4+ T cells 10.
Endothelial cell modification of leukocytes for subsequent alloantigen-dependent activation may be of great physiologic importance for allograft rejection. We have recently shown that ECs enhance the ability of CD4+ T cells to respond to intragraft cytokines including IL-2, IL-4 and IL-12 via induction of the respective cytokine receptor (39 and our unpublished observations). Therefore, ECs may indirectly influence the differentiation of CD4+ T cells within the graft. In addition, these studies show that ECs may enhance the expression of costimulatory cytokines and cell surface molecules by transmigrating leukocytes and promote alloactivation of T cells within the allograft. Although antigen-specific T cell activation is considered to occur predominantly in primary lymphoid tissue, recent studies have established a requirement for B7 costimulatory signals at sites of inflammation 26. We suggest that in vivo, EC-modified leukocytes provide B7-dependent costimulation in trans, allowing for local activation of CD4+ T cells via the direct pathway of allorecognition.
By enhancing the antigen presenting and costimulatory capacity of transmigrating monocytes, ECs may promote CD4+ T cell activation through the indirect pathway of allorecognition. Indeed, recent studies have demonstrated that ECs may promote monocyte differentiation into dendritic cells, which may subsequently emigrate back to lymph nodes as efficient APCs 37 38. In support of a role for ECs in promoting indirect allorecognition, Vallee et al. demonstrated that indirect presentation of xenoantigens by human APCs is crucial in the proliferative response of human CD4+ T cells to porcine endothelial cells 44. We find that EC-modified monocytes promote greater proliferation of CD4+ T cells than do resting monocytes. This data is consistent with the ability of ECs to modify monocytes for enhanced antigen presenting and costimulatory function. Furthermore, the ability of EC-modified monocytes to promote proliferation of autologous CD4+ T cells may suggest that ECs donate alloantigen to monocytes for indirect activation of CD4+ T cells.
In summary, we show that CD28–B7 interactions are functional in the antigen-specific alloactivation of CD4+ T cells by ECs. Moreover by inducing CD86 and cytokine expression, human ECs modify the costimulatory capacity of infiltrating CD4+ T cells and monocytes, which may provide costimulation in trans to CD4+ T cells. Our new findings provide evidence for a unique function of the endothelium in allograft rejection in the direct activation of CD4+ T cells. In addition, the modification of monocytes by ECs may provide a mechanism whereby ECs promote indirect allorecognition.
Acknowledgments
We wish to acknowledge Dr. Andrew Lichtman for helpful discussions, and Professor Raif Geha and Dr. Frank Boriello for kindly reviewing the manuscript.
This work was supported by National Institutes of Health grants HL03518 to D.M. Briscoe and F32 DK09754 to M.D. Denton.
Footnotes
1used in this paper: CHO, Chinese hamster ovary; CTLA4, cytolytic T lymphocyte–associated antigen 4; EC, endothelial cell; HUVEC, human umbilical vein EC; ICAM-1, intercellular adhesion molecule 1; LFA-3, lymphocyte function–associated antigen 3; U, abbitor units
References
- Lechler R., Lombardi G., Batchelor J., Reinsmoen N., Bach F. The molecular basis of alloreactivity. Immunol. Today. 1990;11:83–88. doi: 10.1016/0167-5699(90)90033-6. [DOI] [PubMed] [Google Scholar]
- Janeway C.A., Bottomly K. Signals and signs for lymphocyte responses. Cell. 1994;76:275–285. doi: 10.1016/0092-8674(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Lenschow D., Walunas T., Bluestone J. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996;14:233–258. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- Lucas P., Negishi I., Nakayama K., Field L., Loh D. Naive CD28-deficient T cells can initiate but not sustain an in vitro antigen-specific immune response. J. Immunol. 1995;154:5757–5768. [PubMed] [Google Scholar]
- Noel P., Boise L., Green J., Thompson C. CD28 costimulation prevents cell death during primary T cell activation. J. Immunol. 1996;157:636–642. [PubMed] [Google Scholar]
- Walunas T., Bakker C., Bluestone J. CTLA-4 ligation blocks CD28-dependent T cell activation. J. Exp. Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T., Korman A., Wake C. Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc. Natl. Acad. Sci. USA. 1984;81:4917–4921. doi: 10.1073/pnas.81.15.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M., Coles M., Griffin R., Promerance A., Yacoub M. Expression of class I and class II major histocompatibility antigens in normal and transplanted human heart. Transplantation. 1986;41:776–781. doi: 10.1097/00007890-198606000-00021. [DOI] [PubMed] [Google Scholar]
- Page C., Rose M., Yacoub M., Pigotti R. Antigenic heterogeneity of vascular endothelium. Am. J. Pathol. 1992;141:673–683. [PMC free article] [PubMed] [Google Scholar]
- Briscoe D., DesRoches L., Kielly J., Lederer J., Lichtman A. Antigen-dependent activation of T helper cell subsets by endothelium. Transplantation. 1995;59:1638–1641. [PubMed] [Google Scholar]
- Briscoe D., Ganz P., Alexander S., Melder R., Jain R., Cotran R., Lichtman A. The problem of chronic rejectioninfluence of leukocyte–endothelial interactions. Kidney Int. 1997;58:S22–S27. [PubMed] [Google Scholar]
- Pober J., Orosz C., Rose M., Savage C. Can graft endothelial cells initiate a host anti-graft response. Transplantation. 1996;61:343–349. doi: 10.1097/00007890-199602150-00001. [DOI] [PubMed] [Google Scholar]
- Savage C., Hughes C., Pepinsky R., Wallner B., Freedman A., Pober J. Endothelial cell lymphocyte function-associated antigen-3 and an unidentified ligand act in concert to provide costimulation to human peripheral blood CD4+ T cells. Cell. Immunol. 1991;137:150–163. doi: 10.1016/0008-8749(91)90065-j. [DOI] [PubMed] [Google Scholar]
- Karmann K., Pober J., Hughes C. Endothelial cell-induced resistance to cyclosporin A in human peripheral blood T cells requires contact-dependent interactions involving CD2 and LFA3. J. Immunol. 1994;153:3929–3937. [PubMed] [Google Scholar]
- Ma W., Pober J. Human endothelial cells costimulate cytokine production by but not differentiation of naive CD4+ T cells. J. Immunol. 1998;165:2158–2167. [PubMed] [Google Scholar]
- Marelli-Berg F., Hargreaves R., Carmichael P., Dorling A., Lombardi G., Lechler R. Major histocompatibility complex class II–expressing endothelial cells induce allospecific nonresponsiveness in naive T cells. J. Exp. Med. 1996;183:1603–1612. doi: 10.1084/jem.183.4.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka H., Surh C., Sprent J. Stimulation of mature unprimed CD8+ T cells by semiprofessional antigen-presenting cells in vivo. J. Exp. Med. 1992;176:1291–1302. doi: 10.1084/jem.176.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan P., Schechner J., McNiff J., Hochman P., Hughes C., Lorber M., Askenase P., Pober J. Blockade of CD2-LFA-3 interactions protects human skin allografts in immunodeficient mouse/human chimeras. Nat. Biotechnol. 1997;15:759–762. doi: 10.1038/nbt0897-759. [DOI] [PubMed] [Google Scholar]
- Perez V., Henault L., Lichtman A. Endothelial antigen presentationstimulation of previously activated but not naive TCR-transgenic mouse T cells. Cell. Immunol. 1998;189:31–40. doi: 10.1006/cimm.1998.1362. [DOI] [PubMed] [Google Scholar]
- Bravery C., Batten P., Yacoub M., Rose M. Direct recognition of SLA- and HLA-like class II antigens on porcine endothelium by human T cells results in T cell activation and release of IL-2. Transplantation. 1995;60:1024–1033. [PubMed] [Google Scholar]
- Maher S., Karmann K., Min W., Hughes C., Pober J., Bothwell A. Porcine endothelial CD86 is a major costimulator of xenogeneic human T cellscloning, sequencing, and functional expression in human endothelial cells. J. Immunol. 1996;157:3838–3844. [PubMed] [Google Scholar]
- Kuiper H., Brouwer M., de Boer M., Parren P., van Lier R. Differences in responsiveness to CD3 stimulation between naive and memory CD4+ T cells cannot be overcome by CD28 costimulation. Eur. J. Immunol. 1994;24:1956–1960. doi: 10.1002/eji.1830240903. [DOI] [PubMed] [Google Scholar]
- Bluestone J. New perspectives of CD28-B7 mediated T cell costimulation. Immunity. 1995;2:555–559. doi: 10.1016/1074-7613(95)90000-4. [DOI] [PubMed] [Google Scholar]
- Gimmi C., Freeman G., Gribben J., Gray G., Nadler L. Human T cell anergy is induced by antigen presentation in the absence of B7 costimulation. Proc. Natl. Acad. Sci. USA. 1993;90:6586–6590. doi: 10.1073/pnas.90.14.6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer A., Sharpe A. Studies using antigen presenting cells lacking expression of both B7-1 (CD80) and B7-2 (CD86) show distinct requirements for B7 molecules during priming versus restimulation of Th2 but not Th1 cytokine production. J. Immunol. 1998;161:2762–2771. [PubMed] [Google Scholar]
- Chen H., Hendricks R. B7 costimulatory requirements of T cells at an inflammatory site. J. Immunol. 1998;160:5045–5052. [PubMed] [Google Scholar]
- Gimbrone M.J. Culture of human vascular endothelium. Prog. Hemostasis Thromb. 1976;3:1–28. [PubMed] [Google Scholar]
- Mach F., Schonbeck U., Sukhova G.K., Bourcier T., Bonnefoy J.Y., Pober J.S., Libby P. Functional CD40 ligand is expressed on human vascular endothelial cells, smooth muscle cells, and macrophagesimplications for CD40-CD40 ligand signaling in atherosclerosis. Proc. Natl. Acad. Sci. USA. 1997;94:1931–1936. doi: 10.1073/pnas.94.5.1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik N., Greenfield B., Wahl A., Kiener P. Activation of human monocytes through CD40 induces matrix metalloproteinases. J. Immunol. 1996;156:3952–3960. [PubMed] [Google Scholar]
- Lichtman A., Ding H., Henault L., Vachino G., Camphausen R., Cumming D., Luschinskas F. CD45RA−RO+ (memory) but not CD45RA+RO− (naive) T cells roll efficiently on E-selectin and P-selectin and vascular cell adhesion molecule-1 under flow. J. Immunol. 1997;158:3640–3650. [PubMed] [Google Scholar]
- Luscinskas F., Brock A., Arnaout M., Gimbrone M. Endothelial-leukocyte adhesion molecule-1 (ELAM-1)-dependent and leukocyte (CD11/CD18)-dependent mechanisms contribute to polymorphonuclear leukocyte adhesion to cytokine activated endothelium. J. Immunol. 1989;142:2257–2263. [PubMed] [Google Scholar]
- Roth S., Woldemar Car M., Rose S., Springer T. Characterization of transendothelial chemotaxis of T lymphocytes. J. Immunol. Methods. 1995;188:97–116. doi: 10.1016/0022-1759(95)00208-1. [DOI] [PubMed] [Google Scholar]
- Azuma M., Yssel H., Phillips J., Spits H., Lanier L. Functional expression of B7/BB1 on activated T lymphocytes. J. Exp. Med. 1993;177:845–850. doi: 10.1084/jem.177.3.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeannin P., Herbault N., Delneste Y., Magistrelli G., Lecoanet-Henchez S., Caron G., Aubry J., Bonnefoy J. Human effector memory T cells express CD86a functional role in naive T cell priming. J. Immunol. 1999;162:2044–2048. [PubMed] [Google Scholar]
- Hughes C., Savage C., Pober J. Endothelial cells augment T cell IL-2 production by a contact-dependent mechanism involving CD2:LFA3 interaction. J. Exp. Med. 1990;171:1453–1467. doi: 10.1084/jem.171.5.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Doherty U., Steinman R., Peng M., Cameron P., Gezelter S., Kopeloff I., Swiggard W., Pope M., Bhardwaj N. Dendritic cells freshly isolated from human blood express CD4 and mature into typical immunostimulatory dendritic cells after culture in monocyte-conditioned medium. J. Exp. Med. 1993;178:1067–1078. doi: 10.1084/jem.178.3.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrero E., Bondanza A., Leone B., Manici S., Poggi A., Zocchi M. CD14+CD34+ peripheral blood mononuclear cells migrate across endothelium and give rise to immunostimulatory dendritic cells. J. Immunol. 1998;160:2675–2683. [PubMed] [Google Scholar]
- Randolph G., Beaulieu S., Lebecque S., Steinman R., Muller W. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- Briscoe D., Henault L., Geehan C., Alexander S., Lichtman A. Human endothelial cell costimulation of T cell IFN-γ production. J. Immunol. 1997;159:3247. [PubMed] [Google Scholar]
- Greeenfield E., Howard E., Paradis T., Nguyen K., Benazzo F., McLean P., Hollsberg P., Davis G., Hafler D., Sharpe A. B7-2 expressed by T cells does not induce CD28-mediated costimulatory activity but retains CTLA4 bindingimplications for induction of antitumor immunity to T cell tumors. J. Immunol. 1997;158:2025–2034. [PubMed] [Google Scholar]
- Kennedy M., Mohler K., Shanebeck K., Baum B., Picha K.E., Janeway C.A., Grabstein K. Induction of B cell costimulatory function by recombinant murine CD40 ligand. Eur. J. Immunol. 1994;24:116–123. doi: 10.1002/eji.1830240118. [DOI] [PubMed] [Google Scholar]
- Caux C., Massacrier C., Vanbervliet B., Dubois C., van Kooten I., Durand I., Banchereau J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994;180:1263–1272. doi: 10.1084/jem.180.4.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P., Lee H., Waldman W., Sedmak D., Morgan C., Ward J., Orosz C. Allogenicity of human endothelial cells. I. Frequency and phenotype of human T helper lymphocytes that can react to allogeneic endothelial cells. J. Immunol. 1992;148:3753–3760. [PubMed] [Google Scholar]
- Vallee I., Guillaumin J., Thibault G., Lebranchu Y., Bardos P., Watier H. Human T lymphocyte proliferative response to resting porcine endothelial cells results from an HLA-restricted, IL-10-sensitive, indirect presentation pathway but also depends on endothelial-specific costimulatory factors. J. Immunol. 1998;161:1652–1658. [PubMed] [Google Scholar]