

Abstract
Optimal T cell differentiation into effector cells with specialized functions requires the participation of cytokine receptor signals. In T helper cells, this process is controlled by chromatin changes and distal and proximal regulatory elements as well as specific transcription factors. Analogous events during cytotoxic T lymphocyte (CTL) differentiation remain to be identified. This process is known, however, to be crucially regulated by interleukin (IL)-2 receptor (R) signals. It is accompanied by the induction of perforin expression via a mechanism that does not entail proximal regulatory elements. In this report, transgenically expressed human perforin gene locus DNAs demonstrate that IL-2R signals target two IL-2–dependent enhancers ∼15 and 1 kilobase upstream of the promoter. The most distal enhancer may also respond to TCR signals. In transient transfections, both enhancers required two identically spaced Stat-like elements for their activation, which was abolished by expression of a dominant negative signal transducer and activator of transcription (Stat)5 molecule, whereas a constitutively active Stat5 molecule bypassed the requirement for IL-2R signals. These results provide a molecular explanation for the activation of the perforin gene during CTL differentiation and complement the analysis of animals deficient in the activation of the IL-2R Stat signaling pathway by establishing perforin as a target gene.
Keywords: cytotoxic T lymphocyte, IL-2 receptor, T cell activation, perforin, transgenic mouse
Recognition of intracellular pathogens by the immune system leads to the differentiation of peripheral CD8+ T cells into effector CTLs with lytic capacity. This process involves signals from both the TCR and cytokine receptors. TCR signals assure the specificity of a CTL response. Cytokine signals not only sustain the proliferation of CTLs but also induce and fine tune the expression of genes required for the cytotoxic function, such as the expression of perforin.
Perforin is an inducible component of the lytic machinery of CTLs 1 2 3 and thus part of the adaptive immune system. As a constitutive component of NK cells 2, it is also part of the innate immune system. The release of perforin onto target cells results in the formation of large transmembrane channels 4. The pore formation may be lytic on its own, or it can provide conduits for other effector molecules, such as granzymes 5. Despite other cytotoxic mechanisms 6, perforin expression plays a nonredundant role for the clearance of certain noncytopathic viral infections 7 8. It also participates in the clearance of infections with intracellular bacteria 9 and in the surveillance of emerging tumors and the rejection of established tumors 10.
Perforin expression is undetectable in naive CD8+ cells, but it is upregulated in virtually all CTLs of the effector phenotype 11. Perforin is also expressed by some activated CD4+ T cells 12 13. The concise activation requirements for perforin induction in CD4+ T cells and their biological role in vivo remain to be clarified. On the other hand, IL-2 and IL-15 have been shown to directly induce perforin gene expression in CD8+ T cells 1 2 3 14 15. Both cytokines lead to identical signals because their receptors are comprised of identical signaling chains, namely IL-2Rβ and IL-2Rγ 16. It is important to emphasize that naive CD8+ T cells express both chains, in contrast to naive CD4+ T cells, which lack detectable levels of IL-2Rβ 17. This differential expression of IL-2Rβ provides one molecular explanation as to why naive CD8+ cells, but not naive CD4+ cells, are able to respond to high doses of IL-2 without prior TCR signals 13. The important role of IL-2R or IL-15R signals for the generation of effector CTLs in vivo is consistent with the inability of IL-2Rβ–deficient animals to generate virus-specific effector CTLs 18. Hence, the molecular characterization of the inducible expression of perforin by IL-2R signals may offer insights into the activation/differentiation processes of CTLs.
Previous investigations by several laboratories have focused on transcriptional events mediated by the mouse perforin promoter. Using different approaches, three distinct mechanisms were implied: Cooperation between Sp-1 and Ets-related transcription factors acting on the proximal promoter 19; an effector CTL–specific posttranslational modification of a ubiquitous Ets-related transcription factor binding to an Ets-consensus in the distal promoter 20 21; and gene derepression by two distal promoter elements and two nuclear proteins expressed by noncytolytic cells 22 23. These mechanisms involving the perforin promoter may complement each other, providing a reasonable explanation for the T and NK cell–restricted expression of perforin in vivo in normal cells, based on the analysis of the perforin promoter in transgenic mice 24. Unexpectedly, however, they could not provide insight into how perforin expression is induced upon T cell activation, because transgene expression by perforin promoter–transgenic mice is constitutive and not upregulated by IL-2 or other T cell activation signals 24. Our study was undertaken to reveal the molecular basis for how IL-2R signaling regulates the perforin gene.
Materials and Methods
Identification and Characterization of SAM-19 Cells.
SAM-19 cells were derived from the autonomously growing mouse CTL/rat thymoma hybrid PC60 25 that cannot be activated by IL-2 on its own to express perforin 26. We noticed a weak IL-2 response of the perforin gene in a subline designated SA. Further analysis indicated that the SA line, but not the other lines, contained cells constitutively expressing the mouse IL-2Rβ chain. Constitutive expression of mouse IL-2Rγ was equivalent in all lines. After cloning these cells at the single-cell level, one clone, SAM-19, was obtained that expressed IL-2Rβ mRNA at levels equivalent to those in the CTL effector–like CTLL-2 line. Neither resting nor IL-2–stimulated cells expressed detectable levels of mouse IL-2Rα protein or mRNA. As described for T cells exclusively expressing IL-2Rβ and IL-2Rγ 27, SAM-19 responded to recombinant mouse IL-2 at concentrations >100 U/ml with the induction of the mouse perforin and granzyme B genes. Half-maximal levels of perforin mRNA were detected after ∼14 h of induction. Maximum levels were induced after ∼48 h. Cell counts of SAM-19 cultures maintained in the presence of 500 U/ml IL-2 versus its absence did not significantly differ from each other, suggesting that IL-2R signaling has little or no effect on the growth and survival of SAM-19. The cells were maintained in IMDM supplemented with 10% FBS, 5 × 10−5 M β-ME, and 50 μg/ml gentamycin.
Northern Blot and Nuclear Run-On Analysis.
Total RNA was extracted and analyzed as described 28. Purification and RNase treatment of nuclei as well as the elongation reactions and their purifications were performed as described 29 using ∼2.5 × 107 nuclei and 240 μCi of α-[32P]UTP (800 Ci/mmol) per reaction (15 min at 30°C). Hybridizations were carried out with 1.6 × 107/ml TCA-precipitable counts for 36 h at 65°C in 10 mM Tris, pH 7.5, 10 mM EDTA, pH7.5, 0.3 M NaCl, 1% SDS, 250 μg/ml tRNA, 1× Denhardt's solution, and 0.5% nonfat dry milk. Filter washes, including an RNase A digestion, have been described 29. Target probes were obtained by PCR of genomic DNA and cloned as follows. The β2-microglobulin probe (nucleotides 3,942–4,384 of sequence available from EMBL/GenBank/DDBJ under accession number M18837) and the 5S ribosomal RNA probe (nucleotides 268–449 of sequence available from EMBL/GenBank/DDBJ under accession number X51545) were cloned into pCRII (Invitrogen Corp.). The perforin 5′ probe containing the short first exon and the beginning of the first intron (nucleotides 830–1,156 available from EMBL/GenBank/DDBJ under accession number M95527) and the perforin 3′ probe containing part of the third exon (nucleotides 101–464 of sequence available from EMBL/GenBank/DDBJ under accession number X51446) were cloned into M13 mp18, and single-stranded phage DNA containing perforin antisense DNA was produced. 10 μg denatured DNA was slot blotted for each target. All constructs were confirmed by sequencing.
Human Genomic Perforin Clones and Reporter Gene Constructs.
The two genomic clones depicted (see Fig. 2) were obtained by screening a commercially available human placenta pWE15 cosmid library (Stratagene Inc.) on duplicate filters with a human perforin exon III probe and a human perforin promoter probe. They were further analyzed by restriction mapping and Southern hybridization to these two probes. All analyzed fragments matched the results of genomic Southern blots. Also, the sequence of a promoter fragment of both clones was identical to the one we reported previously 30. The initial reporter gene construct was assembled by cloning a KpnI–SfiI fragment (−15,600 to −277) into the promoterless pGL3 basic reporter vector (Promega Corp.), in which the remaining human perforin promoter, exon I, intron I, and the untranslated sequences of exon II had been fused to the firefly luciferase gene. Constructs without the first intron used an EcoRV site at +59. The progressive deletion constructs (see Fig. 3) were obtained by exonuclease treatment of a KpnI (−15,600)- and HindIII (−13,300)-digested plasmid, followed by Klenow and T4 DNA polymerase treatment, gel purification, and religation. All constructs were transformed by electroporation into DH10B cells (Life Technologies). Other deletions, including internal deletions, were created by restriction enzyme digestion followed by T4 DNA polymerase treatment and religation of the vector. The analysis of the upstream enhancer (see Fig. 4 B) also involved the cloning of PCR fragments, all of which were verified by sequencing both strands of the enhancer. Mutations of the signal transducer and activator of transcription (Stat)1 elements were introduced using the QuickChange site-directed mutagenesis kit from Stratagene Inc. After verification of the sequences on both strands of the enhancers, they were recloned into a fresh vector to eliminate second-site mutations outside of the sequenced DNA. The dominant negative Stat5a expression vector was generated similarly.
Figure 2.

Structure and cis-acting function of transgenic human perforin genes in SAM-19 cells. The human perforin gene locus consisting of three exons (reference 30) is depicted at top (UT, untranslated sequence). Below are the genomic DNAs and chimeric reporter constructs whose expression was analyzed before and after overnight stimulation with IL-2 in the indicated number of stably transfected, independent clones. Expression of the human perforin gene was analyzed by Northern blot, and a representative is shown for each genomic DNA. Note that the human perforin probe did not crosshybridize with the endogenous SAM-19 murine mRNA under our experimental conditions. Values under inducibility represent the mean and standard deviation after correction of the densitometrically quantitated Northern blot data for G3PDH or respectively after correction of the luciferase activity for the protein content of the extracts. Three clones did not express human perforin detectable by Northern blot analysis (two clones for the –16,500 DNA and one clone for the −20,700 DNA). They were excluded from the statistical analysis.
Figure 3.

Identification of two IL-2–responsive regulatory domains in the human perforin 5′ flank. (A) Transient transfection analysis of 5′ progressive and internal deletions. The genomic DNAs depicted at left were used to control firefly luciferase expression in SAM-19 that were split 1–3 h after electroporation and cultured overnight for 15–17 h with and without IL-2. The data are given as the ratio of reporter activity of activated versus unactivated cells and represent the averages and standard deviations of the indicated numbers of independent transfections. 20 μg of DNA was used, comprising a 12:1 molar excess of the experimental plasmid over an internal control vector. Its CMV promoter–driven renilla luciferase expression, which did not respond significantly to IL-2 in SAM-19, served to correct for sample differences. In the absence of IL-2, the normalized expression of all perforin promoter–bearing constructs was 10–40 times the levels obtained from the promoterless construct, with no reproducible correlation to any particular construct. (B) Further characterization of the far-upstream regulatory DNA. Transient transfection analysis of the depicted DNAs in the context of the perforin promoter (−277 to +59) were performed as described for A, with the exception of electroporating an 18:1 molar ratio of the experimental plasmids to the internal control vector.
Figure 4.

IL-2–dependent, enhancer-like activities of the far-upstream (A) and upstream (B) regulatory DNA. Fragments of the human perforin gene locus identified in Fig. 3 were cloned upstream of an SV40 promoter. The data summarize four independent transient transfections of SAM-19 using 20 μg of DNA comprising an 18:1 molar excess of the experimental plasmid over the internal control vector as described in Fig. 3. The expression of the experimental reporter in the absence or presence of IL-2R signals is given after it was normalized to that derived from the control reporter to account for variations in the transfection efficiency and sample differences. These data were used to calculate the induction.
Stable and Transient Expression Studies.
Vector backbone–free DNA for stable transfections employed NotI sites flanking the inserts of the pWE15 cosmid clones or the NotI and SalI sites of the pGL3 reporter vectors. For stable transfections, an ∼5:1 molar excess of the perforin transgene over a selection cartridge containing a TK promoter driving the neomycin gene in a total of 40 μg DNA was transfected into 400 μl of 2.5 × 107/ml SAM-19 in DMEM supplemented with 4.5 g/liter glucose and 25 mM Hepes by electroporation of a 4-mm gap cuvette with an ECM600 system (BTX) set to 230 V, 3,000 μF, and 24 Ω. The washed cells were cultured for 15–17 h in 2 ml of complete growth medium, washed again, and selected as 2 × 104/ml live cells (1.0 ml per well in a 48-well plate) in the presence of 1.3 mg/ml active G418 (Life Technologies). 2–3 wk later, G418-resistant clones were expanded and screened by PCR for the presence of human perforin promoter sequences. PCR-positive clones were further analyzed by genomic Southern blot of restriction-digested DNA side-by-side with digested human genomic DNA to determine the integrity of the transgenic constructs and to estimate the copy numbers of the transgene. Transgenic mice, generated as we described previously 24, were analyzed accordingly.
Electroporation conditions for transient transfections were identical but used less DNA (indicated in the figure legends) and included an internal control plasmid, pRL-CMV (Promega Corp.) in which a CMV promoter drives the renilla luciferase gene. After the electroporated cells were washed, they were split into two 500-μl aliquots (three aliquots for activations using pharmacological agents), rested for 1–3 h, and then supplemented with 500 μl of medium with or without recombinant mouse IL-2 (and/or pharmacological agents), providing a final concentration of 600 U/ml. After overnight incubation for 15–17 h, the cells were washed with PBS and lysed and extracted in 50–80 μl passive lysis buffer (Promega Corp.) by two rounds of freeze thawing. Reporter gene activities were determined in three 10-μl aliquots of each extract using the dual luciferase assay system from Promega Corp. and a ML2250 96-well plate luminometer (Dynex Technologies). Signals were integrated for 10 s for both luciferase activities. The ratios of firefly luciferase activity to renilla luciferase activity varied by <5% in the triplicate measurements. Their average was used to represent the analysis of each independently transfected sample.
Results
Perforin Gene Regulation in the SAM-19 Model.
An important limitation in the study of antigen- and/or growth factor–dependent CTL clones is that they downregulate their overall RNA and protein synthesis and begin to apoptose when deprived of stimuli. In fact, the time periods required to “downregulate” the perforin gene to study its “activation” considerably overlap with this generalized cell shutdown. Therefore, we established a novel CTL tissue culture model, designated SAM-19, to facilitate the study of perforin gene induction by IL-2R signaling. SAM-19 is an autonomously growing, growth factor–independent clone derived from a mouse CTL–rat thymoma hybrid (see Materials and Methods). These cells respond to high doses of mouse IL-2 with the induction of mouse perforin and granzyme B mRNAs with kinetics similar to those observed in primary cells 3. The induction of the perforin gene by IL-2 did not depend on newly synthesized proteins (Fig. 1 A), and the mRNA induction was not regulated at the posttranscriptional level (data not shown), properties analogous to those of primary CTLs 3. In addition, perforin mRNA induction in SAM-19 was not blocked by rapamycin (Fig. 1 B), as reported for primary cells 31. Taken together, SAM-19 appears to comprise a reasonable in vitro model with which to study perforin gene regulation by IL-2R signals.
Figure 1.
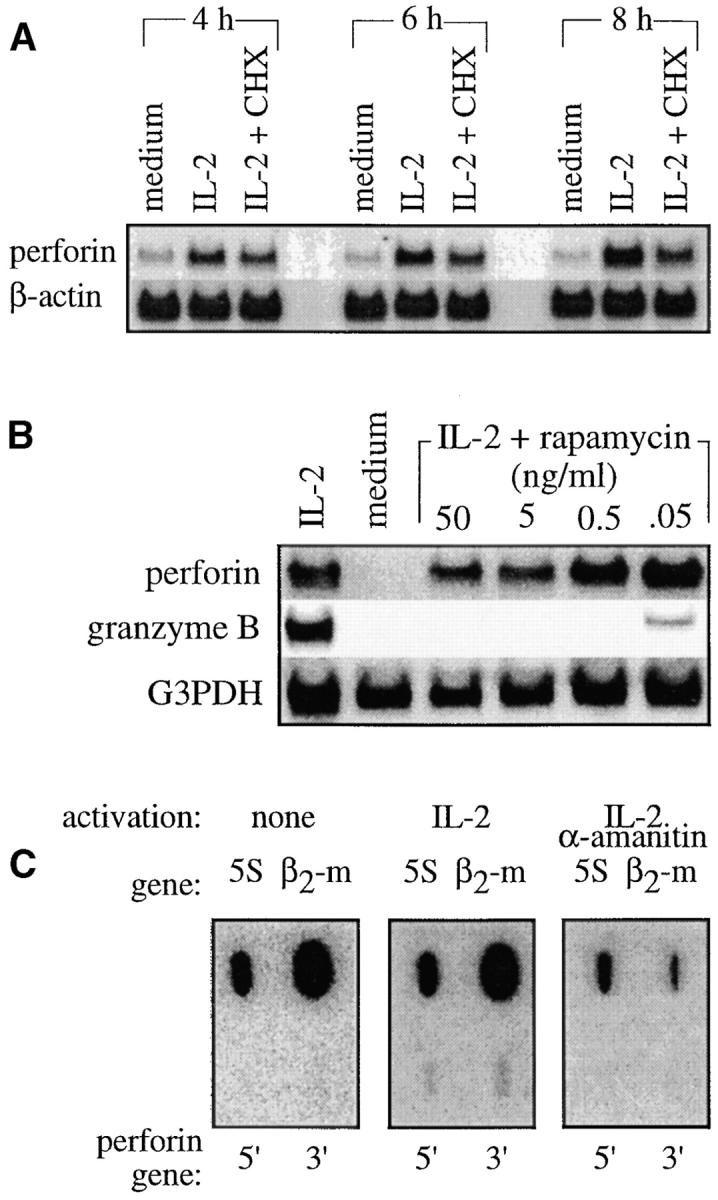
Regulation of mouse perforin mRNA induction by IL-2 in SAM-19. (A) Perforin mRNA induction does not require newly synthesized proteins. SAM-19 cells were activated for the indicated times with 1,000 U/ml IL-2 in the presence or absence of 40 μM cycloheximide (CHX), which blocked >95% of protein synthesis (data not shown). Total RNA was analyzed by Northern blot analysis. (B) Perforin mRNA induction is not inhibited by rapamycin. Total RNA was extracted from SAM-19 activated for 24 h with 1,000 U/ml IL-2 in the presence of the indicated concentrations of rapamycin and analyzed by Northern blot analysis. G3PDH denotes the glyceraldehyde-3-phosphate dehydrogenase mRNA. (C) Perforin mRNA induction occurs at the level of transcription initiation. Nuclei of SAM-19 cells maintained in medium versus 1,000 U/ml IL-2 for 24 h were analyzed by run-on assays in both the absence and presence of 1 μg/ml α-amanitin, which was included to document that transcripts detected for perforin (5′ vs. 3′ end of the transcribed gene; see Materials and Methods) and β2-microglobulin (β2-m) but not for the 5S ribosomal RNA gene (5S; transcribed by RNA polymerase III) were elongated by RNA polymerase II. Identical numbers of incorporated counts were hybridized to each filter.
Before undertaking more detailed studies, we considered the possibility that IL-2–unresponsive constitutive expression by the mouse perforin promoter in transgenic mice 24 could have been due to a regulation at the level of transcription elongation. As assayed by nuclear run-on analysis, however, transcription of the 5′ and 3′ ends of the perforin gene were induced by IL-2 to similar levels (Fig. 1 C). These data indicate that IL-2R signals regulate the perforin gene at the level of transcription initiation via regulatory domains other than its promoter.
Transgenic Perforin Gene Regulation in SAM-19 and in Primary T Cells.
An expression screening strategy was used to identify putative regulatory domains other than the promoter. This approach was facilitated by the compact dimension of the human perforin gene (Fig. 2), the ability to distinguish the human and the mouse perforin mRNAs from each other, and the transfectability of SAM-19. In initial experiments, two suitable human gene locus DNAs from a cosmid library were stably transfected into SAM-19. The expression of both the −16,500 and −20,700 DNAs was regulated by IL-2 in several independent clones (Fig. 2). This suggested that these DNAs contained IL-2–responsive cis-acting sequences in their overlapping regions. Further efforts focused on their 5′ flanks, because we failed to detect transcriptionally relevant DNA in transient transfections of the intragenic DNA and the 3′ flank of the −20,700 gene locus DNA. Indeed, a restriction fragment comprising most of the cloned 5′ flank fragment also drove a transgenic luciferase reporter gene in an IL-2–responsive manner (Fig. 2, −15,600), in contrast to a promoter construct (−277). Interestingly, an intermediate construct (−5,300) responded at intermediate levels, which was shown in further investigations to be due to the presence of two regulatory domains rather than one. Unlike the inducibility of the transgenes, their levels of expression in relation to the transgenic copy numbers varied considerably in all clones, suggesting that these DNAs do not contain additional constitutive enhancers, silencers, or a locus control region.
To extend the results from the stable transfections of SAM-19 to normal T cells, a −15,600 to +59 perforin/luciferase construct was analyzed in transgenic mice. Two founders did not express luciferase activity at all, neither in resting nor activated peripheral lymphocytes. The other two founders expressed the transgene, with its expression regulated by IL-2R as well as TCR signals, including the cross-linking of CD3 or PMA plus ionomycin (Table ). The rates of induction for luciferase protein observed in the transgenic mice were higher than those in the SAM-19 system. Based on our experience with a transgenic CD4 reporter gene 24, this may reflect a global increase in the translation efficiency upon activation of freshly obtained primary T cells. More importantly and unlike the previously investigated promoter transgenes, the −15,600 to +59 perforin/luciferase-transgenic T cells clearly responded to activation signals.
Table 1.
The Human Perforin 5′ Flank (−15,600 to +59) Promotes Regulated Reporter Expression in Transgenic Mice
| Time | Medium | P | P + I | TCR | IL-2 |
|---|---|---|---|---|---|
| h | |||||
| 0 | 1 (standard) | – | – | – | – |
| 24 | 1 | 3 | 19 | 21 | 6 |
| 48 | 1 | 1 | 14 | 16 | 18 |
Splenic T cells of transgenic offspring (eight copies) were purified by magnetic microparticles conjugated to Thy1.2 mAb (MiniMACS™ system; Miltenyi Biotec). Luciferase was extracted from 106 purified cells (>95% CD3+ and ∼40% CD8+) immediately and after culture for 24 and 48 h. Activations involved 10 ng/ml PMA (P) plus 1 μM ionomycin (P + I), cross-linking of CD3 (TCR; plate-bound 2C11 mAb), and 1,000 U/ml IL-2 (IL-2). Aliquots were measured in duplicate, and their average was used to calculate the fold inductions in reference to the activity obtained from cells prior to their in vitro culture (set as 1).
Characterization of Two IL-2–responsive Enhancers.
To address exactly where the IL-2–responsive cis-acting DNA was located, progressive deletions of the construct analyzed in transgenic mice were assayed in transient transfections. A consistent pattern for their IL-2 responses was noted (Fig. 3 A). The most 5′ deletion (from −15,600 to −13,300) resulted in an impaired IL-2 response. This response was abolished once sequences downstream of −1,450 were deleted. This finding suggested the presence of two cis-acting DNAs, consistent with the intermediate levels of response seen previously in the stable transfections of the reporter construct of intermediate length (Fig. 2). This interpretation was also consistent with the reporter analysis of internal deletions (Fig. 3 A, bottom). The deletion of the sequences residing between the two putative domains as well as the spacing between the two domains did not impair the levels of regulation observed by the entire 5′ flank. Finally, the far-upstream regulatory domain on its own also conferred an IL-2 response to the unresponsive perforin promoter (Fig. 3 A, last construct). Both regulatory domains required the context of a promoter (data not shown), indicating that neither comprised a second promoter. The responses by each individual domain appeared to be additive, suggesting that their functions were independent of each other within the experimental context.
The far-upstream regulatory DNA identified in the transient transfections comprised the very end of the −15,600 construct, whereas both of the original gene locus DNAs extended farther upstream and appeared to respond somewhat more to IL-2 (Fig. 2). Therefore, the remainder of the cloned DNA was analyzed to delineate the 5′ and 3′ borders of the far-upstream regulatory DNA (Fig. 3 B). This led to an NsiI–BalI fragment that retained the maximal response of the far-upstream regulatory DNA (Fig. 3 B, bottom). This fragment also functioned in the context of the heterologous SV40 promoter (Fig. 4 A). IL-2 increased the transcriptional activity of the constructs over the promoter levels irrespective of the orientation of the regulatory DNA. These attributes were consistent with an IL-2–inducible enhancer, whose required core was contained within ∼150 bp (Fig. 4 A, bottom). The analogous experiments for the upstream regulatory domain recapitulated these findings and localized this enhancer within ∼130 bp (Fig. 4 B).
Second Messenger Pathways Involved in the Activation of the Perforin Enhancers.
Consistent with the rapamycin insensitivity of perforin mRNA induction by IL-2 (Fig. 1 B), the activation of the enhancers was not blocked by rapamycin (Fig. 5 A) at concentrations that abolished granzyme B induction (Fig. 1 B).
Figure 5.

Second messenger pathway requirements for activation of the perforin enhancers. (A) Enhancer activation by IL-2R signals is not blocked by rapamycin. SAM-19 cells were activated in both the presence and absence of 0.5 ng/ml rapamycin, which was shown to abrogate granzyme B but not perforin mRNA induction (Fig. 1 B). The data summarize four independent transient transfections of 20 μg of DNA comprising an 18:1 molar excess of the minimal enhancer SV40 reporter vectors (Fig. 3) over the internal control vector. (B) Selective activation of the far-upstream enhancer by PMA and ionomycin in SAM-19 CTLs but not in J558L B cells nor L929 fibroblasts. The indicated cells were activated with 10 ng/ml PMA plus 1 μM ionomycin (P + I) in the absence or presence of 200 ng/ml CsA. The data summarize three independent transient transfections (four for SAM-19) of 8 μg of DNA comprising a 10:1 molar excess of the minimal enhancer SV40 plasmids over the internal control vector, as described for Fig. 4. The data are expressed in relation to the activity of the promoterless reporter vector, whose normalized expression in media alone was set as one for each individual cell line. The data for the activated cells were corrected for their protein content rather than expression of the cotransfected marker, because the CMV promoter responded to PMA and ionomycin. (C) Activation of the perforin enhancers by IL-2 is blocked by coexpression of a dominant negative Stat5 molecule. The data summarize three independent transient transfections of SAM-19 using 15 μg of DNA comprising the tabulated molar ratios of the indicated expression vectors to the minimal enhancer SV40 plasmids (Fig. 3). The two CMV promoter–driven expression constructs are identical, except that amino acid 750 of murine Stat5a had been mutated into a stop codon to generate a dominant negative molecule as described 32. Transfected cells were split and cultured in the absence or presence of IL-2. Reporter expression was quantitated and corrected for the protein content of the samples.
Because perforin mRNA can also be induced in primary CTLs by TCR signals 2, SAM-19 was activated with pharmacological agents known to mimic TCR signals, namely phorbol ester and ionomycin. These agents induce perforin mRNA in SAM-19 in the absence of detectable IL-2 (data not shown). Regarding the involvement of the identified enhancers in this process, the far-upstream enhancer, but not the upstream enhancer, was significantly activated in SAM-19 (Fig. 5 B, top). This activation was sensitive to cyclosporin A (CsA),1 indicating that calcineurin participated in the activation of the enhancer. These findings suggest that the far-upstream enhancer may also respond to TCR signals.
The T and NK cell–restricted expression of perforin raised the additional question of whether activation of the enhancers by pharmacological agents might be lineage specific. There was no substantial activation of either enhancer in the J588L B cell or the L929 fibroblast model (Fig. 5 B, center and bottom). These data suggest that the enhancers can be activated primarily in T cells.
Lastly, participation of the Jak/Stat signaling pathway from the IL-2R was investigated, because both enhancers contained Stat-like elements. Coexpression of a dominant negative signal transducer and activator of transcription (Stat)5 molecule 32 blocked the activation of both enhancers by IL-2R signals in a dose-dependent manner, in contrast to the cotransfections with the parental wild-type Stat5 expression vector (Fig. 5 C). These data imply that the activation of Stat5 plays an important direct or indirect role in the activation of the perforin enhancers by IL-2R signals.
Both Enhancers Contain Functionally Important Tandem Stat-like Elements and Can Be Transactivated by Stat5.
Visual and computer-aided inspection 33 of the sequenced enhancer cores revealed several similarities to known binding sites or their cores (Fig. 6 A). Potential binding sites present in both enhancers at similar positions included two Ets cores, an activator protein (AP)-1 site and, most strikingly, two identically spaced Stat-like elements. One closely resembled the consensus dyad symmetry (Fig. 6 A, STAT). The other element was less well conserved (Fig. 6, Fig. 3′ STAT-n). A third less well conserved Stat-like element occurred only in the far-upstream enhancer (Fig. 6, Fig. 5′ STAT-n) and overlapped with the highly conserved element (STAT).
Figure 6.

Both perforin enhancers contain tandem Stat-like elements that are required for their IL-2 response and transactivation by Stat5. (A) Sequence and putative regulatory elements of the perforin enhancers. Names next to the boxed residues indicate either a potential transcription factor or the name of the core binding sequence. Boxes extending through the middle indicate potential binding sites that are present in both enhancers. Asterisks indicate the mutations functionally analyzed in B and C. TTTC of the element indicated as 5′ STAT-n was changed to CGCT; all other mutations involved A↔C and G↔T substitutions. The sequence data are available from EMBL/GenBank/DDBJ under accession numbers AF152113 (far-upstream enhancer) and M31951 (upstream enhancer; nucleotides 311–427 of the deposited sequence). (B) Activation of the upstream enhancer (left) and the far-upstream enhancer (right) by IL-2 and a constitutively active, chimeric Stat5 molecule. The data summarize four independent transfections of SAM-19 using 22 μg of DNA comprising a 1:2:0.25 molar ratio of the SV40 promoter vector with or without (none) the minimal enhancers shown in A, the indicated expression vector, and the internal control vector. STAT5-VP16-JAK2 is a CMV promoter–driven, constitutively active Stat5 molecule. It denotes a fusion of amino acids 1–750 of sheep Stat5 (i.e., Stat5 lacking its endogenous transactivation domain) to the VP16 transactivation domain and the kinase domain of Jak2 34. The assayed mutations of each enhancer are indicated in A.
To determine whether these elements were indeed of functional relevance, mutant enhancers were analyzed. Minimal mutations were designed to selectively interfere with the potential Stat elements (Fig. 6 A, asterisks). Regarding both enhancers, a mutation of their 3′ STAT-n sites reduced their transcriptional activation by IL-2 nearly to the levels of unactivated cells (compare data in Fig. 6 B for constructs cotransfected with an empty expression vector). A mutation of their STAT sites or both sites together (double mutant) completely abolished their enhancer function. In contrast, mutation of the 5′ STAT-n site present only in the far-upstream enhancer did not impair enhancer activation by IL-2 (Fig. 6 B). These results demonstrate a cooperative requirement for both the STAT and the 3′ STAT-n site in each enhancer. Combined with the observed inhibition of enhancer activation by a dominant negative Stat5 molecule, these results suggest that Stat molecules may directly target these elements.
To address whether Stat molecules could transactivate the enhancers via the putative elements, a constitutively active Stat5 molecule was coexpressed with the perforin enhancer reporter constructs. We focused on Stat5 rather than Stat3 because we could not detect the activation of Stat3 in SAM-19 upon IL-2R signaling under conditions where activated Stat5 molecules were readily detectable (data not shown). To that end, a chimeric Stat5-VP16-Jak2 molecule that is autoactivated and leads to Stat5-specific DNA binding and strong transactivation was used 34. Cotransfection with the respective expression vector, but not the empty expression vector, strongly hyperactivated the wild-type enhancer, but not the SV40 promoter, in the absence of IL-2 (Fig. 6 B). Regarding the upstream enhancer, the transactivation was absolutely dependent on the intact STAT site and to a lesser extent on the 3′ STAT-n site (Fig. 6 B, left). Additional IL-2R signaling provided somewhat higher levels, suggesting that additional transcription factors were induced. The analogous transactivation studies of the far-upstream enhancer (Fig. 6 B, right) were complicated by its additional 5′ STAT-n site. As described above, this site was physiologically irrelevant because its mutation did not at all impair the IL-2 response. It may have served, however, as an alternative binding site for Stat5-VP16-Jak2 when the partially overlapping STAT site had been mutated, because all three sites had to be mutated to completely abolish the transactivation (triple mutant). Regardless, the data presented for both enhancers strongly suggest that their activation by IL-2R signals is dependent on the binding of activated Stat molecules to a tandem element, which may be comprised of a higher affinity site (STAT) and a lower affinity site (3′ STAT-n).
Discussion
Scrutiny of the regulation of the perforin gene is biologically important and offers a valuable model to shed molecular light on the activation/differentiation of CTLs. This line of investigation comprises a largely unexplored area in comparison to analogous investigations of cytokine genes, which have become widely used as a paradigm toward an understanding of Th activation/differentiation. In our study toward deciphering the control of perforin expression by IL-2, >45 kb of the human perforin gene locus was surveyed in a transgenic tissue culture system (Fig. 2) that mimics important aspects of perforin gene induction in primary CTLs (Fig. 1). This analysis led to two IL-2–responsive enhancers in the perforin gene locus (Fig. 3 and Fig. 4), which also promoted inducibility in T cells derived from transgenic mice, at least in the context of their intervening genomic sequences (Table ). The activation of both enhancers by IL-2 required not only the activation of the Stat pathway (Fig. 5 C) but also the participation of tandem Stat-like elements that could be transactivated by Stat5 molecules in our CTL differentiation model (Fig. 6). These findings suggest that the activation of Stats by IL-2R signals, in particular the activation of Stat5 molecules, plays an important role for the generation of CTLs.
Regulation of Perforin Gene Expression and Cytotoxicity by Stat5.
The regulation of the perforin gene by IL-2R signals via Stat proteins is consistent with several observations besides the well documented activation of Stat5 and Stat3 by IL-2R signals 35. Stat proteins are latent transcription factors 36 and, accordingly, the onset of perforin mRNA induction by IL-2 does not require newly synthesized proteins in our model cell line (Fig. 1 A) nor in primary cells 3. Similarly, the activation of Stat5 37, the induction of perforin mRNA (Fig. 1 B), and the activation of its enhancers (Fig. 5 A), as well as perforin mRNA induction by IL-2 in primary cells 31, are all resistant to rapamycin. Conversely, TGF-β, which has been suggested to block the activation of the Jak/Stat pathway in T cells, including the activation of Stat5 38 39, also prevents the induction of perforin by IL-2 40.
Importantly, the analysis of Stat5 knockout animals, as well as the phenotypes of animals lacking the Stat docking sites of IL-2Rβ, have recently indicated that this IL-2R signaling pathway may target the perforin gene in vivo and may be essential for the generation of cytotoxicity. Stat5 exists in humans and mice as two closely related genes with overlapping expression patterns and, therefore, has an often redundant role in vivo for both nonlymphoid and lymphoid tissues 41 42. Nevertheless, immunologically relevant phenotypes, albeit perhaps rather discrete, have been reported for certain single-deficient animals. Stat5a-deficient animals fail to upregulate IL-2Rα in response to IL-2R signals 43. Stat5b-deficient splenocytes, on the other hand, poorly generate CTLs in response to IL-2 or IL-15. This defect, which is accompanied by profoundly reduced but still detectable levels of perforin mRNA, led to the proposal that perforin is a Stat5-regulated gene 44. This notion is supported by our results. Similarly, splenocytes of mice expressing an IL-2Rβ lacking the Stat docking sites fail to generate CTLs in response to IL-2 45.
Redundancy of Stats Activated by IL-2R Signaling.
The redundancy of Stat5a and Stat5b may relate to the incomplete block of perforin gene induction by IL-2 or IL-15 in Stat5b-deficient splenocytes inasmuch as Stat5a-deficient cells also expressed slightly reduced levels of perforin 44. The inability of double-deficient T cells to enter the cell cycle 42 does not allow us to readily address this issue experimentally due to the relatively long stimulations required for significant inductions. It is also possible that Stat5 molecules are facilitators rather than the essential players for perforin gene activation or that they are redundantly used with Stat3 molecules. Whereas the latter pathway was not functional in our SAM-19 model (see Results), another report published while this manuscript was in preparation indicates that the upstream enhancer of the perforin gene could also be activated by Stat3 molecules 46. The authors identified a constitutively enhancing fragment similar to the upstream enhancer of our analysis by transient transfections of ∼1,400 bp of the 5′ flank into YT cells, a constitutively perforin-expressing NK cell–like lymphoma. The function of this DNA in YT cells was dependent on what is referred to in Fig. 6 as the STAT element, whereas the 3′ STAT-n site was not investigated. Regardless, the STAT element was shown to bind constitutively activated Stat3 molecules present in the YT lymphoma and Stat5 molecules when extracts of primary NK cells exposed to IL-2 were applied, suggesting that Stat5 and Stat3 molecules may regulate the upstream enhancer of the perforin gene. It is conceivable that genes are regulated redundantly or activation stage specifically by Stat5a, Stat5b, and Stat3 molecules in T cells, because an impaired upregulation of IL-2Rα in response to IL-2R signals has been described not only for Stat5-deficient animals 42 43 but also for Stat3-deficient T cells 47.
Role of Stat5 for Lymphoid Lineages Constitutively Expressing Perforin.
Notably, Stat5 proteins may also play an essential role in the development and/or maintenance of lineages that constitutively express perforin, i.e., NK cells 2 and γ/δ T cells 48 49 50. NK cells are absent in animals deficient in both Stat5 proteins 42, as well as in animals deficient in the ability to activate Stats in response to IL-2R or IL-15R signals 45. The latter animals also lack γ/δ intraepithelial lymphocytes, a lineage that so far has not been investigated in the Stat5 double-deficient animals. The biological relevance of Stat5 molecules and the identified enhancers for the constitutive expression of perforin by these lineages remains to be analyzed. A constitutively active Stat signaling pathway in NK cells in vivo, and thereby also an activation of the perforin gene by the identified enhancers, could be envisioned based on the expression of IL-15 in virtually all tissues and the absolute requirement of IL-15R signals for NK development and/or maintenance 51 52.
Similar Organization of the Perforin and IL-2Rα Enhancers.
The perforin enhancers and the enhancer of the human and mouse IL-2Rα genes 53 54 55 56 57 are presently the only suspected Stat5 and Stat3 targets in vivo among genes specifically expressed by lymphocytes. Interestingly, they are also similarly organized. Just like the perforin enhancers, the IL-2Rα enhancer requires a tandem Stat element that serves as a composite binding site for a tetrameric Stat5 complex binding individual sites of weaker affinity 55 57. We are presently addressing whether the perforin sites, each of which can bind Stat5 in vitro (Schindler, U., and M.G. Lichtenheld, unpublished observation), also serve as targets for a tetrameric Stat5 complex, because the spacing of the Stat-like elements in the perforin enhancers exceeds the spacing of those in the IL-2Rα enhancers (17 vs. 11 bp). The loss of enhancer function by an individual mutation of either site (Fig. 6 B) is consistent with a tetrameric assembly, but these experiments do not address the mechanism for the observed cooperation. The other element with a major function in the mouse and human IL-2Rα enhancers entails an Elf-1–binding Ets core located 14 bp 3′ of their tandem Stat elements 54 56. This organization is exactly like that of the far-upstream enhancer of the perforin gene (Fig. 6 A). An analogous element in the upstream enhancer is located 10 bp 3′ of the tandem Stat-like elements. The STAT sites of both perforin enhancers as well as the 3′ STAT-n site of the upstream enhancer incorporate an Ets binding motif sequence core similar to that of the human IL-2Rα enhancer. This element has been suggested to repress the enhancer in unactivated cells via an unidentified Ets family protein 54.
Unlike the IL-2Rα enhancer and the upstream enhancer of the perforin gene, the far-upstream enhancer may respond not only to IL-2R but also to TCR signals that have been implied as a second pathway for perforin gene induction 1 2 3, because this enhancer also responded to PMA and ionomycin in a CsA-sensitive manner in SAM-19 (Fig. 5 B). This observation could suggest a possible role for a nuclear factor of activated T cells (NFAT)-like element in the far-upstream enhancer whose sequence resembles the “IL140” element of the IL-3 enhancer, which is known to strongly activate transcription independently of AP-1 proteins 58. The physiological activation of the perforin enhancers is likely to involve transcription factors in addition to Stat5. The combined control of the perforin enhancers remains to be experimentally established and compared with that of the IL-2Rα enhancer. Based on the inspection of the perforin sequences (Fig. 6 A), other participating transcription factors could be IL-2 inducible, namely AP-1, cAMP-responsive element binding proteins, and nuclear factor κb 59 60 61, or differentially expressed during lymphoid development, namely Ikaros proteins 62 and proteins recognizing the E-box 63.
In summary, this study investigates the differentiation processes of CTLs by working backwards from the perforin gene. It indicates that the induction of the perforin gene by IL-2R signals involves at least two enhancers whose activation is dependent on Stat elements that can be targeted by Stat5 molecules. These results are consistent with and complement the ongoing findings of the reverse genetics analysis of the IL-2R Jak/Stat signaling pathway.
Acknowledgments
We are indebted to M. Nabholz for providing PC60 cells, B. Groner for providing the chimeric Stat5-VP16-Jak2 expression vector, and T. Malek for the Stat5a expression vector. We thank M. Gutierrez and E. Cepero for their technical assistance in the transgene experiments. We also thank L. Boise, T. Malek, and U. Schindler for helpful discussions and critical reading of the manuscript.
This work was supported by National Institutes of Health grant R01 CA55811 to M.G. Lichtenheld. The Transgene and Sequencing Facilities of the University of Miami School of Medicine are supported by the Sylvester Comprehensive Cancer Center.
Footnotes
1used in this paper: AP, activator protein; CsA, cyclosporin A; Stat, signal transducer and activator of transcription
References
- Liu C.C., Rafii S., Granelli-Piperno A., Trapani J.A., Young J.D. Perforin and serine esterase gene expression in stimulated human T cells. Kinetics, mitogen requirements, and effects of cyclosporin A. J. Exp. Med. 1989;170:2105–2118. doi: 10.1084/jem.170.6.2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth M.J., Ortaldo J.R., Shinkai Y., Yagita H., Nakata M., Okumura K., Young H.A. Interleukin 2 induction of pore-forming protein gene expression in human peripheral blood CD8+ T cells. J. Exp. Med. 1990;171:1269–1281. doi: 10.1084/jem.171.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu P., Garcia-Sanz J.A., Lichtenheld M.G., Podack E.R. Perforin expression in human peripheral blood mononuclear cells. Definition of an IL-2-independent pathway of perforin induction in CD8+ T cells. J. Immunol. 1992;148:3354–3360. [PubMed] [Google Scholar]
- Dennert G., Podack E.R. Cytolysis by H-2–specific T killer cells. Assembly of tubular complexes on target membranes. J. Exp. Med. 1983;157:1483–1495. doi: 10.1084/jem.157.5.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiver J.W., Su L., Henkart P.A. Cytotoxicity with target DNA breakdown by rat basophilic leukemia cells expressing both cytolysin and granzyme A. Cell. 1992;71:315–322. doi: 10.1016/0092-8674(92)90359-k. [DOI] [PubMed] [Google Scholar]
- Henkart P.A. Lymphocyte-mediated cytotoxicitytwo pathways and multiple effector molecules. Immunity. 1994;1:343–346. doi: 10.1016/1074-7613(94)90063-9. [DOI] [PubMed] [Google Scholar]
- Kagi D., Ledermann B., Burki K., Seiler P., Odermatt B., Olsen K.J., Podack E.R., Zinkernagel R.M., Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Rossi C.P., McAllister A., Tanguy M., Kagi D., Brahic M. Theiler's virus infection of perforin-deficient mice. J. Virol. 1998;72:4515–4519. doi: 10.1128/jvi.72.5.4515-4519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagi D., Ledermann B., Burki K., Hengartner H., Zinkernagel R.M. CD8+ T cell-mediated protection against an intracellular bacterium by perforin-dependent cytotoxicity. Eur. J. Immunol. 1994;24:3068–3072. doi: 10.1002/eji.1830241223. [DOI] [PubMed] [Google Scholar]
- van den Broek M.E., Kagi D., Ossendorp F., Toes R., Vamvakas S., Lutz W.K., Melief C.J., Zinkernagel R.M., Hengartner H. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 1996;184:1781–1790. doi: 10.1084/jem.184.5.1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann D., Baars P.A., Rep M.H., Hooibrink B., Kerkhof-Garde S.R., Klein M.R., van Lier R.A. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 1997;186:1407–1418. doi: 10.1084/jem.186.9.1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisberg M., Terry L.A., Flomenberg N., Dupont B. Cytotoxic and proliferative allospecific T-cell clones contain perforin and mediate anti-CD3-induced cytotoxicity. Hum. Immunol. 1992;35:239–245. doi: 10.1016/0198-8859(92)90005-8. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Norihisa Y., Ortaldo J.R. Multiple cytolytic mechanisms displayed by activated human peripheral blood T cell subsets. J. Immunol. 1992;148:55–62. [PubMed] [Google Scholar]
- Gamero A.M., Ussery D., Reintgen D.S., Puleo C.A., Djeu J.Y. Interleukin 15 induction of lymphokine-activated killer cell function against autologous tumor cells in melanoma patient lymphocytes by a CD18-dependent, perforin-related mechanism. Cancer Res. 1995;55:4988–4994. [PubMed] [Google Scholar]
- Ye W., Young J.D., Liu C.C. Interleukin-15 induces the expression of mRNAs of cytolytic mediators and augments cytotoxic activities in primary murine lymphocytes. Cell. Immunol. 1996;174:54–62. doi: 10.1006/cimm.1996.0293. [DOI] [PubMed] [Google Scholar]
- Waldmann T., Tagaya Y., Bamford R. Interleukin-2, interleukin-15, and their receptors. Int. Rev. Immunol. 1998;16:205–226. doi: 10.3109/08830189809042995. [DOI] [PubMed] [Google Scholar]
- Zhang X., Sun S., Hwang I., Tough D.F., Sprent J. Potent and selective stimulation of memory-phenotype CD8+ T cells in vivo by IL-15. Immunity. 1998;8:591–599. doi: 10.1016/s1074-7613(00)80564-6. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Kundig T.M., Furlonger C., Wakeham A., Timms E., Matsuyama T., Schmits R., Simard J.J., Ohashi P.S., Griesser H. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science. 1995;268:1472–1476. doi: 10.1126/science.7770771. [DOI] [PubMed] [Google Scholar]
- Youn B.S., Kim K.K., Kwon B.S. A critical role of Sp1- and Ets-related transcription factors in maintaining CTL-specific expression of the mouse perforin gene. J. Immunol. 1996;157:3499–3509. [PubMed] [Google Scholar]
- Koizumi H., Horta M.F., Youn B.S., Fu K.C., Kwon B.S., Young J.D., Liu C.C. Identification of a killer cell-specific regulatory element of the mouse perforin genean Ets-binding site-homologous motif that interacts with Ets-related proteins. Mol. Cell. Biol. 1993;13:6690–6701. doi: 10.1128/mcb.13.11.6690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horta M.F., Fu K.C., Koizumi H., Young J.D., Liu C.C. Cell-free conversion of a ubiquitous nuclear protein into a killer-cell-specific form that binds to the NF-P enhancer element of the mouse perforin gene. Eur. J. Biochem. 1996;238:639–646. doi: 10.1111/j.1432-1033.1996.0639w.x. [DOI] [PubMed] [Google Scholar]
- Lichtenheld M.G., Podack E.R. Structure and function of the murine perforin promoter and upstream region. Reciprocal gene activation or silencing in perforin positive and negative cells. J. Immunol. 1992;149:2619–2626. [PubMed] [Google Scholar]
- Zhang Y., Lichtenheld M.G. Non-killer cell-specific transcription factors silence the perforin promoter. J. Immunol. 1997;158:1734–1741. [PubMed] [Google Scholar]
- Lichtenheld M.G., Podack E.R., Levy R.B. Transgenic control of perforin gene expression. Functional evidence for two separate control regions. J. Immunol. 1995;154:2153–2163. [PubMed] [Google Scholar]
- Conzelmann A., Corthesy P., Cianfriglia M., Silva A., Nabholz M. Hybrids between rat lymphoma and mouse T cells with inducible cytolytic activity. Nature. 1982;298:170–172. doi: 10.1038/298170a0. [DOI] [PubMed] [Google Scholar]
- Garcia-Sanz J.A., Podack E.R. Regulation of perforin gene expression in a T cell hybrid with inducible cytolytic activity. Eur. J. Immunol. 1993;23:1877–1883. doi: 10.1002/eji.1830230822. [DOI] [PubMed] [Google Scholar]
- Willerford D.M., Chen J., Ferry J.A., Davidson L., Ma A., Alt F.W. Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity. 1995;3:521–530. doi: 10.1016/1074-7613(95)90180-9. [DOI] [PubMed] [Google Scholar]
- Cepero E., Hnatyszyn H.J., Kraus G., Lichtenheld M.G. Potent inhibition of CTLA-4 expression by an anti-CTLA-4 ribozyme. Biochem. Biophys. Res. Commun. 1998;247:838–843. doi: 10.1006/bbrc.1998.8889. [DOI] [PubMed] [Google Scholar]
- Roberts S., Bentley D.L. Distinct modes of transcription read through or terminate at the c-myc attenuator. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:1085–1093. doi: 10.1002/j.1460-2075.1992.tb05147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenheld M.G., Podack E.R. Structure of the human perforin gene. A simple gene organization with interesting potential regulatory sequences. J. Immunol. 1989;143:4267–4274. [PubMed] [Google Scholar]
- Makrigiannis A.P., Hoskin D.W. Inhibition of CTL induction by rapamycinIL-2 rescues granzyme B and perforin expression but only partially restores cytotoxic activity. J. Immunol. 1997;159:4700–4707. [PubMed] [Google Scholar]
- Moriggl R., Gouilleux-Gruart V., Jahne R., Berchtold S., Gartmann C., Liu X., Hennighausen L., Sotiropoulos A., Groner B., Gouilleux F. Deletion of the carboxyl-terminal transactivation domain of MGF-Stat5 results in sustained DNA binding and a dominant negative phenotype. Mol. Cell. Biol. 1996;16:5691–5700. doi: 10.1128/mcb.16.10.5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quandt K., Frech K., Karas H., Wingender E., Werner T. MatInd and MatInspectornew fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold S., Moriggl R., Gouilleux F., Silvennoinen O., Beisenherz C., Pfitzner E., Wissler M., Stocklin E., Groner B. Cytokine receptor-independent, constitutively active variants of STAT5. J. Biol. Chem. 1997;272:30237–30243. doi: 10.1074/jbc.272.48.30237. [DOI] [PubMed] [Google Scholar]
- Leonard W.J., O'Shea J.J. Jaks and STATsbiological implications. Annu. Rev. Immunol. 1998;16:293–322. doi: 10.1146/annurev.immunol.16.1.293. [DOI] [PubMed] [Google Scholar]
- Horvath C.M., Darnell J.E. The state of the STATsrecent developments in the study of signal transduction to the nucleus. Curr. Opin. Cell. Biol. 1997;9:233–239. doi: 10.1016/s0955-0674(97)80067-1. [DOI] [PubMed] [Google Scholar]
- Gaffen S.L., Lai S.Y., Xu W., Gouilleux F., Groner B., Goldsmith M.A., Greene W.C. Signaling through the interleukin 2 receptor β chain activates a STAT-5-like DNA-binding activity. Proc. Natl. Acad. Sci. USA. 1995;92:7192–7196. doi: 10.1073/pnas.92.16.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright J.J., Kerr L.D., Sriram S. TGF-β inhibits IL-2-induced tyrosine phosphorylation and activation of Jak-1 and Stat 5 in T lymphocytes. J. Immunol. 1997;159:175–183. [PubMed] [Google Scholar]
- Han H.S., Jun H.S., Utsugi T., Yoon J.W. Molecular role of TGF-β, secreted from a new type of CD4+ suppressor T cell, NY4.2, in the prevention of autoimmune IDDM in NOD mice. J. Autoimmun. 1997;10:299–307. doi: 10.1006/jaut.1997.0137. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Strobl S.L., Young H.A., Ortaldo J.R., Ochoa A.C. Regulation of lymphokine-activated killer activity and pore-forming protein gene expression in human peripheral blood CD8+ T lymphocytes. Inhibition by transforming growth factor-β. J. Immunol. 1991;146:3289–3297. [PubMed] [Google Scholar]
- Teglund S., McKay C., Schuetz E., van Deursen J.M., Stravopodis D., Wang D., Brown M., Bodner S., Grosveld G., Ihle J.N. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93:841–850. doi: 10.1016/s0092-8674(00)81444-0. [DOI] [PubMed] [Google Scholar]
- Moriggl R., Topham D.J., Teglund S., Sexl V., McKay C., Wang D., Hoffmeyer A., van Deursen J., Sangster M.Y., Bunting K.D. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Nakajima H., Liu X.W., Wynshaw-Boris A., Rosenthal L.A., Imada K., Finbloom D.S., Hennighausen L., Leonard W.J. An indirect effect of Stat5a in IL-2-induced proliferationa critical role for Stat5a in IL-2-mediated IL-2 receptor α chain induction. Immunity. 1997;7:691–701. doi: 10.1016/s1074-7613(00)80389-1. [DOI] [PubMed] [Google Scholar]
- Imada K., Bloom E.T., Nakajima H., Horvath-Arcidiacono J.A., Udy G.B., Davey H.W., Leonard W.J. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J. Exp. Med. 1998;188:2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H., Ogasawara K., Otsuka H., Suzuki M., Yamamura K., Yokochi T., Miyazaki T., Suzuki H., Mak T.W., Taki S. Functional dissection of the cytoplasmic subregions of the IL-2 receptor β chain in primary lymphocyte populations. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6551–6557. doi: 10.1093/emboj/17.22.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C.R., Ortaldo J.R., Curiel R.E., Young H.A., Anderson S.K., Gosselin P. Role of a STAT binding site in the regulation of the human perforin promoter. J. Immunol. 1999;162:2785–2790. [PubMed] [Google Scholar]
- Akaishi H., Takeda K., Kaisho T., Shineha R., Satomi S., Takeda J., Akira S. Defective IL-2-mediated IL-2 receptor α chain expression in Stat3-deficient T lymphocytes. Int. Immunol. 1998;10:1747–1751. doi: 10.1093/intimm/10.11.1747. [DOI] [PubMed] [Google Scholar]
- Nakata M., Smyth M.J., Norihisa Y., Kawasaki A., Shinkai Y., Okumura K., Yagita H. Constitutive expression of pore-forming protein in peripheral blood γ/δ T cellsimplication for their cytotoxic role in vivo. J. Exp. Med. 1990;172:1877–1880. doi: 10.1084/jem.172.6.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi H., Liu C.C., Zheng L.M., Joag S.V., Bayne N.K., Holoshitz J., Young J.D. Expression of perforin and serine esterases by human γ/δ T cells. J. Exp. Med. 1991;173:499–502. doi: 10.1084/jem.173.2.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy-Grand D., Malassis-Seris M., Briottet C., Vassalli P. Cytotoxic differentiation of mouse gut thymodependent and independent intraepithelial T lymphocytes is induced locally. Correlation between functional assays, presence of perforin and granzyme transcripts, and cytoplasmic granules. J. Exp. Med. 1991;173:1549–1552. doi: 10.1084/jem.173.6.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H., Duncan G.S., Takimoto H., Mak T.W. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor β chain. J. Exp. Med. 1997;185:499–505. doi: 10.1084/jem.185.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodolce J.P., Boone D.L., Chai S., Swain R.E., Dassopoulos T., Trettin S., Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- Sperisen P., Wang S.M., Soldaini E., Pla M., Rusterholz C., Bucher P., Corthesy P., Reichenbach P., Nabholz M. Mouse interleukin-2 receptor α gene expression. Interleukin-1 and interleukin-2 control transcription via distinct cis-acting elements. J. Biol. Chem. 1995;270:10743–10753. doi: 10.1074/jbc.270.18.10743. [DOI] [PubMed] [Google Scholar]
- John S., Robbins C.M., Leonard W.J. An IL-2 response element in the human IL-2 receptor α chain promoter is a composite element that binds Stat5, Elf-1, HMG-I(Y) and a GATA family protein. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:5627–5635. [PMC free article] [PubMed] [Google Scholar]
- Meyer W.K., Reichenbach P., Schindler U., Soldaini E., Nabholz M. Interaction of STAT5 dimers on two low affinity binding sites mediates interleukin 2 (IL-2) stimulation of IL-2 receptor α gene transcription. J. Biol. Chem. 1997;272:31821–31828. doi: 10.1074/jbc.272.50.31821. [DOI] [PubMed] [Google Scholar]
- Serdobova I., Pla M., Reichenbach P., Sperisen P., Ghysdael J., Wilson A., Freeman J., Nabholz M. Elf-1 contributes to the function of the complex interleukin (IL)-2–responsive enhancer in the mouse IL-2 receptor α gene. J. Exp. Med. 1997;185:1211–1221. doi: 10.1084/jem.185.7.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S., Vinkemeier U., Soldaini E., Darnell J.E., Jr., Leonard W.J. The significance of tetramerization in promoter recruitment by Stat5. Mol. Cell. Biol. 1999;19:1910–1918. doi: 10.1128/mcb.19.3.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncliffe K.N., Bert A.G., Vadas M.A., Cockerill P.N. A T cell-specific enhancer in the interleukin-3 locus is activated cooperatively by Oct and NFAT elements within a DNase I-hypersensitive site. Immunity. 1997;6:175–185. doi: 10.1016/s1074-7613(00)80424-0. [DOI] [PubMed] [Google Scholar]
- Trouche D., Robin P., Robillard O., Sassone-Corsi P., Harel-Bellan A. c-fos transcriptional activation by IL-2 in mouse CTL-L2 cells is mediated through two distinct signal transduction pathways converging on the same enhancer element. J. Immunol. 1991;147:2398–2403. [PubMed] [Google Scholar]
- Feuerstein N., Huang D., Hinrichs S.H., Orten D.J., Aiyar N., Prystowsky M.B. Regulation of cAMP-responsive enhancer binding proteins during cell cycle progression in T lymphocytes stimulated by IL-2. J. Immunol. 1995;154:68–79. [PubMed] [Google Scholar]
- Arima N., Kuziel W.A., Grdina T.A., Greene W.C. IL-2-induced signal transduction involves the activation of nuclear NF-κB expression. J. Immunol. 1992;149:83–91. [PubMed] [Google Scholar]
- Georgopoulos K., Winandy S., Avitahl N. The role of the Ikaros gene in lymphocyte development and homeostasis. Annu. Rev. Immunol. 1997;15:155–176. doi: 10.1146/annurev.immunol.15.1.155. [DOI] [PubMed] [Google Scholar]
- Spits H., Blom B., Jaleco A.C., Weijer K., Verschuren M.C., van Dongen J.J., Heemskerk M.H., Res P.C. Early stages in the development of human T, natural killer and thymic dendritic cells. Immunol. Rev. 1998;165:75–86. doi: 10.1111/j.1600-065x.1998.tb01231.x. [DOI] [PubMed] [Google Scholar]