

Abstract
Viral dynamics were intensively investigated in eight patients with acute HIV infection to define the earliest rates of change in plasma HIV RNA before and after the start of antiretroviral therapy. We report the first estimates of the basic reproductive number (R 0), the number of cells infected by the progeny of an infected cell during its lifetime when target cells are not depleted. The mean initial viral doubling time was 10 h, and the peak of viremia occurred 21 d after reported HIV exposure. The spontaneous rate of decline (α) was highly variable among individuals. The phase 1 viral decay rate (δI = 0.3/day) in subjects initiating potent antiretroviral therapy during acute HIV infection was similar to estimates from treated subjects with chronic HIV infection. The doubling time in two subjects who discontinued antiretroviral therapy was almost five times slower than during acute infection. The mean basic reproductive number (R 0) of 19.3 during the logarithmic growth phase of primary HIV infection suggested that a vaccine or postexposure prophylaxis of at least 95% efficacy would be needed to extinguish productive viral infection in the absence of drug resistance or viral latency. These measurements provide a basis for comparison of vaccine and other strategies and support the validity of the simian immunodeficiency virus macaque model of acute HIV infection.
Keywords: basic reproductive number, viral decay, primary HIV, lymphocyte dynamics, modeling
Acute infection with HIV is characterized by an exponential rise in viral titers to extraordinary levels in the plasma 1 2. In many individuals, this dynamic phase of viral infection is accompanied by an acute syndrome resembling infectious mononucleosis 3 4. Viral titers spontaneously decline over a period of months to variable steady state levels that predict HIV disease progression 5 6.
The dynamics of viral replication in acute HIV infection provide important insights into the initial events of HIV pathogenesis. The basic reproductive number (the number of infected cells that arise from a single HIV-infected cell before any depletion of target cells) 7 can be estimated from the initial rise in levels of HIV RNA in plasma 8. An estimate of efficacy for an intervention, which prevents sustained propagation of HIV infection from productively infected cells, can be calculated from the basic reproductive number. This estimate has particular relevance for vaccine development, the only intervention likely to influence the global HIV epidemic. It also has important implications for postexposure prophylaxis routinely recommended after occupational exposure to HIV 9 10 and certain empirically defined sexual exposures 11.
The viral dynamics of primary infection have been characterized for simian immunodeficiency virus (SIV)1 infection in the macaque 12 and demonstrated the first in vivo estimates of R 0 ranging from 2.2 to 68. It is critical, however, to establish estimates of viral production and clearance in acute HIV infection in humans both for potential therapeutic applications and for validation of the SIV model. In this report, viral dynamics before and after the initiation of potent antiretroviral therapy were characterized in eight subjects with acute HIV infection who were identified before development of an HIV antibody response or during exponential viral decay.
Materials and Methods
Human Subjects.
Eight adults with a symptomatic viral illness after sexual exposure to HIV were referred to our clinic for evaluation. Seven subjects had acute HIV infection, as defined by detectable HIV RNA in the plasma and nonreactive HIV antibody test or indeterminate Western blot (see Table ). One subject presented with a positive HIV enzyme immunoassay and a positive Western blot (two bands; p24 and gp120/160 were positive) in the setting of exponential decline of plasma HIV RNA. All study subjects signed an informed consent approved by the University of California San Diego Human Subjects Committee. Baseline (day 0) demographic information, risk assessment for HIV exposure, clinical features, and duration of signs and symptoms compatible with an acute retroviral syndrome were recorded. The date of HIV infection was ascertained from patient history by Dr. S.J. Little. For individuals unable to identify a single high-risk exposure, the range of potential exposure dates was recorded and the midpoint in this range was assigned as the HIV exposure date.
Table 1.
Subject Characteristics
| Subject (PID) | Age/ sex | HIV EIA/WB | Interval between HIV exposure and presentation | Baseline HIV RNA | Time from exposure to peak | Peak HIV RNA | CD4 | Total symptom duration | Interval between HIV exposure and start of ARV Rx | Antiretroviral regimen |
|---|---|---|---|---|---|---|---|---|---|---|
| d | copies/ml | d | copies/ml | cells/mm3 | d | d | ||||
| 001 | 45/M | Neg/ND | 23 | 44,443,601 | – | – | 937 | 14 | 35 | ZDV/3TC/IDV |
| 002 | 21/M | Neg/ND | 49 | 226,820 | – | – | 163 | 35 | 58 | ZDV/3TC/IDV |
| 004 | 32/M | Neg/ND | 21 | 738,240 | 23 | 10,328,200 | 450 | 14 | 29 | ZDV/3TC/IDV |
| 044 | 28/M | Neg/ND | 16 | 19,200,000 | – | – | 397 | 6 | 26 | ZDV/3TC/NFV |
| 045 | 28/F | Pos/Ind. | 26 | 1,767,250 | 31 | 6,235,500 | 314 | 9 | 53 | ABC/APV |
| 054 | 38/M | Pos/Pos | 50 | 438,501 | – | – | 255 | 14 | 63 | ZDV/3TC/NFV |
| 056 | 29/M | Neg/ND | 12 | 534,600 | 16 | 2,491,300 | 354 | 3 | 57 | IDV/EFV |
| 059 | 39/M | Neg/ND | 12 | 21,617,100 | 12 | 21,617,100 | 288 | 4 | N/A | – |
| Mean | 33 | 26 | 2,445,601 | 21 | 7,674,142 | 395 | 12 | 46 |
Patient characteristics are shown. HIV exposure date was estimated for all study subjects. The intervals between HIV exposure and baseline (presentation) HIV RNA, peak HIV RNA, and start of antiretroviral therapy are shown. CD4 and CD8 cell counts were collected at presentation for subjects 004, 044, 045, 056, and 059. The first CD4 cell subsets were collected 11 and 37 d after presentation of subjects 001 and 002, respectively. Mean values for plasma HIV RNA are geometric means; all other means are arithmetic. ABC, abacavir; APV, amprenavir; ARV Rx, antiretroviral therapy; EFV, efavirenz; IDV, indinavir; Ind., indeterminate; N/A, not applicable; NFV, nelfinavir; 3TC, lamivudine; ZDV, zidovudine.
This was an observational study, and participants chose to initiate antiretroviral therapy in conjunction with their physicians. Subjects 001 and 002 subsequently interrupted their antiretroviral therapy against advice 160 and 34 d, respectively, after treatment initiation (see Fig. 1). Frequent whole blood samples were obtained for isolation of plasma and PBMCs. Samples were collected every 1–2 d for weeks 1 and 2, twice per week for week 3, weekly for weeks 4–6, and every 4 wk thereafter. Whole blood was collected in acid-citrate dextrose tubes and processed within 6 h of collection. Plasma was separated and stored at −70°C. PBMCs were separated by established density gradient methods and prepared by controlled rate freezing for storage at −150°C.
Figure 1.


Plasma viral load was measured in the eight study subjects (A and B) from the estimated HIV exposure date through day 150 of follow-up. (A) Patients identified during rising viremia. (B) Patients identified after the peak of plasma viremia. Patients are indicated by the following symbols: P001, □; P002, ○; P004, □; P044, ▵; P045, ○; P054, ⋄; P056, ▵; and P059, ⋄. Open symbols indicate RNA measurements before treatment (or after treatment interruption), and closed symbols indicate RNA measurements while on therapy. Arrows mark the start of antiretroviral therapy for the seven treated subjects. Subject P059 did not receive antiretroviral therapy during the follow-up period and is identified by the ⋄ in A.
Virologic Studies.
HIV enzyme immunoassay (Abbott Labs.) and Western blot (Cambridge Biotech Corp.) were performed at presentation and repeated at regular intervals until a positive antibody and Western blot were documented. Plasma HIV RNA measures were performed by the Roche Amplicor Assay (Roche Molecular Systems) according to the manufacturer's instructions. The Roche ultrasensitive assay was used to determine HIV RNA copy number on a subset of samples. A virologic “plateau” was defined as at least two measures of HIV RNA, which varied by less than threefold and occurred before the initiation of antiretroviral therapy.
T Lymphocyte Studies.
Assessment of T cell subsets was performed on whole blood within 24 h of specimen collection by dual color FACS™ analysis (FACScan™; Becton Dickinson Cytometry Systems). Baseline (day 0) T cell subsets were collected within 7 d of the day 0 visit, except for subjects 001 and 002, for which the first CD4 assessments were performed 11 and 37 d after presentation, respectively.
Modeling of Acute HIV Infection.
Fig. 2 schematically illustrates the viral dynamics of acute HIV infection and is adapted from a study of acute SIV dynamics 12. This figure demonstrates the viral parameters measured in our study population. These parameters are based on a standard model of infected cell dynamics (I) where free virus is used as a surrogate for infected cells: dIdt=βIT−δI I, where T represents the target cell number, δI the death rate of infected cells, β the rate of new infection of uninfected cells, and I the number of infected cells. We do not explicitly include free virus; our model assumes that direct cell–cell transmission is the most important mode of spread of infection. If this were not true, we would use the same model, but the definition of β would change to include a constant of proportionality relating free virus density to infected cell density.
Figure 2.

A schematic illustration of the viral dynamics of acute HIV-1 infection and the viral parameters we measured is shown and adapted from an article by Nowak et al. 12 in the Journal of Virology, 1997, vol. 71, pp. 7518–7525 by copyright permission of the American Society for Microbiology. An initial exponential rise of viremia, r 0, is observed followed by a spontaneous decline of viremia, α. The rate of rise of HIV RNA, r̂, which occurs within days of the peak of viremia, is less than the rate, r 0, observed initially. Upon initiation of potent antiretroviral therapy, the phase 1 decay rate, δI, is measured. The rate of rise of viremia after interruption of therapy, r, is measured in two of our subjects.
The inferred growth rate, r 0, represents the initial rate of rise of viremia per cell per day and is related to the model equation by setting r 0=βT 0−δI so that dIdt=r 0 I and I t=I 0er0t. The inferred growth rate, r 0, was measured directly in one of our study subjects (patient identification number [PID] 059). The observed rate of rise of viremia, r̂, is the measured slope of rising viremia within days of the peak of viremia. Measurements of r̂ are obtained at a time when the slope of the observed rate of rise r̂ of HIV RNA is likely to be slower than that observed initially (r 0). The inferred r 0 assumes that (a) the CD4 cell count before HIV infection (T 0) is 1,000 cells/mm3, (b) the efficiency of infection, β, does not change between the time of infection and the observed time, and (c) the life span of an infected cell remains constant. The rationale underlying an inferred calculation for r 0 was that part of the slowdown in growth rate could be attributed to depletion of target cells 13 14. This assumption is supported by the recent observation that in a comparison of SIV-infected monkeys with and without CD8+ cells, viral dynamics up to and including the time of peak viremia were not affected by the presence or absence of CTLs 15. Using total CD4 counts as the best available surrogate for target cells and a nominal normal value of 1,000 cells/mm3, we corrected the observed rate of rise for the degree of target cell depletion. Based on the observed growth rate for the four study subjects identified with rising viremia r̂, the following formula was used to calculate an inferred initial growth rate (r 0):
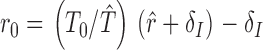 |
1 |
from the observed growth rate, r̂, where T̂ represents the CD4 cell count at (or close to) the first viremia observation. If the infected cell death rate had already grown larger by the time we made our observations, this formula would underestimate r 0 by the amount of that change.
The initial viral doubling time, t 2, is calculated using the slope of rising viremia over time. The slope of declining viremia over time is used to measure the rate of decline of viremia during the subsequent spontaneous decline of viremia, α, and the phase 1 (first order) viral decay rate after initiation of potent antiretroviral therapy, δI. In the two subjects who discontinued antiretroviral therapy against advice, the rate of rise of viremia, r, after interruption of therapy was also measured.
The number of secondary infections arising from one infected cell over the course of its life span, when target cells are not depleted, is the basic reproductive number, R 0. We estimated R 0 using a model that incorporates a fixed time delay, τ, between the infection of a target cell and the subsequent production of progeny virions. This fixed delay model gives R 0 as:
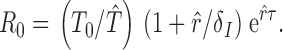 |
The actual duration of the intercellular lag between infection of a target cell and production of progeny virions is likely to be between 12 and 24 h, as determined by in vitro and in vivo estimates 16 17 18. We used a 24-h delay (eclipse period) from the infection of one target cell and the subsequent production of progeny virions for the fixed delay model.
A second model without delay was also investigated. Although this is not a biologically plausible model, it was included for comparison with prior studies in macaques 12. The model without delay is a special case of the fixed delay model with the delay set to zero (τ = 0):
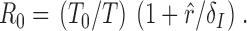 |
This model assumes the instantaneous production of progeny virions from the moment of initial infection of a target cell and almost certainly generates minimal estimates of R 0.
These methods of inferring r 0 and R 0 from r̂ and T̂ allow us to combine information on viral growth rate and degree of target cell depletion. They are sensitive to the allotted values of τ and T 0. Thus, for patient 004, if T 0 takes values of 500, 1,000 or 1,500, then R 0 = 4.5, 9.1, or 13.6, respectively. If τ was 12 rather than 24 h, values of R 0 would be intermediate between those shown for the two different models.
Results
Acute Retroviral Illness.
Eight subjects (seven men and one woman) were referred to our clinic 2–35 d after the onset of an acute retroviral illness and 12–50 d after high-risk sexual exposure to HIV (Table ). Patient recall of HIV exposure date placed the interval between HIV exposure and the onset of symptoms between 8 and 47 d in this cohort (mean, 22 d). Seven of the eight subjects initiated potent antiretroviral therapy 26–63 d after their reported HIV exposure (Table ).
All subjects exhibited diffuse lymphadenopathy at presentation. Myalgias, fever, headache, and fatigue were reported in 63–88% of subjects. CD4 cell numbers at initial presentation were highly variable, ranging from 163 to 937 cells/mm3 (Table ). The CD4/CD8 T cell ratio was within a normal range (2.0 ± 0.8) in two of eight patients who presented less than 2 wk after HIV exposure but was low (0.2–1.0) in the remaining six subjects identified more than 2 wk after HIV exposure (data not shown).
Viral Doubling Time during Rising Viremia.
Four of the eight subjects were identified before attaining peak levels of acute viremia, which ranged from 2.5 to 22 million HIV RNA copies per milliliter of plasma (Fig. 1). The mean time from reported HIV exposure to the peak measure of viremia was 21 d (range, 12–31 d; Table ). Sequential measures of plasma viremia were obtained to estimate the rates of rising HIV RNA (r 0) before the initiation of antiretroviral therapy (Fig. 2). Subject 059 had a serum HIV RNA of 30 copies/ml serendipitously obtained 3 d after his reported HIV exposure. His plasma HIV RNA copy number upon presentation with an acute retroviral illness 9 d later (12 d after reported exposure) was 21,617,100 copies/ml. The observed initial rate of rise of HIV RNA (r 0) was 1.5/day in this subject, which corresponds to a viral doubling time (t 2) of 12 h (Table ).
Table 2.
Initial Viral Replication Dynamics and Calculated R0 Values
| Subject (PID) | Observed initial rise | Observed rise | Inferred initial rise | Calculated R 0 value | ||||
|---|---|---|---|---|---|---|---|---|
| r 0/d | t 2(d) | r̂/d | t 2(d) | r 0/d | t 2(d) | Standard model (no delay) | Fixed delay (24 h) | |
| 059 | 1.5 | 0.5 | – | – | – | – | 5.2 | 23.2 |
| 004 | – | – | 1.3 | 0.5 | 3.5 | 0.2 | 9.1 | 34.0 |
| 045 | – | – | 0.2 | 2.9 | 1.4 | 0.5 | 5.9 | 7.4 |
| 056 | – | – | 0.4 | 1.6 | 1.6 | 0.4 | 8.1 | 12.4 |
| Mean | – | – | 0.5 | 1.3 | 2.0 | 0.3 | 7.1 | 19.3 |
The mean inferred r 0 was 2.0 among the three subjects with an observed r̂ value. The mean r 0 for all four study subjects was 1.9 (three inferred rates and one observed), which corresponds to a viral doubling time (t 2) of 0.4 d. Viral dynamic parameters include: r 0, the initial rate of rise of plasma viremia per cell per day, shown either as an observed (directly measured) or inferred (calculated from r̂) parameter; t 2, the initial viral doubling time; r̂, the rate of rise of plasma viremia per cell per day within days of the peak of HIV RNA; and R 0, the basic reproductive number. The mean R 0 (no delay) was 7.1, with a 95% confidence interval of 4.3–13.2. The mean R 0 (24-h fixed delay) was 19.3, with a 95% confidence interval of 5.4–54.3. 95% confidence intervals for R 0 were calculated by fitting a slope to all data points from 3 d before the peak to the peak of viremia and using the variability in the estimate of this slope as the sole source of uncertainty in estimating R 0. All means are geometric, except those for R 0, which are arithmetic.
The observed rate of rise of viremia r̂ for the other three subjects ranged from 0.2 to 1.3/day (Table ). Because these plasma HIV RNA measurements were collected within days of the peak and likely reflect a reduced exponential growth rate r̂ compared with the initial rate (r 0), we calculated an inferred initial rate (Fig. 2). These inferred r 0 values ranged from 1.4 to 3.5/day (mean, 2.0), and the mean doubling time (t 2) was 0.3 d, or 7 h. Comparison of the observed initial rate of rise of viremia (r 0) for subject 059 and the mean inferred rate, r 0, for the remaining subjects revealed similar estimates of r 0 (1.5 and 2.0/day, respectively) and a mean viral doubling time (t 2) of 10 h.
The Basic Reproductive Number.
The basic reproductive number (R 0) is the estimate of the number of secondary infections arising from one infected cell over the course of its life span when target cells are not a limiting resource. Using a standard model, which assumes no delay between the infection of a cell and the production of progeny virions, R 0 values ranged from 5.2 to 9.1, with a mean value of 7.1 (95% confidence interval, 4.3–13.2; Table ). Slightly greater values were obtained using a fixed delay model for R 0, which incorporates a 24-h delay between the infection of a target cell and the subsequent production of progeny. The fixed delay model, which is more biologically plausible, generated estimates of R 0 ranging from 7.4 to 34.0, with a mean value of 19.3 (95% confidence interval, 5.4–54.3). These estimates suggest that ∼19 infected cells will arise from each HIV-infected cell over the course of its life span when CD4 target cells are not limiting. Furthermore, these R 0 estimates predict that an antiretroviral regimen or vaccine, which is at least 95% effective, will prevent sustained viral propagation in the absence of drug resistance or viral latency. These are probably worst case estimates for the required efficacy of a prophylactic vaccine, as they are derived from viremic patients. This estimate of the value of the basic reproductive number must be treated as an attempt to gain an order of magnitude estimate rather than a definitive value.
Spontaneous Decline in Viral Titers.
A spontaneous decline of viremia was observed in all subjects before initiation of antiretroviral therapy (Fig. 1). The measured rates of decline of viremia (α), measured over the first 10 d of spontaneous viral decay, were highly variable, with a range of 0.1 to 0.8/day (mean, 0.3; Table ). The accompanying t 1/2 of spontaneous decline ranged from 0.9 to 12.3 d (mean, 2.4). An apparent virologic plateau, defined as at least two measures of HIV RNA that varied by less than threefold, was observed a mean of 33 d after the peak measure of viremia in seven of the study subjects (data not shown). In the absence of antiretroviral therapy, these early plateau values for HIV RNA ranged from 7,074 to 436,354 copies/ml (mean, ∼165,000 copies/ml) and were sustained for 2–4 wk in the absence of therapy. A subsequent further spontaneous decline of HIV RNA was noted in subjects 056 and 059, who did not initiate therapy during the plateau period.
Table 3.
Spontaneous Decline of Viremia and Viral Clearance after Initiation of Potent Antiretroviral Therapy
| Subject (PID) | Spontaneous viral decay | Potent ARV Rx (down slope) | Off ARV Rx (up slope) | |||
|---|---|---|---|---|---|---|
| α/d | t 1/2(d) | δI/d | t 1/2(d) | r/d | t 2(d) | |
| 001 | 0.8 | 0.9 | 0.3 | 2.3 | 0.5 | 1.3 |
| 002 | 0.1 | 12.3 | 0.5 | 1.3 | 0.4 | 1.5 |
| 004 | 0.4 | 1.6 | 0.4 | 1.6 | – | – |
| 044 | 0.3 | 2.1 | 0.5 | 1.4 | – | – |
| 045 | 0.4 | 1.9 | 0.3 | 2.5 | – | – |
| 054 | 0.3 | 2.1 | 0.3 | 2.1 | – | – |
| 056 | 0.5 | 1.5 | 0.2 | 3.0 | – | – |
| 059 | 0.1 | 5.2 | – | – | – | – |
| Mean | 0.3 | 2.4 | 0.3 | 1.9 | 0.4 | 1.4 |
Spontaneous decay (α) was measured from the slope of observed viral decay between the peak of viremia and subsequent 10 d of follow-up. t 1/2, half life; t 2, doubling time. Phase 1 viral decay rate in the setting of potent therapy (δI) is measured from the slope of the observed viral decay between the start date of therapy and subsequent 14 d of follow-up. The rate of rise of viremia (r) after treatment interruption was measured from the slope of observed relapse of viremia between the date of therapy interruption and the next 7–14 d of follow-up. All means are geometric means.
Treatment Effects.
Seven of eight subjects initiated potent antiretroviral therapy during the first 2 mo after reported HIV exposure. The initiation of potent antiretroviral therapy in these subjects permitted estimation of the death rate of productively infected cells (δI) and the t 1/2 of virus during the decline of plasma viremia (Fig. 2). Values for the phase 1 viral decay rate (δI) were remarkably consistent and ranged from 0.2 to 0.5/day, with clearance t 1/2 of 1.3–3.0 d for the subjects (Table ). Relapse of viremia was observed after voluntary interruption of antiretroviral therapy in two subjects (Table ). The mean rate of viral replication upon relapse of viremia (r) was 0.4/day, corresponding to a mean viral doubling time (t 2) of 1.4 d. Comparison of the rate of viral replication after withdrawal of treatment (r) to the rate of viral replication during initial acute infection (r 0) showed that the initial rate was nearly five times faster (0.4 vs. 1.9/day).
Comparison of Viral Dynamics in HIV to the SIV Model.
A comparison of the viral replication dynamics during primary infection revealed remarkably similar parameters between the macaque model 12 with acute SIV infection and human subjects with acute HIV infection (Table ). The mean initial rate of rise of viremia (r 0) in 12 SIVsmE660-infected macaques was 2.2/day (range, 1.7–2.7), with a viral doubling time of 0.32 d (8 h). This measure is similar to a mean r 0 value of 1.9/day (range, 1.4–3.5) and doubling time of 0.4 d (10 h) observed in our study population. Both the magnitude and the time to peak of viremia in the macaque and human infections were similar. As observed in HIV-infected humans, a period of spontaneous decay of SIV after the peak of viremia is observed in macaques with primary SIV infection. The mean rate of spontaneous SIV decay (α) was 0.52/day, with a mean clearance t 1/2 of 1.33 d, compared with a mean rate of spontaneous decay (α) of 0.3/day, with a mean viral clearance t 1/2 of 2.4 d in humans. Similar lower limit estimates of R 0 were observed in macaques (5.4) and humans (7.4) using the fixed delay model 12, with more varied upper limit estimates in the macaques, perhaps attributable to the incorporation of data on target cell availability in the human study. Comparison of phase 1 decay rates and relapse rates of viremia are limited by the very different treatment regimens and durations of therapy used in the different hosts as well as small sample numbers.
Table 4.
Acute HIV Infection Dynamics: HIV versus SIV
| Parameter | Human mean (range) | Macaque mean (range) |
|---|---|---|
| Early increase in viral load, r 0 | 2.0 (1.4–3.5) | 2.2 (1.7–2.7) |
| Doubling time (days), t 2 | 0.3 (0.2–0.5) | 0.32 (0.26–0.42) |
| Peak HIV RNA (copies/ml) | 7.7 × 106 (2.5–22) | 2.0 × 106 (0.8–7.0) |
| Time to peak HIV RNA (days) | 21 (12–31) | 17 (10–21) |
| Spontaneous decline, α | 0.3 (0.1–0.8) | 0.52 (0.18–0.86) |
| Clearance half life (days), t 1/2 | 2.4 (0.9–12.3) | 1.33 (0.8–3.81) |
| Treatment decline, δI | 0.3 (0.2–0.5) | 0.74 (0.51–0.96) |
| Treatment cessation rebound, r | 0.4 (0.4–0.5) | 1.23 (0.45–1.77) |
| Reproductive number (standard), R 0 | 7.1 (5.2–9.1) | 3.6 (2.2–4.6) |
| Reproductive number (fixed delay), R 0 | 19.3 (7.4–34.0) | 36.5 (5.4–68) |
Discussion
Viral dynamics of acute HIV infection were described nearly a decade ago 1 2, yet only recently has the potential value of precisely defining viral and host immune responses in acute HIV infection been appreciated. The increased recognition of the clinical syndrome associated with primary HIV infection coupled with the potential benefits of early intervention with potent antiretroviral therapy have facilitated investigation of acute HIV infection 19 20. Identifying patients with exponentially increasing viral titers in this study permitted the first estimates of viral doubling time associated with acute HIV infection.
The viral doubling time during acute HIV infection was estimated to be 10 h. The abundance of permissive target cells during acute HIV infection and the rapid viral doubling time resulted in extraordinarily high levels of HIV RNA (2.5–44 million copies per milliliter of plasma) within 3 wk of infection. This estimate of the doubling time in acute infection is nearly five times faster than that measured after treatment interruption (t 2 = 1.4 d) and what has been observed after interruption of treatment of chronic infection (t 2 = 1.7 d; reference 21). The viral doubling time after treatment interruption may represent an objective parameter to evaluate the restoration of HIV-specific immune responses generated during therapy. A delay before the emergence of detectable plasma HIV RNA after therapy withdrawal or a slower viral doubling time than was observed with our patients may provide a quantitative estimate of acquired host immunity. We cannot, however, exclude the possibility that greater target cell availability during acute infection supports the observed more rapid viral doubling.
The quantitation of viral titers during acute infection also permitted estimation of the basic reproductive number (R 0). Any intervention that reduces R 0 to <1 results in less than one secondary infected cell arising from each HIV infected cell. This parameter provides an estimate of how effective an intervention must be to result in extinction of viral replication and thus has significant implications for vaccine design and postexposure therapies. To extinguish ongoing viral replication, any intervention must reduce the basic reproductive number to below one. At least two important qualifications must be considered before claiming that such an intervention will successfully control HIV infection. First, ongoing replication in the presence of the selective pressure of an immune response or drug therapy could permit the outgrowth of resistant (escape) mutants. Consideration of resistance is relevant because both zidovudine 22 23 and multidrug-resistant virus 24 have already been reported in acute HIV infection. Furthermore, mathematical models suggest that drug resistance is more likely to develop if treatment is initiated during the very high titer viremia of acute HIV infection 25. Second, even during acute infection, latently infected CD4 cells are being generated 26, which upon later reactivation could provide the spark to rekindle viral replication. Our estimates of R 0 do not consider the impact of long-lived pools of latently infected cells 27 28, which have been detected within the first week after the onset of an acute retroviral syndrome 26.
Our estimates of the basic reproductive number ranged from 7.4 to 34.0 (mean, 19.3) using the model with fixed delay and suggest that after acute HIV infection, an intervention at least 95% effective will be needed to prevent sustained viral propagation. The presence of drug-resistant virus or an established pool of latently infected cells could make such a target extremely difficult to attain.
Our estimates of the basic reproductive number depend on several additional assumptions. First, although biologically implausible, a no delay model was evaluated for the purpose of comparison with prior studies of SIV dynamics in acutely infected macaques. Second, our model assumes that CD4 lymphocytes are a good surrogate for target cells. This amounts to assuming that the fraction of CD4 cells that are target cells is constant, an assumption that is born out by our own preliminary observations (data not shown). The lymphocyte populations, which are productively infected in vivo, have not been precisely defined. Based upon our observations that HIV replication in vitro is selectively favored in CD4 cells of the memory phenotype (CD4/CD45RO) 29, we and others speculated that activated CD4 cells 30 31 32 33, such as memory cells expressing the IL-2 receptor (CD4/CD45RO/CD25) might represent a target cell population for HIV infection in vivo. However, in the absence of data defining the rate of production and clearance of these cells (both infected and uninfected), we were unable to fit a model that included these variables and used absolute CD4 counts as the target cell population for our estimates of R 0. If the population of true target cells for HIV infection was present as a stable proportion of the total CD4 cell population, then our estimates of R 0 would be unbiased. Supporting this assumption, we observed that the proportion of CD4 cells that coexpressed CD45RO/CD25 remained stable during the first 6 mo of follow-up in all study subjects (data not shown). Third, we assumed a preinfection CD4 count of 1,000 cells/mm3. This would underestimate R 0 for subjects with preinfection T cell counts (T 0) > 1,000 and overestimate R 0 for subjects with T 0 < 1,000.
Spontaneous decline of viremia was highly variable both within and among patients. It is possible that the explanation for this variability is attributable to host immune responses; however, there are yet no strong data to support this assumption. Prior observations by Riggs et al. in a single subject showed that the viral burst size appeared to be stable during the decline of acute viremia, arguing against the hypothesis that the life span of HIV-infected cells was being shortened by effective CTL responses 34.
The rate of viral decay over the first 10 d of declining viremia (α) was identical to the mean phase 1 viral decay rate after initiation of potent antiretroviral therapy (0.3/d). In one patient with peak HIV RNA counts over 10 million, the rate of spontaneous decline was actually faster than that seen after potent antiretroviral therapy (data not shown). This observation is difficult to reconcile with the assumption that during spontaneous decline, some new infections are still being produced, whereas in the setting of potent antiretroviral therapy, virtually all new infections are blocked.
The initiation of potent antiretroviral therapy in the setting of acute HIV infection resulted in measures of phase 1 viral decay rates remarkably similar to reported measures in subjects with chronic HIV infection across a range of treatment regimens 18 21 35 36. In contrast to previous reports 21 37, no association between baseline HIV RNA or CD4 cell count (or percentage) and the rate of phase 1 viral decay was observed. These data suggest that either an acquired host immune response develops very early in the course of acute HIV infection and remains relatively constant thereafter, or, conversely, that the contribution of host immunity to the clearance of productively infected cells is minimal in both acute and chronic HIV infection 14 34 38.
Frequent measurements of plasma HIV RNA in these subjects demonstrated the appearance of a relative virologic plateau a mean of 33 d after the peak of viremia that has not been identified in prior publications 14 39. This virologic plateau occurred several months before the virologic “set point” shown to be predictive of disease progression 5. Additional studies are needed to define the exact timing, duration, and etiology of this observation.
Our measures of the initial rate of rise of viremia, magnitude of the peak, and subsequent spontaneous decline of viremia are in close approximation to reported ranges for these variables in a macaque model of primary SIV infection 12. Although the lower limit estimates for R 0 are similar for the human (7.4) and simian models (5.4), the range for the basic reproductive number in the macaque model was somewhat greater (5.4–68) than in the human model (7.4–34.0). The more limited range of our estimate is related to the use of both the viral growth rate and the degree of target cell depletion to estimate R 0, whereas the SIV estimate of R 0 used only viral dynamic parameters.
Similar viral replication dynamics between humans and macaques support the use of the SIV model to study HIV pathogenesis. Studies evaluating postexposure prophylaxis as well as the establishment of viral latency will almost certainly be addressed more efficiently in the macaque model, where route, inoculum, and viral strain are specified. Because candidate HIV vaccines will be evaluated first in the SIV model, our estimates of the basic reproductive number will provide a rational means to select vaccines that merit testing in humans.
Acknowledgments
We thank J. Santangelo for her outstanding efforts in patient recruitment and counseling, D. Mosier for his contribution of clinical specimens for subject 059, and S. Albanil, N. Keating, B. Denison, K. Ignacio, L. Terry, A. Rigby, S. Maddox-Gomez, and B. Wright for technical assistance. We also thank J. Guatelli for his thoughtful review of this manuscript.
This research was supported by funds from the following: University-wide AIDS Research Program, University of California, grant no. PH97-SD-201; Center for AIDS Research grant no. AI 36214; grant nos. AI 29164, AI 38858, AI 43638, and AI 27670 from the National Institutes of Health; and the Research Center for AIDS and HIV Infection of the San Diego Veterans Affairs Medical Center. A.R. McLean gratefully acknowledges funding from the Royal Society and the BBSRC.
Footnotes
1used in this paper: PID, patient identification number; SIV, simian immunodeficiency virus
References
- Clark S.J., Saag M.S., Decker W.D., Campbell-Hill S., Roberson J.L., Veldkamp P.J., Kappes J.C., Hahn B.H., Shaw G.M. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N. Engl. J. Med. 1991;324:954–960. doi: 10.1056/NEJM199104043241404. [DOI] [PubMed] [Google Scholar]
- Daar E.S., Moudgil T., Meyer R.D., Ho D.D. Transient high levels of viremia in patients with primary human immunodeficiency virus type 1 infection. N. Engl. J. Med. 1991;324:961–964. doi: 10.1056/NEJM199104043241405. [DOI] [PubMed] [Google Scholar]
- Cooper D.A., Gold J., Maclean P., Donovan B., Finlayson R., Barnes T.G., Michelmore H.M., Brooke P., Penny R. Acute AIDS retrovirus infection. Definition of a clinical illness associated with seroconversion. Lancet. 1985;1:537–540. doi: 10.1016/s0140-6736(85)91205-x. [DOI] [PubMed] [Google Scholar]
- Ho D.D., Sarngadharan M.G., Resnick L., Dimarzoveronese F., Rota T.R., Hirsch M.S. Primary human T-lymphotropic virus type III infection. Ann. Intern. Med. 1985;103:880–883. doi: 10.7326/0003-4819-103-6-880. [DOI] [PubMed] [Google Scholar]
- Mellors J.W., Kingsley L.A., Rinaldo C.R.J., Todd J.A., Hoo B.S., Kokka R.P., Gupta P. Quantitation of HIV-1 RNA in plasma predicts outcome after seroconversion. Ann. Intern. Med. 1995;122:573–579. doi: 10.7326/0003-4819-122-8-199504150-00003. [DOI] [PubMed] [Google Scholar]
- O'Brien T.R., Blattner W.A., Waters D., Eyster E., Hilgartner M.W., Cohen A.R., Luban N., Hatzakis A., Aledort L.M., Rosenberg P.S. Serum HIV-1 RNA levels and time to development of AIDS in the Multicenter Hemophilia Cohort Study. JAMA. 1996;276:105–110. [PubMed] [Google Scholar]
- Macdonald G. The analysis of equilibrium in malaria. Trop. Dis. Bull. 1952;49:813–829. [PubMed] [Google Scholar]
- McLean A. Can HIV be eradicated from an infected individual? Antiviral Therapy 1Suppl. 4–5. (Abstr.)1996. [Google Scholar]
- 1995. Case-control study of HIV seroconversion in health-care workers after percutaneous exposure to HIV-infected blood—France, United Kingdom, and United States, January 1988-August 1994. MMWR Morb. Mortal. Wkly. Rep. 44:929–933. [PubMed]
- 1998. Public Health Service guidelines for the management of health-care worker exposures to HIV and recommendations for postexposure prophylaxis. Centers for Disease Control and Prevention. MMWR Morb. Mortal. Wkly. Rep. 47(RR-7):1–33. [PubMed]
- Katz M.H., Gerberding J.L. The care of persons with recent sexual exposure to HIV. Ann. Intern. Med. 1998;128:306–312. doi: 10.7326/0003-4819-128-4-199802150-00012. [DOI] [PubMed] [Google Scholar]
- Nowak M.A., Lloyd A.L., Vasquez G.M., Wiltrout T.A., Wahl L.M., Bischofberger N., Williams J., Kinter A., Fauci A.S., Hirsch V.M. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 1997;71:7518–7525. doi: 10.1128/jvi.71.10.7518-7525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean A.R., Emery V.C., Webster A., Griffiths P.D. Population dynamics of HIV within an individual after treatment with zidovudine. AIDS. 1991;5:485–489. doi: 10.1097/00002030-199105000-00002. [DOI] [PubMed] [Google Scholar]
- Phillips A.N. Reduction of HIV concentration during acute infectionindependence from a specific immune response. Science. 1996;271:497–499. doi: 10.1126/science.271.5248.497. [DOI] [PubMed] [Google Scholar]
- Schmitz J.E., Kuroda M.J., Santra S., Sasseville V.G., Simon M.A., Lifton M.A., Racz P., Tenner-Racz K., Dalesandro M., Scallon B.J. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Kim S.Y., Byrn R., Groopman J., Baltimore D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infectionevidence for differential gene expression. J. Virol. 1989;63:3708–3713. doi: 10.1128/jvi.63.9.3708-3713.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guatelli J.C., Gingeras T.R., Richman D.D. Alternative splice acceptor utilization during human immunodeficiency virus type 1 infection of cultured cells. J. Virol. 1990;64:4093–4098. doi: 10.1128/jvi.64.9.4093-4098.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelson A.S., Neumann A.U., Markowitz M., Leonard J.M., Ho D.D. HIV-1 dynamics in vivovirion clearance rate, infected cell lifetime, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Schacker, T.W., J. Hughes, T. Shea, and L. Corey. 1996. Virologic course of primary HIV infection. 3rd Conference on Retroviruses and Opportunistic Infections. 480. (Abstr.)
- Rosenberg E.S., Billingsley J.M., Caliendo A.M., Boswell S.L., Sax P.E., Kalams S.A., Walker B.D. Vigorous HIV-1-specific CD4+ T cell responses associated with control of viremia. Science. 1997;278:1447–1450. doi: 10.1126/science.278.5342.1447. [DOI] [PubMed] [Google Scholar]
- Neumann A.U., Tubiana R., Calvez V., Robert C., Li T.-S., Agut H., Autran B., Katlama C. HIV-1 rebound during interruption of highly active antiretroviral therapy has no deleterious effect on reinitiated treatment. Comet Study Group. AIDS. 1999;13:677–683. doi: 10.1097/00002030-199904160-00008. [DOI] [PubMed] [Google Scholar]
- Erice A., Mayers D.L., Strike D.G., Sannerud K.J., McCutchan F.E., Henry K., Balfour H.H., Jr. Brief reportprimary infection with zidovudine-resistant human immunodeficiency virus type 1. N. Engl. J. Med. 1993;328:1163–1165. doi: 10.1056/NEJM199304223281605. [DOI] [PubMed] [Google Scholar]
- Imrie A., Carr A., Duncombe C., Finlayson R., Vizzard J., Law M., Kaldor J., Penny R., Cooper D.A. Primary infection with zidovudine-resistant human immunodeficiency virus type 1 does not adversely affect outcome at 1 year. Sydney Primary HIV Infection Study Group. J. Infect. Dis. 1996;174:195–198. doi: 10.1093/infdis/174.1.195. [DOI] [PubMed] [Google Scholar]
- Imrie A., Beveridge A., Genn W., Vizzard J., Cooper D.A. Transmission of human immunodeficiency virus type 1 resistant to nevirapine and zidovudine. Sydney Primary HIV Infection Study Group. J. Infect. Dis. 1997;175:1502–1506. doi: 10.1086/516487. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer S., May R.M., Shaw G.M., Nowak M.A. Virus dynamics and drug therapy. Proc. Natl. Acad. Sci. USA. 1997;94:6971–6976. doi: 10.1073/pnas.94.13.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D., Blankson J., Siliciano J.D., Margolick J.B., Chadwick K., Pierson T., Smith K., Lisziewicz J., Lori F., Flexner C. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Wong J., Hezareh M., Gunthard H., Havlir D.V., Ignacio C.C., Spina C.A., Richman D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. doi: 10.1126/science.278.5341.1291. [DOI] [PubMed] [Google Scholar]
- Finzi D., Hermankova M., Pierson T., Carruth L.M., Buck C., Chaisson R.E., Quinn T.C., Chadwick K., Margolick J., Brookmeyer R. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- Spina C.A., Prince H.E., Richman D.D. Preferential replication of HIV-1 in the CD45RO memory cell subset of primary CD4 lymphocytes in vitro. J. Clin. Invest. 1997;99:1774–1785. doi: 10.1172/JCI119342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal J.S., Mawle A., Cort S.P., Nicholson J.K., Cross G.D., Scheppler-Campbell J.A., Hicks D., Sligh J. Cellular tropism of the human retrovirus HTLV-III/LAV. I. Role of T cell activation and expression of the T4 antigen. J. Immunol. 1985;135:3151–3162. [PubMed] [Google Scholar]
- Schnittman S.M., Lane H.C., Greenhouse J., Justement J.S., Baseler M., Fauci A.S. Preferential infection of CD4+ memory T cells by human immunodeficiency virus type 1evidence for a role in the selective T-cell functional defects observed in infected individuals. Proc. Natl. Acad. Sci. USA. 1990;87:6058–6062. doi: 10.1073/pnas.87.16.6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson M., Stanwick T.L., Dempsey M.P., Lamonica C.A. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO (Eur. Mol. Biol. Organ.) J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staprans S.I., Hamilton B.L., Follansbee S.E., Elbeik T., Barbosa P., Grant R.M., Feinberg M.B. Activation of virus replication after vaccination of HIV-1–infected individuals. J. Exp. Med. 1995;182:1727–1737. doi: 10.1084/jem.182.6.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs N.L., Little S.J., Richman D.D., Guatelli J.C. Cell-associated viral RNA expression during acute infection with HIV type 1. AIDS Res. Hum. Retroviruses. 1998;14:1141–1149. doi: 10.1089/aid.1998.14.1141. [DOI] [PubMed] [Google Scholar]
- Ho, D. 1998. Novel Approaches for the Evaluation of New Drugs: Approaches Using Viral Dynamics. 5th Conference on Retroviruses and Opportunistic Infections. S49. (Abstr.)
- DeWolf, F., V. Lukashov, S. Danner, J. Goudsmit, and J. Lange. 1998. Clearance of HIV-1 Following Treatment with Three, Four and Five Anti-HIV Drugs. 5th Conference on Retroviruses and Opportunistic Infections. 384. (Abstr.)
- Notermans D.W., Goudsmit J., Danner S.A., de Wolf F., Perelson A.S., Mittler J. Rate of HIV-1 decline following antiretroviral therapy is related to viral load at baseline and drug regimen. AIDS. 1998;12:1483–1490. doi: 10.1097/00002030-199812000-00010. [DOI] [PubMed] [Google Scholar]
- McLean, A. 1998. CTL Effectiveness In Vivo. 5th Annual HIV Dynamics and Evolution Conference. 31. (Abstr.)
- Mellors J.W., Rinaldo C.R., Jr., Gupta P., White R.M., Todd J.A., Kingsley L.A. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]