

Abstract
In vivo manipulation of cytokine and/or cytokine receptor expression has previously shown that resistance to infection with the caecum-dwelling helminth Trichuris muris is dependent on interleukin (IL)-4 and IL-13 while susceptibility is associated with a T helper cell type 1 (Th1) cytokine response. Using gene-targeted mice deficient in tumor necrosis factor (TNF) receptor signaling and anti–TNF-α monoclonal antibody treatment, we have extended these studies to reveal a critical role for TNF-α in regulation of Th2 cytokine–mediated host protection. In vivo blockade of TNF-α in normally resistant mice, although not altering IL-4, IL-5, or IL-13 production in the draining lymph node, significantly delayed worm expulsion for the duration of treatment. IL-13–mediated worm expulsion in IL-4 knockout (KO) mice was also shown to be TNF-α dependent, and could be enhanced by administration of recombinant TNF-α. Furthermore, TNF receptor KO mice failed to expel T. muris, producing high levels of parasite-specific immunoglobulin G2a and the generation of a predominantly Th1 response, suggesting that the absence of TNF function from the onset of infection dramatically alters the phenotype of the response. These results provide the first demonstration of the role of TNF-α in regulating Th2 cytokine–mediated responses at mucosal sites, and have implications for the design of rational therapies against helminth infection and allergy.
Keywords: tumor necrosis factor α, T helper cell type 2 cytokines, helminth infection, interleukin 13, mucosal immunology
Initiation of an appropriate Th response is critical in determining the outcome of infection with the caecum-dwelling nematode Trichuris muris. After antigenic stimulation, naive T cells differentiate into distinct Th cell subsets that produce characteristic cytokine profiles and determine the nature of the effector response. Th1 cells are defined by their secretion of IFN-γ, IL-2, and lymphotoxin (LT),1 and stimulate IgG2a production and cell-mediated effector responses, while Th2 cells produce IL-4, IL-5, IL-9, and IL-13 and promote mastocytosis, eosinophilia, and the production of IgE and IgG1 (for reviews, see references 1 2 3). Inbred mouse strains that naturally mount a Th1 response are unable to expel T. muris and are susceptible to infection 4. Induction of a Th2 response in susceptible strains, either by blocking IFN-γ or IL-12 production or by the administration of IL-4 (5; Bancroft, A.J., manuscript in preparation), converts them to a resistant phenotype. Conversely, in mouse strains that are resistant to infection, administration of IL-12, disruption of the IL-4 gene, or blockade of the IL-4 receptor (IL-4R) will effectively ablate protection 5 6 7. Similar in vivo manipulations in other helminth models, including Heligmosomoides polygyrus and Nippostrongylus brasiliensis infections, have identified the role of Th1 responses in prolonging parasite survival and Th2 responses in regulation of host protection (8 9 10 11; for a review, see reference 12). A vital and distinct role for IL-13 in resistance to T. muris has recently been identified in IL-13 knockout (KO) mice, which are unable to clear infection despite generating parasite-specific Th2 responses 7.
Although the effector mechanisms operating in T. muris infection have yet to be defined, it is clear that IL-4 and IL-13 regulate immunological and physiological events in the intestinal mucosa which mediate host protection. Th2 cells are also known to be important in the regulation of mucosal inflammation and exacerbation of disease in models of airway hyperresponsiveness. Transgenic mice overexpressing IL-4 or IL-5 in the lung develop mucus hypersecretion and similar pathology to that observed during allergic asthma 13 14. IL-13 has also been identified as a key regulatory cytokine in airway inflammation 15 16. Using an OVA-specific TCR transgenic CD4 cell transfer model, administration of TNF-α has been shown to enhance Th2 cell transendothelial migration and to potentiate mucosal inflammation in the airway epithelium 17, suggesting that TNF-α may also be important in mucosal Th2 responses during intestinal helminth infection.
In this study, we have identified a novel role for TNF-α in the downstream regulation of Th2 cytokine effector responses in the intestinal mucosa. Both in vivo neutralization of TNF-α and use of TNFR-deficient mice have shown that TNF-α is critical in regulating host protection to helminth infection. Blockade of TNF-α in normally resistant C57BL/6 mice prevents worm expulsion for the duration of treatment despite equivalent Th2 responses in anti–TNF-α and control treated mice. In the complete absence of TNFR signaling, T. muris infection proceeds to chronicity with the generation of a nonprotective Th1 response. Furthermore, IL-13–dependent expulsion in IL-4 KO mice is also impaired during blockade of TNF-α, whereas clearance of infection is enhanced in these mice by the administration of recombinant TNF-α. This is the first study to identify a role for TNF-α in protection during intestinal helminth infection and has important implications for our understanding of the initiation and regulation of Th2 effector responses at mucosal sites.
Materials and Methods
Animals.
Mice doubly deficient in both the TNFR p55 and p75 genes (TNFR KO; from Dr. J. Peschon, Immunex Corp., Seattle, WA) were generated as described 18 and maintained as random C57BL/6 × 129 hybrids at the University of Manchester. Age- and sex-matched C57BL/6 × 129 F2 mice were bred from F1 littermates (Harlan Olac) and used as wild-type (WT) controls. BALB/c IL-4 KO mice were generated by Noben-Trauth et al. 19 and bred at the University of Manchester. Age- and sex-matched C57BL/6 and BALB/c mice were purchased from Harlan Olac. In all experiments, mice were infected when 6–9 wk old, and experimental groups contained four to six animals. All experiments were performed under the regulations of the Home Office Scientific Procedures Act (1986).
Parasites.
The maintenance, infection, and recovery of T. muris were as described previously 20. Mice were infected on day 0 with ∼200 embryonated eggs, and numbers of larvae were counted on day 10 postinfection (p.i.) to ensure equivalent establishment of infection in different groups. Worm burdens were assessed on various days p.i. as described previously 21. T. muris excretory/secretory antigen (ES Ag) was prepared as detailed previously 7.
Antibody and Cytokine Reagents.
In vivo depletion of TNF-α was carried out using purified rat IgG1 mAb XT22 (neutralizing TNF-α; from Dr. R. Coffman, DNAX Research Institute, Palo Alto, CA) injected intraperitoneally as detailed in the text. Control groups were treated with either isotype-matched control (GL113; from Dr. F. Finkelman, University of Cincinnati, Cincinnati, OH) or purified rat IgG (Sigma Chemical Co.). Recombinant TNF-α (Dr. G. Luheshi, University of Manchester) was delivered intraperitoneally as described in the text.
Cell Culture and Cytokine Analysis.
Mesenteric lymph node cells were removed from uninfected and infected mice and resuspended in RPMI 1640 supplemented with 10% FCS, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin (all from GIBCO BRL), and 60 μM monothioglycerol (Sigma Chemical Co.). Cultures were stimulated with a predetermined optimum concentration of T. muris ES Ag (50 μg/ml) at 37°C and 5% CO2. Anti–IL-4R (M1) mAb (5 μg/ml; from Dr. C. Maliszewski, Immunex Corp., Seattle, WA) was added to cultures to increase detection of IL-4. Cell-free supernatants were harvested after 24 h and stored at −20°C.
Cytokine analysis was carried out by sandwich ELISA using paired mAbs to detect IL-4 (BVC4-1D11 and BVD6-24G2.3; PharMingen), IL-5 (TRFK.5 and TRFK.4; PharMingen), IL-9 (249.2 and biotinylated 1C10/2C12; from Dr. J. van Snick, Ludwig Institute of Cancer Research, Brussels, Belgium), IFN-γ (R46A2 and XMG.2; PharMingen), and IL-12 (C15.6 and C17.8; from Dr. G. Trinchieri, Wistar Institute, Philadelphia, PA). TNF-α and IL-13 production was assessed using an R&D Systems ELISA kit. Quantification of cytokines was determined by reference to commercially available recombinant murine standards, and the sensitivity of the assays was determined by taking the mean + 3 SD of 16 control wells containing medium alone.
Antibody Analysis.
Analysis of parasite-specific IgG1 and IgG2a production was carried out by capture ELISA as described 22. In brief, Immulon IV plates (Dynatech) were coated with T. muris ES Ag (5 μg/ml) in carbonate/bicarbonate buffer, pH 9.6, overnight at 4°C. After blocking (3% BSA in PBS, 0.05% Tween), eight serial 2-fold dilutions of sera (from an initial 20-fold dilution) were added to the plates. Parasite-specific antibody was detected using biotinylated rat anti–mouse IgG1 (Serotec Ltd.) and biotinylated rat anti–mouse IgG2a (PharMingen).
Statistics.
Significant differences (P < 0.05) between experimental groups were determined using the Mann-Whitney U test.
Results
In Vivo Blockade of TNF-α in C57BL/6 Mice Prevents Worm Expulsion.
We have previously demonstrated that C57BL/6 mice mount Th2 responses and expel T. muris 7. The role of TNF-α in regulating this host protection was investigated by administering 2 mg of anti–TNF-α or control antibody (GL113) intraperitoneally every 3 d between days 7 and 19 p.i. Blockade of TNF-α significantly prevented worm expulsion at day 21 p.i. compared with control treated groups (Fig. 1 A). With the exception of depressing IL-9 production, anti–TNF-α treatment did not significantly alter the magnitude of the Th2 response generated at day 21 p.i. (Fig. 2).
Figure 1.

Mean worm burden ± SEM in C57BL/6 mice treated with either PBS, anti–TNF-α, or control Ig. Mice were infected on day 0 with 200 T. muris eggs and treated intraperitoneally with 2 mg mAb every 3 d between days 7 and 19 p.i. Worm burdens from four mice per group were determined on days 21 (A) and 35 (B). *Significantly higher worm burden than control treated mice (P < 0.05).
Figure 2.
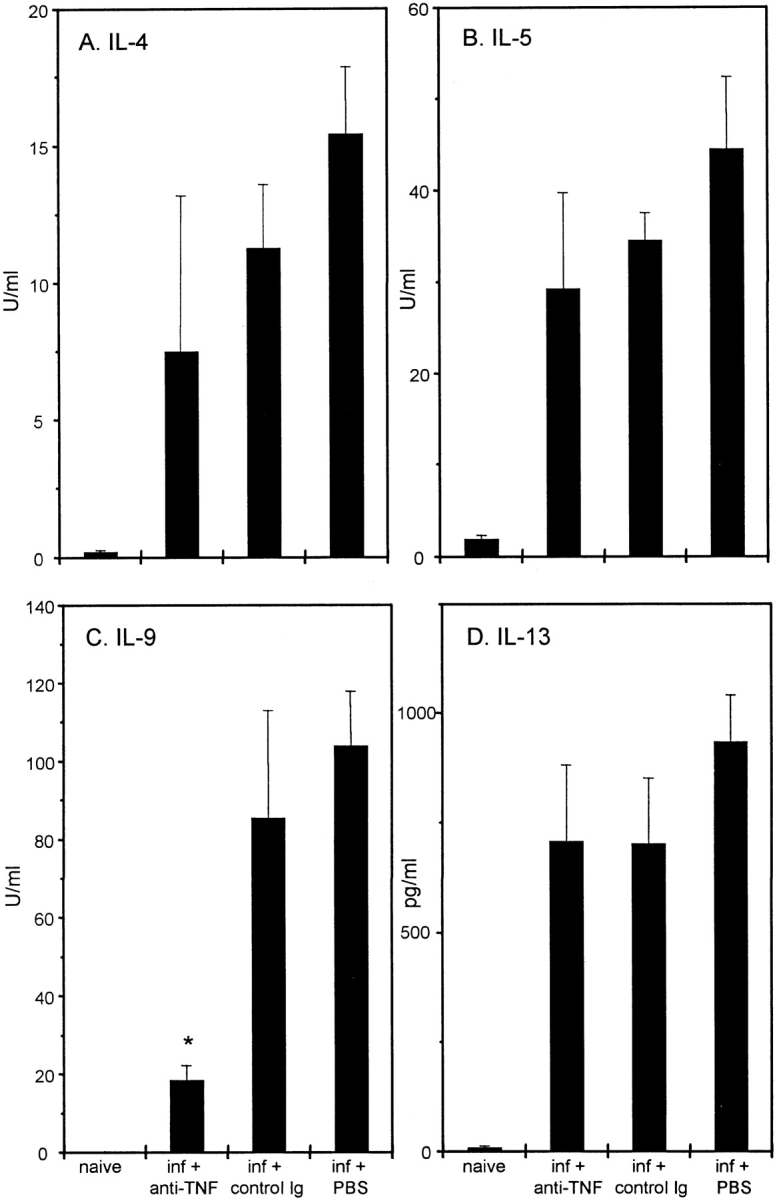
Cytokine production from T. muris–infected C57BL/6 mice after treatment with PBS, anti–TNF-α, or control Ig. Mesenteric lymph node cells were removed on day 21 p.i., stimulated in vitro with 50 μg T. muris Ag for 24 h, and supernatants were analyzed by sandwich ELISA for the presence of IL-4 (A), IL-5 (B), IL-9 (C), and IL-13 (D). Results represent the mean values of four mice per group ± SEM. *Significantly lower than control treated groups (P < 0.05).
Prevention of worm expulsion is transient, and cessation of anti–TNF-α treatment resulted in the clearance of infection in all treatment groups by day 35 p.i. (Fig. 1 B). As the cytokine response has diminished by day 35 p.i. (data not shown), this suggests that a sufficient Th2 response was generated in the mesenteric lymph nodes of anti–TNF-α treated mice to induce worm expulsion (Fig. 2). Therefore, although depletion of TNF-α has no demonstrable effect on the initiation of Th2 responses, it appears to be critical in the regulation of Th2 cell effector function in the intestine. This hypothesis is reinforced by the parasite-specific IgG isotype response at day 35 p.i. Expulsion of T. muris occurs in the absence of antibody 23; however, assessment of IgG isotype production provides an indication of the polarization of the Th response after infection. IgG1 production is known to be under the control of Th2 cytokines 24, and there was no significant difference in this response between treatment groups (Fig. 3 A), substantiating the fact that there is no reduction in the magnitude of the Th2 response after anti–TNF-α treatment. The elevated IgG2a response (under the control of IFN-γ 25) observed after blockade of TNF-α (Fig. 3 B) may reflect elevated IFN-γ production in these mice resulting from protracted exposure to infection.
Figure 3.
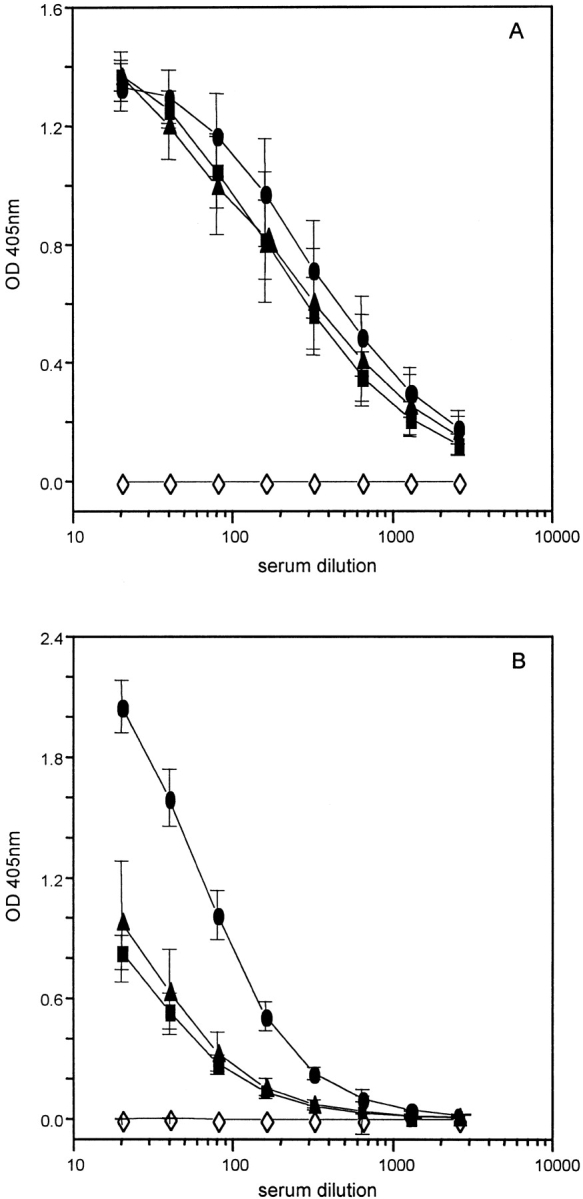
Ag-specific IgG isotype responses from naive (⋄) and T. muris–infected C57BL/6 mice after treatment with PBS (▴), anti–TNF-α (•), or control Ig (▪). Serum was collected from naive and infected mice at day 35 p.i. and assayed by capture ELISA for the presence of IgG1 (A) and IgG2a (B). Results represent the mean of four mice per group ± SEM.
TNFR KO Mice Are Highly Susceptible to T. muris Infection.
As anti–TNF-α treatment blocked protection in C57BL/6 mice, we investigated the kinetics of infection in mice completely deficient in TNFR signaling. We consistently found that TNFR KO mice were highly susceptible to T. muris and unable to expel worms by day 35 p.i. (Fig. 4 A). Previous data have shown that mature worms (day 32 p.i.) can promote their own survival and can only be expelled through drug intervention after this time point 4. WT mice began worm expulsion between days 10 and 18 p.i., and had completely cleared infection by day 22 (Fig. 4 B). Surprisingly, analysis of Ag-specific cytokine production after restimulation of mesenteric lymph node cells showed that TNFR KO mice made little or no Th2 cytokines, whereas WT mice made a strong Th2 response (characteristic of resistant strains [4, 7, 22]) with high levels of IL-4, IL-5, IL-9, and IL-13 at day 18 p.i. (Fig. 5). However, KO mice produced higher levels of Ag-specific IFN-γ (KO, 221.32 ± 121.17 U/ml; WT, 153.5 ± 50.0 U/ml) and IL-12 (KO, 585.51 ± 139.09 ng/ml; WT, 123.03 ± 103.38 ng/ml) after restimulation of mesenteric lymph node cells.
Figure 4.

Mean worm burden ± SEM in TNFR KO (A) and C57BL/6 × 129 WT (B) mice on various days after infection. Mice were infected on day 0 with 200 T. muris eggs, and worm burden was assessed from four to six mice per group. *Significantly lower worm burden than KO mice at corresponding time point (P < 0.05).
Figure 5.

Cytokine production from T. muris–infected TNFR KO and C57BL/6 × 129 WT mice. Mesenteric lymph node cells were removed on day 18 p.i., stimulated in vitro with 50 μg T. muris Ag for 24 h, and supernatants were analyzed by sandwich ELISA for the presence of IL-4 (A), IL-5 (B), IL-9 (C), and IL-13 (D). Results represent the mean values of four to six mice per group ± SEM. *Significantly lower than WT values (P < 0.05).
Parasite-specific IgG isotype responses at day 35 p.i. reflected the resistant and susceptible phenotype of WT and TNFR KO mice and the polarized Th response observed. WT mice made high levels of IgG1 (Fig. 6 A) and relatively low levels of IgG2a (Fig. 6 B) typical of resistant mice. TNFR KO mice made lower IgG1 responses and high levels of IgG2a (Fig. 6). The higher IgG2a response generated in KO mice supported the elevated Th1 cytokine levels observed in the absence of TNFR signaling. This is in accordance with previous observations in other susceptible strains that produced predominantly Th1 cytokines and high levels of IgG2a 4 5 6 7.
Figure 6.

Ag-specific IgG isotype responses from T. muris–infected TNFR KO (•) and C57BL/6 × 129 WT (▪) mice (naive KO [⋄] and naive WT [+]). Serum was collected from naive and infected mice at day 35 p.i. and assayed by capture ELISA for the presence of IgG1 (A) and IgG2a (B). Results represent the mean of four to six mice per group ± SEM.
IL-13–mediated Expulsion of T. muris Is Dependent on TNF-α.
We have previously shown that IL-13 is directly involved in expulsion of T. muris, as IL-13 KO mice were unable to clear infection despite generating equivalent parasite-specific Th2 responses to WT mice at day 21 p.i. 7. Subsequent studies have also found that expulsion of T. muris in female BALB/c IL-4 KO mice is IL-13 dependent. In the absence of IL-4, protection is almost completely abrogated by blockade of IL-13 using a soluble IL-13R fusion protein (Bancroft, A.J., manuscript in preparation). To investigate whether this IL-13–mediated expulsion is also dependent on TNF-α, infected female BALB/c IL-4 KO mice were treated with 2 mg of anti–TNF-α mAb or rat IgG intraperitoneally every 3 d between days 7 and 28 p.i.
Blockade of TNF-α in female BALB/c IL-4 KO mice significantly prevented worm expulsion (Fig. 7 C). In accordance with previously unpublished data, in control treated IL-4 KO mice worm expulsion was initiated around day 22 p.i. (Fig. 7 B), and approximately 60% of worms were cleared by day 35 p.i. (Fig. 7 C). Ag-specific cytokine production by mesenteric lymph node cells at day 18 p.i. showed comparable Th2 responses in the different treatment groups (Fig. 8), although IL-9 and IL-13 production was lower in anti–TNF-α treated mice than in the control Ig group (primarily as a result of the variability observed in this group; Fig. 8b and Fig. c). No difference in the IgG1 response was observed between anti–TNF-α and control treated groups (data not shown), supporting the hypothesis that TNF-α blockade did not alter the generation of Th2 responses, but may be critical at the effector stage in the intestine. Comparable and relatively high IgG2a responses were observed in all groups (data not shown), reflecting the production of Th1 cytokines as previously reported in T. muris–infected C57BL/6 IL-4 KO mice 7.
Figure 7.

Mean worm burden ± SEM in female BALB/c IL-4 KO mice treated with either anti–TNF-α (black bars), control Ig (stippled bars), or PBS (striped bars). Mice were infected on day 0 with 200 T. muris eggs and treated intraperitoneally with 2 mg mAb every 3 d between days 7 and 28 p.i. Worm burdens from four mice per group were determined on days 18, 22, and 35 p.i. *Significantly lower worm burden than anti–TNF-α treated groups (P < 0.05).
Figure 8.



Cytokine production from T. muris–infected female BALB/c IL-4 KO mice after treatment with anti–TNF-α (black bars), control Ig (stippled bars), or PBS (striped bars). Mesenteric lymph node cells were removed on day 18 p.i., stimulated in vitro with 50 μg T. muris Ag for 24 h, and supernatants were analyzed by sandwich ELISA for the presence of IL-5 (A), IL-9 (B), and IL-13 (C). Results represent the mean values from four mice per group ± SEM. *Significantly lower than control treated groups (P < 0.05).
Administration of TNF-α Enhances IL-13–mediated Expulsion of T. muris.
Previous experiments have revealed a sex difference in resistance to T. muris in BALB/c IL-4 KO mice (Bancroft, A.J., and D. Artis, unpublished observations). We have now confirmed these data demonstrating that female mice produce higher parasite-specific Th2 cytokines (IL-5, IL-9, and IL-13) than males (compare Fig. 8 with Table ) and elevated IgG1 responses during infection (data not shown). Therefore, female IL-4 KO mice expel worms around day 35 p.i., whereas males are unable to do so. To investigate whether elevation of TNF-α levels in vivo could promote a resistant phenotype in male BALB/c IL-4 KO mice, 2 μg of TNF-α was administered intraperitoneally to infected mice daily between days 10 and 24 p.i., and infection outcome was monitored at days 21 and 35 p.i.
Table 2.
Th2 Cytokine Production (± SEM) by Ag-stimulated Mesenteric Lymph Node Cells from Male BALB-c IL-4 KO Mice at Day 21 p.i.
| Naive | Infected + PBS | Infected + TNF | |
|---|---|---|---|
| IL-5 (U/ml) | 2.072 | 2.744 (± 0.25) | 2.52 (± 1.03) |
| IL-9 (U/ml) | 2.291 | 7.84 (± 3.28) | 10.63 (± 2.05) |
| IL-13 (pg/ml) | 5.892 | 231.44 (± 44.05) | 278.45 (± 38.87) |
Male BALB/c IL-4 KO mice were infected with 200 T. muris eggs on day 0 and treated intraperitoneally with either 2 μg of TNF-α or an equal volume of PBS daily between days 10 and 24 p.i. Mesenteric lymph node cells were taken on day 21 p.i., restimulated in vitro, and supernatants were assayed for cytokine by ELISA. Results represent the mean values (± SEM) of four animals per group.
Administration of TNF-α to male BALB/c IL-4 KO mice considerably increased worm expulsion by day 35 p.i., with 43% of worms cleared in TNF-α–treated mice compared with no reduction in worm burden in the control group at day 35 p.i. (Table ; P < 0.05). (TNF-α treatment did not have any obvious detrimental effects on the general health of the animals.) Interestingly, treatment with TNF-α did not alter the production of IL-5, IL-9, or IL-13 after in vitro restimulation with ES Ag (Table ), suggesting again that TNF-α may potentiate the effects of Th2 cytokines (in this case IL-13) rather than enhance their production. In addition, no difference in IgG1 production was observed between TNF-α and control treated mice although higher IgG2a production was observed in TNF-α–treated mice, reflecting higher IFN-γ production (data not shown). Administration of TNF-α to male IL-4 KO mice conferred a similar rate of worm expulsion (Table ) to that observed in female mice (Fig. 7), suggesting that impaired protection in male mice may be due to reduced levels of TNF-α during infection. However, analysis of TNF-α production after Ag-specific in vitro restimulation of mesenteric lymph node cells showed no significant difference between male and female mice (Table ). Therefore, impaired clearance of infection in male mice is more likely to be due to lower Th2 responses compared with females (Fig. 8, and Table ) rather than a defect in TNF-α production. Thus, although there is no inherent difference between male and female IL-4 KO mice in the production of TNF-α, administration of this cytokine is able to induce a resistant phenotype in male mice without a significant elevation in parasite-specific Th2 responses.
Table 1.
Mean Worm Burden (± SEM) in Male BALB/c IL-4 KO Mice after Administration of TNF-α
| Mean worm burden (± SEM) | ||
|---|---|---|
| Day 21 | Day 35 | |
| Infected + TNF-α | 100.75 (± 6.92) | 57.0 (± 20.59) |
| Infected + PBS | 96.0 (± 4.97) | 105.0 (± 20.57) |
Male BALB/c IL-4 KO mice were infected with 200 T. muris eggs on day 0 and treated intraperitoneally with either 2 μg of TNF-α or an equal volume of PBS daily between days 10 and 24 p.i. Worm burdens were assessed on days 21 and 35 p.i. Results represent the mean values of four animals (± SEM) per group.
Table 3.
TNF-α Production (± SEM) by Ag-stimulated Mesenteric Lymph Node Cells from Male and Female BALB/c IL-4 KO Mice at Day 18 p.i.
| Naive | Infected | |
|---|---|---|
| pg/ml | pg/ml | |
| Male | 103.19 | 307.58 ± 43.68 |
| Female | 139.23 | 236.54 ± 50.53 |
Male and female BALB/c IL-4 KO mice were infected with 200 T. muris eggs on day 0, and mesenteric lymph node cells were restimulated in vitro with parasite antigen. Supernatants were assayed for cytokine by ELISA. Results represent the mean values (± SEM) of four animals per group.
Discussion
Previous studies have shown that immune-mediated expulsion of the intestinal helminth T. muris is dependent on IL-4 and IL-13 5 7. The results presented here provide the first demonstration of a critical role for TNF-α in host protection against an intestinal helminth infection and extend our understanding of the role of TNF-α as an essential component of Th2 cell–mediated effector responses. TNF-α is known to be a key mediator of pathogenesis in a broad range of infectious, inflammatory, and autoimmune diseases (for a review, see reference 26), although few studies have investigated the role of this prototypic inflammatory cytokine in the regulation of immune responses dominated by Th2 cytokines. The role of TNF-α in the pathogenesis of predominantly Th1-mediated inflammation, including collagen-induced arthritis 27, autoimmune encephalomyelitis 28, and intestinal inflammation 29 30, is well characterized. Our results extend these observations and identify a novel role for TNF-α in the regulation of Th2 cell–mediated protection during helminth infection.
It is significant that immunodepletion of TNF-α in C57BL/6 and BALB/c IL-4 KO mice showed little or no reduction in the magnitude of Ag-specific Th2 cytokine production and IgG1 responses during infection, despite blocking worm expulsion. This demonstrated that blockade of TNF-α had little effect on the initiation of Th2 responses in the draining lymph node. Preliminary analysis of intestinal mast cell hyperplasia (which is under the control of Th2 cytokines) during infection in C57BL/6 mice showed a depressed mastocytosis after anti–TNF-α treatment (data not shown). This supports the hypothesis that although the magnitude of the Th2 response in the draining lymph node is unaltered, in the intestinal microenvironment the Th2 response is impaired, implicating a role for TNF-α in regulation of effector function. Previous experiments have shown that in vivo elevation of IL-9 enhances resistance to T. muris 31, and after depletion of TNF-α in infected C57BL/6 and female IL-4 KO mice the depressed production of IL-9 may be important in preventing normal expulsion (although it is clear that mast cells are not important in host protection 32).
TNF-α is known to regulate expression of a range of cell adhesion molecules on vascular endothelium 33 34 and leukocytes 35 and to control the expression of chemoattractant cytokines 36. In addition, TNF-α has been shown to be necessary for homing of Th2 cells to the site of allergic inflammation 17. Thus, ongoing studies are addressing leukocyte homing and recirculation to the intestine during infection in the presence and absence of TNF-α. TNF-α is a pleiotropic cytokine, and the mechanisms of regulating host protection to T. muris may not be through the recruitment of inflammatory cells, but rather through amplification of the existing Th2 response. Certainly, TNF-α has been shown to enhance Th2 cell–mediated phenomena in other systems, including pathogenesis of allergic inflammation in the gastric mucosa 37, Schistosoma mansoni egg–induced granuloma formation 38 39, and airway hyperresponsiveness 17.
A role for TNF-α in amplifying effector function of Th2 cytokines is recapitulated by our studies in which at least partial protection against T. muris can be conferred on normally susceptible male BALB/c IL-4 KO mice after administration of TNF-α. Comparison between the magnitude of the Th2 response mounted in male and female BALB/c IL-4 KO mice supports previously unpublished data demonstrating a Th2 bias in females and enhanced protection. Furthermore, it is clear that sex differences in host protection are not due to lower TNF-α production in male mice after infection. That enhancing in vivo levels of TNF-α in male IL-4 KO mice increased worm expulsion at day 35 p.i. to levels similar to those observed in female mice (without altering the levels of Th2 cytokines produced) supports the hypothesis that TNF-α is acting downstream of Th2 cell differentiation and is potentiating the protective effects of Th2 cytokines in male mice. Whether TNF-α administration can enhance IL-13–mediated phenomena in other systems such as airway inflammation 15 16, S. mansoni egg–induced pulmonary granuloma 40, and resistance to Nippostrongylus brasiliensis infection 41 remains to be determined.
The development of chronic infection in mice deficient in TNFR p55 and p75 provides the first characterization of intestinal helminth infection in these mice. The inability of TNFR KO mice to expel T. muris may reflect a similar impairment of Th2 cell–mediated effector function as suggested above. However, we were unable to detect any significant Th2 cytokines after restimulation of mesenteric lymph node cells in these mice, suggesting that the absence of TNF-α function at the initiation of infection in TNFR KO mice had a profoundly different effect (i.e., impairing initiation of a Th2 response) than depletion of TNF-α later in infection (see above). Mice deficient in either or both p55 and p75 have been shown to be resistant to lethal shock induced by LPS 42, have attenuated responses to cerebral malaria 43, and were highly susceptible to intracellular bacterial 42 44 45 and protozoan infections 46. TNFR KO mice have also revealed a novel role for these receptors in lymphoid tissue organogenesis and morphogenesis with the reported absence of B cell follicles, germinal center formation, and follicular dendritic cell networks (for reviews, see references 47 and 48). Our results demonstrate no impairment of immune responsiveness per se during T. muris infection, with polarized Ag-specific Th1 cytokine production and strong parasite-specific IgG isotype responses detected at day 35 p.i. It has been suggested that IgG isotype switching in TNFR KO mice is normal 49, and given that expulsion of T. muris can occur in the absence of antibody 23, we feel this is not the basis of susceptibility to infection in these mice. However, we cannot rule out impaired or altered Ag sampling, processing, and presentation in TNFR KO mice, given the reported absence of Peyer's patches in TNFR KO mice 50 and the suggested role for TNF-α in the maturation of dendritic cell function 51 52 and migration to the draining lymph node 53. Such alterations in initiation of the response may result in the polarized Th1 response observed after infection, suggesting a potentially critical role for TNF-α in response induction. (LT-α is also known to bind TNFR with equal affinity and kinetics to TNF-α 54, and may play a role in host protection.)
In this context, there are several conflicting studies on the role of TNF-α in autoimmune diseases such as insulitis 55 56 with the outcome of TNF-α administration and blockade clearly dependent on the cytokine dose, duration of exposure, and site of expression 57, and similar processes may be operating in our system. The data presented here support the hypothesis that TNF-α, when administered or blocked after the first week of infection (and coincident with the generation of a Th2 response), has a clear effect on host protection without altering the magnitude of the Th2 response, while the absence of TNF function from day 0 (in TNFR KO mice) inhibits the induction of Th2 responses. However, the exact molecular and cellular mechanisms through which TNF-α mediates its protective effects remain to be elucidated. For instance, does TNF-α act on lymphocyte populations to augment Th2 effector cell responses 58, on APC function, or on multiple cell types derived from a nonlymphoid lineage? And what contribution do the independently regulated and functionally distinct p55 and p75 receptors make to host protection?
That administration and blockade of TNF-α can modulate infection outcome without altering the production of Th2 cytokines suggests a role for TNF-α in regulating Th2 effector function. An attractive hypothesis is that TNF-α functions through regulation of IL-4 and IL-13 receptor expression on cells in the intestinal microenvironment, a function that has been observed in human endothelial cells 59. However, it is clear that we have identified a novel role for TNF-α in regulating Th2 cytokine responses in the intestine, which has a significant effect on protective immunity to helminth infection. As such, these results extend our understanding of the complex interplay of the cytokine network and have significant implications for the design of rational therapies against helminth infection and allergic reactions in the gut and at other mucosal sites.
Acknowledgments
We would like to thank Dr. R. Coffman, Dr. F. Finkelman, Dr. G. Luheshi, Dr. C. Maliszewski, Dr. J. van Snick, and Dr. G. Trinchieri for mAb and cytokine reagents used. Dr. J. Peschon and Dr. N. Noben-Trauth provided the gene-targeted mice used in these experiments, for which we are also grateful. We would also like to thank Dr. R. Lawrence (University of Manchester, Manchester, UK) for comments on the manuscript.
This work is supported by grants from the Biotechnology and Biological Sciences Research Council (BBSRC), the Cancer Research Campaign (CRC), and The Wellcome Trust.
Footnotes
1used in this paper: ES Ag, excretory/secretory antigen; KO, knockout; LT, lymphotoxin; p.i., postinfection; WT, wild-type
References
- Abbas A.K., Murphy K.M., Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- Mosmann T., Sad S. The expanding universe of T-cell subsetsTh1, Th2 and more. Immunol. Today. 1996;17:138–146. doi: 10.1016/0167-5699(96)80606-2. [DOI] [PubMed] [Google Scholar]
- O'Garra A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity. 1998;8:275–283. doi: 10.1016/s1074-7613(00)80533-6. [DOI] [PubMed] [Google Scholar]
- Else K.J., Hültner L.H., Grencis R.K. Modulation of cytokine production and response phenotypes in murine trichuriasis. Parasite Immunol. 1992;14:441–449. doi: 10.1111/j.1365-3024.1992.tb00018.x. [DOI] [PubMed] [Google Scholar]
- Else K.J., Finkelman F.D., Maliszewski C.R., Grencis R.K. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 1994;179:347–351. doi: 10.1084/jem.179.1.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft A.J., Else K.J., Sypek J.P., Grencis R.K. IL-12 promotes a chronic intestinal nematode infection. Eur. J. Immunol. 1997;27:866–870. doi: 10.1002/eji.1830270410. [DOI] [PubMed] [Google Scholar]
- Bancroft A.J., McKenzie A.N.J., Grencis R.K. A critical role for IL-13 in resistance to intestinal nematode infection. J. Immunol. 1998;160:3453–3461. [PubMed] [Google Scholar]
- Urban J.F., Jr., Katona I.M., Paul W.E., Finkelman F.D. IL-4 is important in protective immunity to a gastrointestinal nematode infection in mice. Proc. Natl. Acad. Sci. USA. 1991;88:5513–5517. doi: 10.1073/pnas.88.13.5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban J.F., Jr., Madden K.B., Cheever A.W., Trotta P.P., Katona I.M., Finkelman F.D. Interferon inhibits inflammatory responses and protective immunity in mice infected with the nematode parasite, Nippostrongylus brasiliensis . J. Immunol. 1993;151:7086–7094. [PubMed] [Google Scholar]
- Urban J.F., Jr., Maliszewski C.R., Madden K.B., Madden I.M., Finkelman F.D. IL-4 treatment can cure established gastrointestinal nematode infections in immunocompetent and immunodeficient mice. J. Immunol. 1995;154:4675–4684. [PubMed] [Google Scholar]
- Urban J.F., Jr., Noben-Trauth N., Donaldson D.D., Madden K.B., Morris S.C., Collins M., Finkelman F.D. IL-13, IL-4Rα, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis . Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- Finkelman F.D., Shea-Donohue T., Goldhill J., Sullivan C.A., Morris S.C., Madden K.B., Gause W.C., Urban J.F., Jr. Cytokine regulation of host defense against parasitic gastrointestinal helminthslessons from studies with rodent models. Annu. Rev. Immunol. 1997;15:505–533. doi: 10.1146/annurev.immunol.15.1.505. [DOI] [PubMed] [Google Scholar]
- Temann U.A., Prasad B.G., Basbaum M.W.C., Ho S.B., Flavell R.A., Rankin J.A. A novel role for murine IL-4 in vivoinduction of MUC5AC gene expression and mucin hypersecretion. Am. J. Respir. Cell Mol. Biol. 1997;16:471–478. doi: 10.1165/ajrcmb.16.4.9115759. [DOI] [PubMed] [Google Scholar]
- Lee J.J., McGarry M.P., Farmer S.C., Denzler K.L., Larson K.A., Carrigan P.E., Brenneise I.E., Horton M.A., Haczku A., Gelfand E.W. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J. Exp. Med. 1997;185:2143–2156. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M., Luyimbazi J., Xu X., Schofield B., Neben T.Y., Karp C.L., Donaldson D.D. Interleukin-13central mediator of allergic asthma. Science. 1998;282:2258–2260. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- Grünig G., Warnock M., Wakil A.E., Venkayya R., Brombacher F., Rennick D.M., Sheppard D., Mohrs M., Donaldson D.D., Locksley R.M., Corry D. Requirement for IL-13 independently of IL-4 in experimental asthma. Science. 1998;282:2261–2263. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L., Homer R.J., Marinov A., Rankin J., Bottomly K. Induction of airway mucus production by T helper 2 (Th2) cellsa critical role for interleukin 4 in cell recruitment but not mucus production. J. Exp. Med. 1997;186:1737–1747. doi: 10.1084/jem.186.10.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon J.J., Torrance D.S., Stocking K.L., Glaccum M.B., Otten C., Willis C.R., Charrier K., Morrissey P.J., Ware C.B., Mohler K.M. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol. 1998;160:943–952. [PubMed] [Google Scholar]
- Noben-Trauth N., Kohler G., Burki K., Ledermann B. Efficient targeting of the IL-4 gene in a BALB/c embryonic stem cell line. Transgenic Res. 1996;5:487–491. doi: 10.1007/BF01980214. [DOI] [PubMed] [Google Scholar]
- Wakelin D. Acquired immunity to Trichuris muris in the albino laboratory mouse. Parasitology. 1967;57:515–524. doi: 10.1017/s0031182000072395. [DOI] [PubMed] [Google Scholar]
- Else K.J., Wakelin D., Wassom D.L., Hauda K.H. The influence of genes mapping within the major histocompatibility complex on resistance to Trichuris muris infections in mice. Parasitology. 1990;101:61–67. doi: 10.1017/s0031182000079762. [DOI] [PubMed] [Google Scholar]
- Else K.J., Entwistle G., Grencis R.K. Correlations between worm burden and markers for Th1 and Th2 cell subset induction in an inbred strain of mouse infected with Trichuris muris . Parasite Immunol. 1993;15:595–600. doi: 10.1111/pim.1993.15.10.595. [DOI] [PubMed] [Google Scholar]
- Else K.J., Grencis R.K. Antibody-independent effector mechanisms in resistance to the intestinal nematode parasite Trichuris muris . Infect. Immun. 1996;64:2950–2954. doi: 10.1128/iai.64.8.2950-2954.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitetta E.S., Ohara J., Myers C.D., Layton J.E., Krammer P.H., Paul W.E. Serological, biochemical, and functional identity of B cell–stimulatory factor 1 and B cell differentiation factor for IgG1. J. Exp. Med. 1985;162:726–731. doi: 10.1084/jem.162.5.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelman F.D., Holmes J., Katona I.M., Urban J.F., Jr., Beckman M.P., Parks L.S., Schooley K.A., Coffman R.L., Mosmann T.R., Paul W.E. Lymphokine control of in vivo immunoglobulin isotype selection. Annu. Rev. Immunol. 1990;8:303–333. doi: 10.1146/annurev.iy.08.040190.001511. [DOI] [PubMed] [Google Scholar]
- Vassalli P. The pathophysiology of tumor necrosis factors. Annu. Rev. Immunol. 1992;10:411–452. doi: 10.1146/annurev.iy.10.040192.002211. [DOI] [PubMed] [Google Scholar]
- Williams R.O., Feldmann M., Maini R.N. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc. Natl. Acad. Sci. USA. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle N.H., Bergman C.M., McGrath K.M., Lingenheld E.G., Grunnet M.L., Padula S.J., Clark R.B. An antibody to lymphotoxin and tumor necrosis factor prevents transfer of experimental allergic encephalomyelitis. J. Exp. Med. 1990;172:1193–2000. doi: 10.1084/jem.172.4.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neurath M.F., Fuss I., Pasparakis M., Alexopoulou L., Haralambous S., Meyer zum Büschenfelde K.H., Strober W., Kollias G. Predominant pathogenic role of tumor necrosis factor in experimental colitis in mice. Eur. J. Immunol. 1997;27:1743–1750. doi: 10.1002/eji.1830270722. [DOI] [PubMed] [Google Scholar]
- Speiser D.E., Bachmann M.F., Frick T.W., McKall-Faienza K., Griffiths E., Pfeffer K., Mak T.W., Ohashi P.S. TNF receptor p55 controls early acute graft-versus-host disease. J. Immunol. 1997;158:5185–5190. [PubMed] [Google Scholar]
- Faulkner H., Renauld J.C., Van Snick J., Grencis R.K. Interleukin 9 enhances resistance to the intestinal nematode Trichuris muris . Infect. Immun. 1998;66:3832–3840. doi: 10.1128/iai.66.8.3832-3840.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betts C.J., Else K.J. Mast cells, eosinophils and antibody-mediated cellular cytotoxicity are not critical in resistance to Trichuris muris . Parasite Immunol. 1999;21:45–52. doi: 10.1046/j.1365-3024.1999.00200.x. [DOI] [PubMed] [Google Scholar]
- Doukas J., Pober P.S. IFN-γ enhances endothelial activation induced by tumor necrosis factor but not IL-1. J. Immunol. 1990;145:1727–1733. [PubMed] [Google Scholar]
- Osborn L. Leukocyte adhesion to endothelium in inflammation. Cell. 1990;62:3–6. doi: 10.1016/0092-8674(90)90230-c. [DOI] [PubMed] [Google Scholar]
- Maiti A., Maki G., Johnson P. TNF-α induction of CD44-mediated leukocyte adhesion by sulfation. Science. 1998;282:941–943. doi: 10.1126/science.282.5390.941. [DOI] [PubMed] [Google Scholar]
- Ohmori Y., Wyner L., Marumi S., Armstrong D., Stoler M., Hamilton T.A. Tumor necrosis factor-α induces cell type and tissue-specific expression of chemoattractant cytokines in vivo. Am. J. Pathol. 1993;142:861–870. [PMC free article] [PubMed] [Google Scholar]
- Furuta G.T., Schmidt-Choudhury A., Wang M.Y., Sheng-Wang Z., Lu L., Furlano R.I., Wershil B.K. Mast cell-dependent tumor necrosis factor α production participates in allergic gastric inflammation in mice. Gastroenterology. 1997;113:1560–1569. doi: 10.1053/gast.1997.v113.pm9352858. [DOI] [PubMed] [Google Scholar]
- Amiri P., Locksely R.M., Parslow T.G., Sadick M., Rector E., Ritter D., McKerrow J.H. Tumor necrosis factor α restores granulomas and induces parasite-egg laying in schistosome-infected SCID mice. Nature. 1992;356:604–607. doi: 10.1038/356604a0. [DOI] [PubMed] [Google Scholar]
- Joseph A.L., Boros D.L. Tumor necrosis factor plays a role in Schistosoma mansoni egg-induced granulomatous inflammation. J. Immunol. 1993;151:5461–5471. [PubMed] [Google Scholar]
- Chiaramonte M.G., Schopf L.R., Neben T.Y., Cheever A.W., Donaldson D.D., Wynn T.A. IL-13 is a key regulatory cytokine for Th2 cell-mediated pulmonary granuloma formation and IgE responses induced by Schistosoma mansoni eggs. J. Immunol. 1998;162:920–930. [PubMed] [Google Scholar]
- McKenzie G.J., Bancroft A.J., Grencis R.K., McKenzie A.N.J. A distinct role for interleukin-13 in Th2-mediated immune responses. Curr. Biol. 1998;8:339–342. doi: 10.1016/s0960-9822(98)70134-4. [DOI] [PubMed] [Google Scholar]
- Pfeffer K., Matsuyama T., Kundig T.M., Wakeham A., Kishihara K., Shahinian A., Weignmann K., Ohashi P.S., Kronke M., Mak T.W. Mice deficient for the p55 kDa tumor necrosis factor receptor are resistant to endotoxic shock, yet succumb to L. monocytogenes infection. Cell. 1993;73:457–467. doi: 10.1016/0092-8674(93)90134-c. [DOI] [PubMed] [Google Scholar]
- Lucas R., Juillard P., Decoster E., Reder M., Burger D., Donati Y., Giroud C., Monso-Hinard C., De Kesel T., Buurman W.A. Crucial role of TNF receptor 2 and membrane bound TNF in experimental cerebral malaria. Eur. J. Immunol. 1997;27:1719–1725. doi: 10.1002/eji.1830270719. [DOI] [PubMed] [Google Scholar]
- Rothe J., Lesslauer W., Loetscher H., Lang Y., Koebel P., Koentgen F., Althage A., Zinkernagel R., Steinmetz M., Bluethmann H. Mice lacking the tumor necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes . Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Flynn J.L., Goldstein M.M., Chan J., Triebold K.J., Pfeffer K., Lowenstein C.J., Schreiber R., Mak T.W., Bloom B.R. Tumor necrosis factor-α is required for the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–572. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- Yap G.S., Scharton-Kersten T., Charest H., Sher A. Decreased resistance of TNF receptor p55- and p75-deficient mice to chronic toxoplasmosis despite normal activation of inducible nitric oxide synthase in vivo. J. Immunol. 1998;160:1340–1345. [PubMed] [Google Scholar]
- Matsumoto M., Fu Y.X., Molina H., Chaplin D.D. Lymphotoxin-α-deficient mice and TNF receptor-I-deficient mice define development and functional characteristics of germinal centers. Immunol. Rev. 1997;156:137–144. doi: 10.1111/j.1600-065x.1997.tb00965.x. [DOI] [PubMed] [Google Scholar]
- Pasparakis M., Alexopoulou L., Douni E., Kollias G. Tumour necrosis factors in immune regulationeverything that's interesting is…new! Cytokine Growth Factors Rev. 1996;7:223–229. doi: 10.1016/s1359-6101(96)00031-7. [DOI] [PubMed] [Google Scholar]
- Le Hir M., Bluethmann H., Kosco-Vilbois M.H., Muller M., di Padora F., Moore M., Ryffel B., Eugster H.P. Differentiation of follicular dendritic cells and full antibody responses require tumor necrosis factor receptor-1 signaling. J. Exp. Med. 1996;183:2367–2372. doi: 10.1084/jem.183.5.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B., Luz A., Pfeffer K., Holzmann B. Defective Peyer's patch organogenesis in mice lacking the 55-kD receptor for tumor necrosis factor. J. Exp. Med. 1996;184:259–264. doi: 10.1084/jem.184.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D.M., Maraskovsky E., Billingsley W.L., Dougall W.C., Tometsko M.E., Roux E.R., Teepe M.C., DuBose R.F., Cosman D., Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Lardon F., Snoeck H.W., Berneman Z.N., Van Tendeloo V.F., Nijs G., Lenjou M., Henckaerts E., Boeckxtaens C.J., Vandenabeele P., Kestens L.L. Generation of dendritic cells from bone marrow progenitors using GM-CSF, TNF-α, and additional cytokinesantagonistic effects of IL-4 and IFN-γ and selective involvement of TNF-α receptor-1. Immunology. 1997;91:553–559. doi: 10.1046/j.1365-2567.1997.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumberbatch M., Kimber I. Tumour necrosis factor-α is required for accumulation of dendritic cells in draining lymph nodes and for optimal contact sensitization. Immunology. 1995;84:31–35. [PMC free article] [PubMed] [Google Scholar]
- Smith C.A., Farrah T., Goodwin R.G. The TNF receptor superfamily of cellular and viral proteinsactivation, costimulation and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Hunger R.E., Carnaud C., Garcia I., Vassalli P., Mueller C. Prevention of autoimmune diabetes mellitus in NOD mice by transgenic expression of soluble tumor necrosis factor receptor p55. Eur. J. Immunol. 1997;27:255–261. doi: 10.1002/eji.1830270138. [DOI] [PubMed] [Google Scholar]
- Jacob C.O., Aiso S., Schreiber R.D., McDevitt H.O. Monoclonal anti-tumor necrosis factor antibody renders non-obese diabetic mice hypersensitive to irradiation and enhances insulitis development. Int. Immunol. 1992;4:611–614. doi: 10.1093/intimm/4.5.611. [DOI] [PubMed] [Google Scholar]
- Cope A.P., Liblau R.S., Yang X.D., Congia M., Laudanna C., Screiber R.D., Probert L., Kollias G., McDevitt H.O. Chronic tumor necrosis factor alters T cell responses by attenuating T cell receptor signaling. J. Exp. Med. 1997;185:1573–1584. doi: 10.1084/jem.185.9.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph S.B., Miner K.T., Croft M. Augmentation of naive, Th1 and Th2 effector CD4 responses by IL-6, IL-1 and TNF. Eur. J. Immunol. 1998;28:277–289. doi: 10.1002/(SICI)1521-4141(199801)28:01<277::AID-IMMU277>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Lugli S.M., Feng N., Heim M.H., Adam M., Schnyder B., Etter H., Yamage M., Eugster H.P., Lutz R.A., Zurawski G., Moser R. Tumor necrosis factor α enhances the expression of the interleukin (IL)-4 receptor α-chain on endothelial cells increasing IL-4 or IL-13-induced Stat6 activation. J. Biol. Chem. 1997;272:5487–5494. doi: 10.1074/jbc.272.9.5487. [DOI] [PubMed] [Google Scholar]