

Abstract
P-selectin glycoprotein ligand 1 (PSGL-1) is a mucin-like selectin counterreceptor that binds to P-selectin, E-selectin, and L-selectin. To determine its physiological role in cell adhesion as a mediator of leukocyte rolling and migration during inflammation, we prepared mice genetically deficient in PSGL-1 by targeted disruption of the PSGL-1 gene. The homozygous PSGL-1–deficient mouse was viable and fertile. The blood neutrophil count was modestly elevated. There was no evidence of spontaneous development of skin ulcerations or infections. Leukocyte infiltration in the chemical peritonitis model was significantly delayed. Leukocyte rolling in vivo, studied by intravital microscopy in postcapillary venules of the cremaster muscle, was markedly decreased 30 min after trauma in the PSGL-1–deficient mouse. In contrast, leukocyte rolling 2 h after tumor necrosis factor α stimulation was only modestly reduced, but blocking antibodies to E-selectin infused into the PSGL-1–deficient mouse almost completely eliminated leukocyte rolling. These results indicate that PSGL-1 is required for the early inflammatory responses but not for E-selectin–mediated responses. These kinetics are consistent with a model in which PSGL-1 is the predominant neutrophil P-selectin ligand but is not a required counterreceptor for E-selectin under in vivo physiological conditions.
Keywords: selectin, leukocyte rolling, cell adhesion, P-selectin, E-selectin
Recruitment of leukocytes from the blood stream to sites of injury or infection is initiated early in the inflammatory response. Cell–cell interactions among leukocytes, platelets, and endothelial cells on the blood vessel wall during this process are mediated by cell adhesion molecules that reside on the plasma membranes of these cells 1. The initial contact and rolling of leukocytes on activated endothelium are mediated by the selectins 2 3. Chemoattractants, together with signals generated by cell–cell interaction, activate integrins on leukocytes, leading to firm attachment and migration of leukocytes into tissues.
The selectin family of adhesion molecules includes E-selectin, L-selectin, and P-selectin. P-selectin, stored in the alpha granules in unstimulated platelets and the Weibel-Palade bodies in resting endothelial cells, is translocated to the cell surface upon cell activation 4 5. In mice lacking P-selectin, neutrophil rolling in venules of the mesentery or the cremaster muscle is nearly absent at early time points and neutrophil influx into the inflamed peritoneum is delayed 6 7 8. Cell surface expression of E-selectin is induced at the transcriptional level after endothelial stimulation by cytokines 9, so E-selectin expression is a late event after de novo synthesis. E-selectin–deficient mice do not show obvious defects in leukocyte function, although slow rolling is impaired 10. L-selectin is expressed constitutively on leukocytes. Significant inhibition in neutrophil rolling at later time points is observed in L-selectin–deficient mice 7 11. Mice deficient in both P-selectin and E-selectin demonstrate minimal neutrophil rolling and delayed and suppressed neutrophil migration 12 13.
The physiologically relevant counterreceptors on cell surfaces that bind specifically to selectins have been difficult to identify, although many putative selectin ligands have been proposed. The selectins are lectins, and the selectin ligands include Lewis X (CD15 14) and sialic acid 15 16. Oligosaccharide sequences of the ligands, predominantly sialyl Lewis X antigen–related sialylated fucosylated lactosamine, are recognized by these selectins but with low affinity. To date, several mucin-like proteins have been reported to bind to L-selectin, P-selectin, and E-selectin. Putative L-selectin ligands include E-selectin and P-selectin 17, glycosylation-dependent cell adhesion molecule 1 (GlyCAM-1 18), CD34 19 20, sialoglycoprotein 200 (Sgp 200 21), mucosal addressin cell adhesion molecule 1 (MAdCAM-1 22), and P-selectin glycoprotein ligand 1 (PSGL-1) 23 24 25 26 27 28 29 30. Putative ligands for E-selectin include E-selectin ligand 1 (ESL-1 [31]), L-selectin 32 33, and PSGL-1 34 35 36 37 38 39 40. Mouse E-selectin also binds PSGL-1 (33; and Hirata, T., B.C. Furie, and B. Furie, unpublished results). Although in vitro studies have indicated binding under certain conditions, none of these ligands has been shown in animal models to mediate physiologically relevant interactions with L-selectin or E-selectin.
The putative P-selectin ligands include CD24 41 42 43, sulfated glycolipids 44, components of the peripheral node addressin (PNAd) complex 45, and PSGL-1 34 46. Of these, only PSGL-1 has been implicated as a biologically important ligand for P-selectin (34; for a review, see Varki [47]). The functional importance of PSGL-1 as a P-selectin ligand was suggested from studies in which blocking antibodies to PSGL-1 inhibited P-selectin–mediated leukocyte rolling in exteriorized rat mesentery venules 48 and prevented the early P-selectin–directed accumulation of neutrophils in a mouse model of peritoneal inflammation 49. Furthermore, infusion of soluble PSGL-1 prevents acute inflammation in transient ischemia in a rat kidney model 50.
PSGL-1 is expressed as a functional ligand of P-selectin on myeloid cells and T lymphocyte subsets 40 51 52 53 54. PSGL-1 was identified by expression cloning from COS cells that were cotransfected with a human HL-60 cDNA library and an α1,3/1,4-fucosyltransferase 34. The mouse homologue of PSGL-1 was subsequently cloned based on sequence similarity 55. The predicted protein sequence reveals a type I integral membrane protein of 402 residues for human PSGL-1 and 397 residues for mouse PSGL-1. The mature PSGL-1 protein begins at residue 42 after proteolysis of a signal peptide and cleavage of a propeptide by a furin-like converting enzyme 52. Human PSGL-1 is a homodimer of ∼210,000 molecular weight, with the two chains covalently linked by a disulfide bond. Formation of the homodimer is required for high-affinity binding of PSGL-1 to P-selectin 56 57. The P-selectin binding domain of PSGL-1 is located within the first 20 residues of the mature PSGL-1 58 59. The NH2-terminal binding domain of human PSGL-1 includes tyrosine sulfate and is also O-glycosylated 58 59 60. PSGL-1 contains a serine/threonine- and proline-rich decameric repeat region. Mouse PSGL-1 contains 10 repeat units 55. Human PSGL-1 from eosinophils 61, HL-60, and U937 cells contains 15 repeat units; PSGL-1 from human polymorphonuclear leukocytes and monocytes contains 16 repeat units 62. PSGL-1 contains O-linked glycans that form highly extended structures that participate with the NH2-terminal binding domain of PSGL-1 to bind the counterreceptor 63 64.
To clarify the physiological role of PSGL-1, we generated mice deficient in PSGL-1. Using this mouse model, we explored the consequences of the absence of PSGL-1 in vivo to leukocyte rolling and leukocyte migration. We demonstrate that PSGL-1 deficiency impairs early neutrophil migration and trauma-induced leukocyte rolling. Our results indicate that in vivo PSGL-1 is required for the earliest steps in the inflammatory response but is not required for cytokine-induced, E-selectin–mediated leukocyte rolling.
Materials and Methods
Cell Lines and Tissue Culture.
The embryonic stem (ES) cell line WW6 65 was generated by F. Poirier and E.J. Robertson (Harvard University) from a mouse (50% 129Sv, 25% C57BL/6, 5% SJL, and 20% unknown) and provided by Dr. P. Stanley (Albert Einstein College of Medicine, New York, NY). Cells were cultured on γ-irradiated STO feeder cells (SNL2; developed by E.J. Robertson) in DMEM (GIBCO BRL) containing 20% FCS (Hyclone).
Construction of the Targeting Vector.
Genomic clones of mouse PSGL-1 were isolated from an adult 129/SvJ mouse liver genomic DNA library (no. 946305; Stratagene) as described previously 55. Two contiguous EcoRI fragments together containing a portion of the 5′ flanking region, the complete coding region, and the complete 3′ untranslated region of the PSGL-1 gene were cloned independently into the pBluescript vector to create A1.6 and A1.1 (see Fig. 1 A). An SmaI-EcoRI fragment from A1.6 and an EcoRI-BamHI fragment from A1.1 were recombined in pBluescript at the EcoRV and BamHI sites to generate ASMB. A HindIII (blunt ended)-BamHI fragment released from ASMB was ligated 3′ to the Neo cassette in pNT at Xbal (blunt ended) and BamHI sites to create KOIII. The vector pNT was provided by Dr. B. Spiegleman (Dana-Farber Cancer Institute, Boston, MA). Primer pairs P1/P2 and P3/P4 were used to amplify the DNA sequence by PCR to be inserted 5′ to the Neo cassette. The PCR product from NotI-tagged P1 (5′-CCCCA ACGGG TTGTT TTGGC CCACC-3′) and EcoRI-tagged P2 (5′-GAAGC CGGAA GGGTC TGGGC ATGG-3′) was subcloned and recombined at the EcoRI site with the PCR product from EcoRI-tagged P3 (5′-AGAATC TCATT GAGTT ACACA GCC-3′) and XhoI-tagged P4 (5′-GGGGT CCTGC AGCTG AAGGC TG-3′). The resulting 1.3-kb fragment was inserted at the NotI and XhoI sites of KOIII to create KO.pNT.
Figure 1.


Disruption of the PSGL-1 gene in mice: targeting vector and Southern blot analysis. (A) Partial restriction maps of the PSGL-1 gene and the targeting vector KO.pNT. Restriction enzyme sites are marked as follows: E, EcoRI; H, HindIII; B, BamHI; S, SmaI; N, NotI. Probes used for Southern analyses (A–D, Neo), primers used for generating the vector (P1–P4), and the repeat elements are indicated. (B) Southern blot analyses of ES clones. Genomic DNA from two clones, W19 and W34, were digested with HindIII. After gel electrophoresis, DNA was transferred to the membrane and hybridized with probe C. The blot was stripped and reprobed with probe Neo. The targeted band (KO) and the wild-type band (WT) are indicated. (C) Southern blot analyses of DNA isolated from F1 and F2 generations. Genomic DNA from tail biopsies was digested with EcoRI. The fragments were separated by gel electrophoresis, transferred to a membrane, and hybridized with probe A. The bands corresponding to the targeted allele (KO) and the wild-type allele (WT) are indicated.
Selection of Homologous Recombinants.
WW6 cells (2 × 107) were electroporated (GenePulser, 500 μF, 240 V; Bio-Rad) with the targeting vector DNA (∼30 μg/ml) that was linearized by NotI digestion. Cells were plated onto monolayers of γ-irradiated feeder cells. After 24 h, medium was replaced with medium containing 200 μg/ml of G418 (GIBCO BRL) and 2 μM ganciclovir (Syntex). Individual colonies were picked 7 d after application of selective media.
Screening of Recombinants and Generation of Homozygous Null Mice.
Genomic DNA from individual colonies was isolated, digested with HindIII, and analyzed by Southern blotting using probes B or C and Neo. One positive clone, W19, was expanded. ES cells from clone W19 were injected into C57BL/6 blastocysts. The blastocysts were transferred to pseudopregnant foster mothers. Four chimeric male mice were obtained and bred with C57BL/6 mice. Germline transmission was identified by Southern analysis of EcoRI-digested DNA using probe A. Heterozygous animals were bred to generate mutant mice. All studies and procedures were approved by the Animal Care and Use Committee of Beth Israel Deaconess Medical Center.
Northern Blot Analysis.
Total RNA samples (20–60 μg) were prepared from spleen and thymus obtained from PSGL-1−/− and PSGL-1+/+ mice using the SV Total RNA Isolation System (Promega). RNA samples were separated by electrophoresis in 0.8% agarose-formaldehyde gels using 1× 3-(N-morpholino)propanesulfonic acid (Mops) buffer (40 mM Mops, 10 mM sodium acetate, and 1 mM EDTA, pH 7.0). The RNA was transferred to a Duralon-UV™ membrane (Stratagene) and affixed to the membrane by incubation at 80°C for 1 h. Prehybridization or hybridization was at 65°C overnight in 50 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (Pipes), pH 6.5, 100 mM sodium chloride, 50 mM sodium phosphate, pH 7.0, 1 mM EDTA, 5% SDS, and 60 μg/ml salmon sperm DNA. The probe was a 1.0-kb PCR fragment amplified from the coding region of mouse PSGL-1 (probe D).
Flow Cytometry Analysis.
Mice were bled from the retroorbital sinus into EDTA-anticoagulated tubes while under anesthesia with Metofane (Mallinckrodt Veterinary). Leukocytes were prepared by red cell lysis followed by washing and centrifugation. Cells were incubated for 10 min with Fc Block (anti-CD32/16; PharMingen) before all staining procedures. Cells were incubated on ice for 30 min with 10 μg/ml of affinity-purified anti–PSGL-1 antibody or rabbit nonimmune IgG in PBS containing 1% FCS and 0.04% azide, washed, and stained with FITC-labeled goat anti–rabbit IgG (Sigma Chemical Co.) and PE-conjugated anti-Ly6G (PharMingen). Analysis was performed on a FACSCalibur™ flow cytometer with CELLQuest™ software (both from Becton Dickinson). Light-scatter gating procedures were used to enrich for granulocytes.
Immunohistochemistry.
Fresh frozen sections were fixed with 80% acetone, and samples were treated with normal goat serum for 1 h at 25°C. The sections were then incubated with rabbit anti–mouse PSGL-1 antibody at 0.5 μg/ml in PBS for 16 h at 4°C. The tissue was washed three times for 5 min with PBS at 23°C. The washed sections were incubated with biotinylated goat anti–rabbit antibody (VectaStain™ Elite ABC staining kit, 1:200 dilution; Vector Labs) for 1 h at 23°C. Sections were washed three times and then incubated with peroxidase-conjugated streptavidin for 1 h at 25°C. After three washes, the tissue sections were stained using 3,3′-diaminobenzidine in 40 mM Tris buffer, pH 7.4, and then counterstained with methylene blue.
Anti–mouse PSGL-1 Antibodies.
A polyclonal antibody directed against PSGL-1 was prepared using a synthetic peptide (QVVGDDDFEDPDYTYC) based on residues 42–56 of mouse PSGL-1 as immunogen 55. A cysteine residue was added at the COOH terminus to facilitate coupling to KLH. The peptide was synthesized using FMOC/N-methylpyrolidone chemistry on a peptide synthesizer (model 430A; Applied Biosystems). After cleavage and deprotection, the peptide was purified to homogeneity by reverse phase HPLC using a C18 column. The peptide was covalently coupled to KLH through the free cysteine, and the conjugate was injected intradermally (550 μg/ml) in complete Freund's adjuvant into a New Zealand White rabbit. Subsequent injections of immunogen (250 μg) were performed weekly for 2 wk, then monthly thereafter. Antipeptide antibodies were purified from rabbit immune serum by immunoaffinity chromatography. The serum was applied to a KLH-Sepharose column to remove antibodies against KLH. The serum proteins that failed to bind to KLH-Sepharose were then applied to a PSGL-1 (residues 42–56)–Sepharose column in which the peptide was covalently attached to Sulfo-Link Resin (Pierce Chemical Co.). Bound antibodies were eluted with 4 M guanidine and dialyzed into PBS.
Peripheral Blood Counts.
Blood was collected from 8–10-wk-old male wild-type and PSGL-1−/− mice as well as P-selectin−/− mice and their matched controls. Complete blood counts were performed using a Coulter Counter. Differential counts were performed on blood smears stained with Wright-Giemsa stain (Sigma Chemical Co.). Differentials were performed in duplicate on 100 leukocytes per slide.
Thioglycollate-induced Peritonitis Model.
PSGL-1−/− mice and wild-type mice of similar mixed genetic background were used at 8–12 wk of age. P-selectin−/− mice (C57BL/6 background) and wild-type C57BL/6 mice (The Jackson Laboratory) were of the same age. Mice were injected intraperitoneally with 1 ml of 4% Brewer thioglycollate medium (Difco). At each time point after stimulation, the mice were killed and 10 ml of PBS containing 1% bovine serum albumin, 0.5 mM EDTA, and 10 U/ml heparin was injected into the peritoneal cavity. After gently massaging the peritoneal wall, the injected wash was withdrawn. Total cell numbers in the peritoneal lavage were determined on a Coulter Counter. Cytospin preparations of the cells were stained with Wright-Giemsa stain, and a differential leukocyte count was determined. From the total cell count in the peritoneal lavage and the percentage of neutrophils on cytospin preparations, the absolute number of neutrophils was calculated.
Leukocyte Rolling In Vivo in the Mouse Cremaster Muscle.
Mice were preanesthetized with an intraperitoneal injection of 125 mg/kg of ketamine HCl (Parke Davis), 12.5 mg/kg xylazine (Phoenix Pharmaceuticals), and 0.25 mg/kg atropine sulfate (American Reagent Laboratories). A trachea tube was inserted to facilitate spontaneous respiration, and the mice were kept on a 37°C thermocontrolled rodent blanket (Thermal Care) during the experiment. To maintain anesthesia and neutral fluid balance, Nembutal (5 mg/ml in saline) was administered in 30–50 μl boluses through a cannula placed in the jugular vein. Mean arterial blood pressure ranged between 60 and 100 mmHg as measured on a physiological pressure transducer (model 60-051; Harvard Apparatus) connected to a carotid artery cannula. When TNF-α was used, mice were treated with an intrascrotal injection of mouse TNF (0.5 μg in 200 μl saline; R&D Systems) 2 h before exteriorization of the cremaster muscle. In experiments where the neutralizing anti–E-selectin mAb 9A9 was used, mice were injected with 80–90 μg of antibody through the jugular cannula just after exteriorization of the cremaster muscle.
The cremaster muscle was prepared for intravital microscopy as described by Ley et al. 7. Through an incision made in the scrotum, the testicle and surrounding cremaster muscle were exteriorized onto an intravital microscopy tray. After removal of the connective tissue, a longitudinal incision was made in the cremaster, and the muscle was carefully stretched and pinned across the intravital microscopy stage while the testis and epididymis were tacked to one side. The cremaster preparation was superfused with thermocontrolled (36°C) and aerated (5% CO2, 95% N2) bicarbonate buffered saline throughout the experiment. The cremaster exteriorization surgery was typically accomplished in 4–7 min.
Microvessel data were obtained using an intravital microscope (Axioskop; Carl Zeiss, Inc.) fitted with an Achroplan (40×, 0.80 numerical aperture) water immersion objective, long distance condenser, and stabilized D.C. power supply. Leukocyte rolling in the venules was recorded for 75–90 s using a Sony CCD camera (model SSC-S20) connected through an IBM-compatible computer to a Sony SVHS video recorder (model SVA-9500MD). The centerline red blood cell velocity (V cl) in each venule was measured in real time with a dual photodiode velocimeter running a digital cross-correlation program on an IBM-compatible computer (Microvessel Velocity OD-RT; CircuSoft Instrumentation). A Coulter Counter (model T890) was used to determine the systemic leukocyte counts from 30–50 μl blood samples taken from the carotid cannula at 30–40-min intervals or after injection of antibodies. After the start of the cremaster surgery, data were acquired for 30 or 40 min in the untreated and TNF-treated mice, respectively.
Analysis of Leukocyte Rolling.
Video recordings from the intravital microscopy experiments were analyzed on a computer-based image acquisition system, and vessel diameter was determined using the IBM-adapted version of NIH Image (Scion Corp.). The centerline red cell velocity recorded from each vessel was used to determine the volumetric blood flow (Q) via the equation Q = (V cl)(0.625)(A cs), where A cs is the cross-sectional area of the cylindrical vessel, and 0.625 is an empirical correction factor 66. For each vessel, wall shear rate (W sr) was determined: W sr = 2.13 × [(8 × 0.625 × V cl)/D v)], where D v is the vessel diameter. Critical velocity (V crit) was also calculated: V crit = (V cl × 0.625)(D cell/D v)[(2 − (D cell/D v)]. Recordings of each vessel were analyzed for 30–60 s, and rolling leukocytes were identified as the visible cells passing through a plane perpendicular to the vessel axis. In certain cases, the rolling velocity of visible high velocity cells (V cell) was estimated in order to determine whether these cells qualified as rolling leukocytes, defined V cell < V crit. Total leukocyte flux was determined as the product of the measured systemic leukocyte concentration and microvessel volumetric blood flow. To compensate for differences in systemic leukocyte count, the rolling behavior of leukocytes in the cremaster muscle is presented as the rolling leukocyte flux fraction, which is the number of rolling leukocytes in the vessel as a percentage of the total leukocyte flux.
Online Supplemental Materials.
We have included six videoclips from representative experiments depicted in Fig. 6 and Fig. 7. These experiments correspond directly to those used to prepare the still photos in Fig. 6 and Fig. 7. Leukocyte rolling in the venules was recorded for 75–90 s using a CCD camera connected through an IBM-compatible computer to an SVHS video recorder. Fig. 7 C was videotaped at an accelerated rate to compress the images into a shorter time span.
Figure 6.
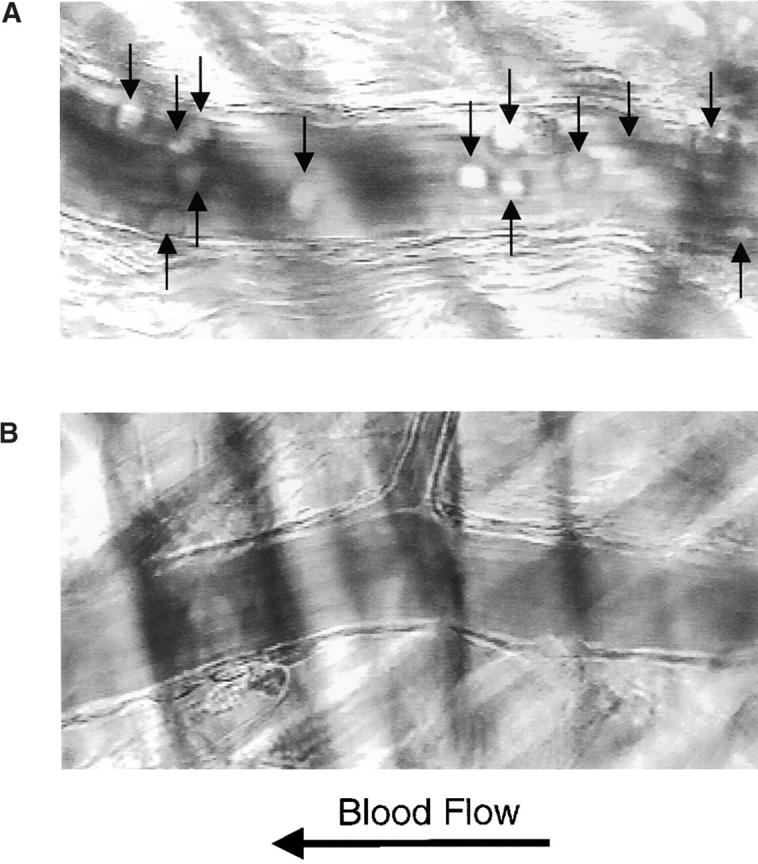

Leukocyte rolling after mild trauma. Intravital microscopy of cremaster muscle venules within 30 min of the initiation of surgery revealed the presence of a large number of rolling leukocytes (arrows) associated with the vascular wall in wild-type mice (A) and no leukocytes associated with the vascular wall in PSGL-1–deficient mice (B). Videos available at http://www.jem.org/cgi/content/full/190/12/1769/F6/DC1 correspond to A and B. The leukocyte rolling flux fraction, measured as the number of rolling leukocytes compared with the total leukocytes in flowing blood per unit time, is compared for PSGL-1+/+, PSGL-1−/−, P-selectin (P-sel)+/+, and P-selectin−/− mice in this assay system (C). Both PSGL-1−/− and P-selectin−/− mice have low but measurable leukocyte rolling flux fractions.
Figure 7.
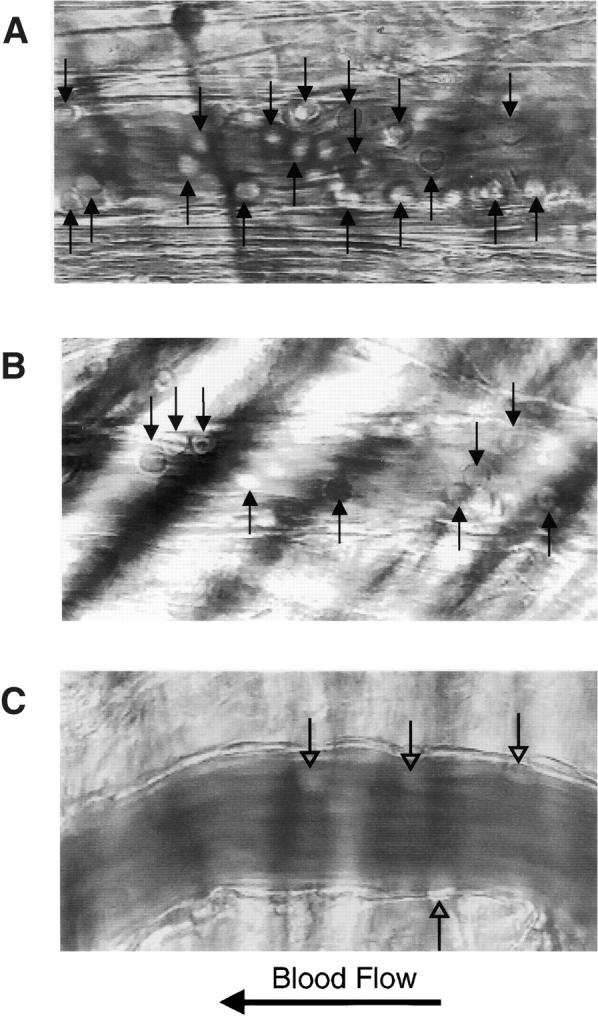

Leukocyte rolling after TNF-α stimulation. Intravital microscopy of cremaster muscle venules 2 h after injection of TNF-α into the scrotal sac revealed the presence of rolling leukocytes (arrows) associated with the vascular wall in wild-type mice (A) and in PSGL-1–deficient mice (B). Infusion of the blocking E-selectin antibody 9A9 into PSGL-1–deficient mice greatly reduced the number of rolling leukocytes; leukocytes visible in C are stationary. Videos available at http://www.jem.org/cgi/content/full/190/12/1769/F7/DC1 correspond to A and B, and to two time points in C, showing leukocyte rolling before infusion of anti–E-selectin antibody, and leukocyte rolling during and after infusion of anti–E-selectin antibody, respectively. The leukocyte rolling flux fraction, measured as the number of rolling leukocytes compared with the total leukocytes in flowing blood per unit time, is compared for PSGL-1+/+ mice, PSGL-1−/− mice, PSGL-1−/− mice treated with anti–E-selectin antibody 9A9, PSGL-1+/+ mice treated with anti–E-selectin antibody 9A9, P-selectin (P-sel)+/+ mice, and P-selectin−/− mice in this assay system (D).
Results
Generation of PSGL-1–deficient Mice.
Several ubiquitous transposon-like repetitive sequences B1 and B2 67 were found in the 5′ and 3′ flanking regions of the PSGL-1 coding region, and two segments containing CT repeats similar to those in ribosomal DNA 68 were located 5′ to the PSGL-1 coding region (Fig. 1 A). These repetitive sequences were excluded from the knockout vector, KO.pNT, in which a small region of the PSGL-1 coding sequence from 69 to 94 nucleotides (nt) was replaced by a neomycin-resistance gene cassette (Fig. 1 A). A deletion of ∼300 bp in the 5′ flanking region of the PSGL-1 coding region was generated in the vector in order to exclude a segment containing CT repeats. A thymidine kinase gene cassette was inserted 3′ of the construct as a negative selection marker. ES cells were transfected with linearized KO.pNT vector, selected with G418 plus ganciclovir, and screened by Southern blot analysis. Of 200 ES cell colonies analyzed, two exhibited a targeted band at 6.1 kb by HindIII digestion (Fig. 1 B) using probe B or probe C (Fig. 1 A) in addition to the expected wild-type band at 4.3 kb. Negative clones including W34, shown in Fig. 1 B, showed only the wild-type band. One of the positive clones, W19, displayed a single band corresponding to the targeted band when the Neo probe was used, indicating a single integration event in the cells (Fig. 1 B). As a result of random integration, the Neo probe hybridized to a band of irrelevant size in W34 (Fig. 1 B). A homologous recombination event in W19 cells was also confirmed by EcoRI digestion followed by Southern blot analyses using probe A or the Neo probe (data not shown). In all cases, a targeted band of the predicted size was detected. Results from the EcoRI digest using probe A (Fig. 1 C) and PCR analyses using primers P1 and P4 (Fig. 1 A) suggested that homologous recombination occurred 3′ of the EcoRI site of the targeting vector. Thus, there is no deletion in the 5′ flanking region of the PSGL-1 coding region in W19 cells.
W19 cells were microinjected into blastocysts from C57BL/6 mice and transferred to recipient female mice, and chimeric mice were obtained. Breeding of the chimera with C57BL/6 mice yielded heterozygous animals. Homozygous mutant mice were obtained from subsequent breeding between heterozygotes. The animals were genotyped by Southern blot analysis as shown in Fig. 1 C. Using probe A, animals with the mutated allele showed an EcoRI band at 7.1 kb, whereas those carrying the wild-type allele yielded a band at 5.3 kb.
Verification of a Null Allele for PSGL-1.
Northern blot analysis of thymus and spleen RNA showed high expression of PSGL-1 messenger RNA in wild-type mice but did not show any transcript in PSGL-1−/− mice (Fig. 2), indicating that the targeted mutation created a null allele.
Figure 2.

Northern blot analysis of tissue for PSGL-1 mRNA. Total RNA isolated from the thymus and spleen of PSGL-1 (−/−) and PSGL-1 (+/+) mice was subjected to electrophoresis, and the RNA was transferred to a Duralon-UV™ membrane. The PSGL-1 messenger RNA was detected with a probe representing most of the coding region. The membrane was stripped and probed with a cDNA probe for β-actin.
To confirm that PSGL-1−/− mice were deficient in cell surface expression of PSGL-1, leukocytes from the blood of wild-type, heterozygous, and homozygous mutant animals were stained with a rabbit anti–PSGL-1 antibody that was raised to an NH2-terminal peptide of PSGL-1, and analyzed by flow cytometry. Granulocytes were identified by light-scatter properties and positive staining with anti–Ly-6G antibody. All granulocytes from wild-type animals expressed high levels of PSGL-1. In contrast, granulocytes from the PSGL-1−/− mice did not show specific staining (Fig. 3), confirming a null mutation. The mean fluorescence intensity of PSGL-1 on granulocytes from heterozygous (+/−) mice was ∼50% of that observed on wild-type granulocytes, consistent with the presence of only one PSGL-1 gene allele in the heterozygotes. Lymphocytes from the blood of PSGL-1−/− mice did not express PSGL-1, as assessed by flow cytometry (data not shown).
Figure 3.
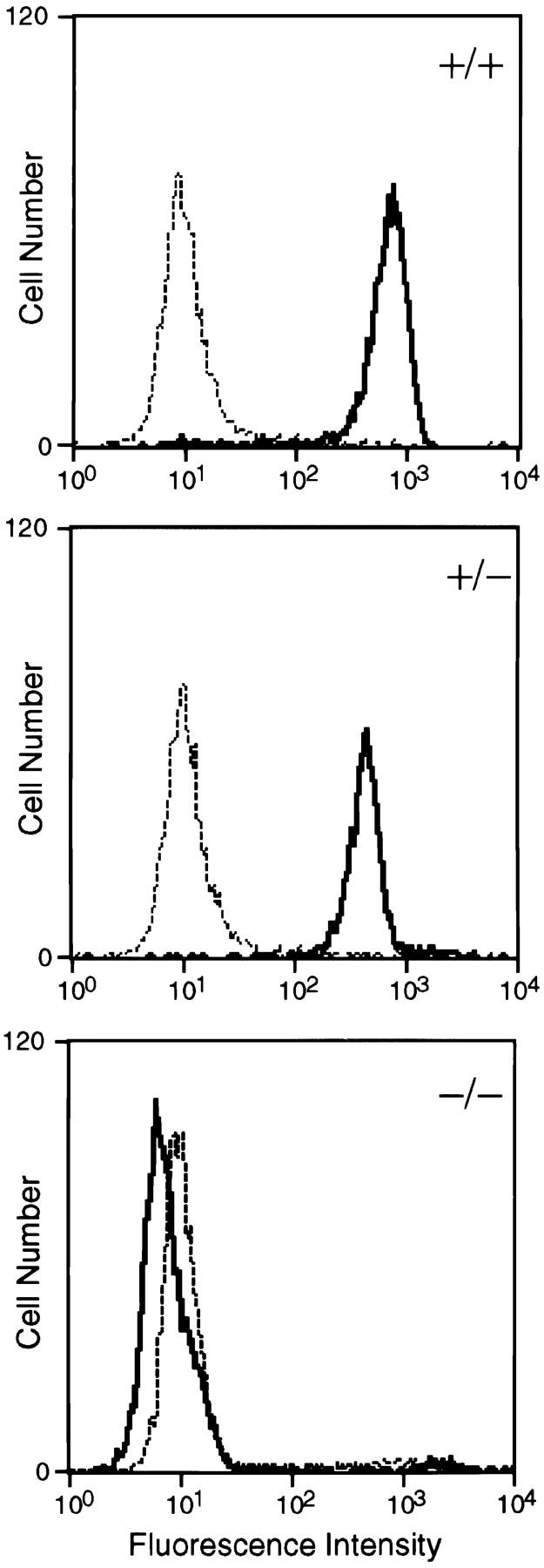
Flow cytometric analysis of PSGL-1 expression on peripheral blood neutrophils. Cells from wild-type (+/+; top), PSGL-1+/− (middle), and PSGL-1−/− (bottom) mice were stained with anti–PSGL-1 antibodies (solid line) or polyclonal rabbit nonimmune IgG (dotted line) followed by FITC-labeled goat anti–rabbit IgG. Fluorescence intensity is shown on the x-axis; cell number is presented on the y-axis.
The absence of PSGL-1 expression in lymphoid organs was also confirmed by immunohistochemistry. Thymus, spleen, and lymph node from wild-type mice showed positive staining on lymphocytes, whereas no specific staining was observed in PSGL-1−/− mice (Fig. 4). Bone marrow was similarly negative for PSGL-1 antigen, in contrast to that from wild-type mice.
Figure 4.
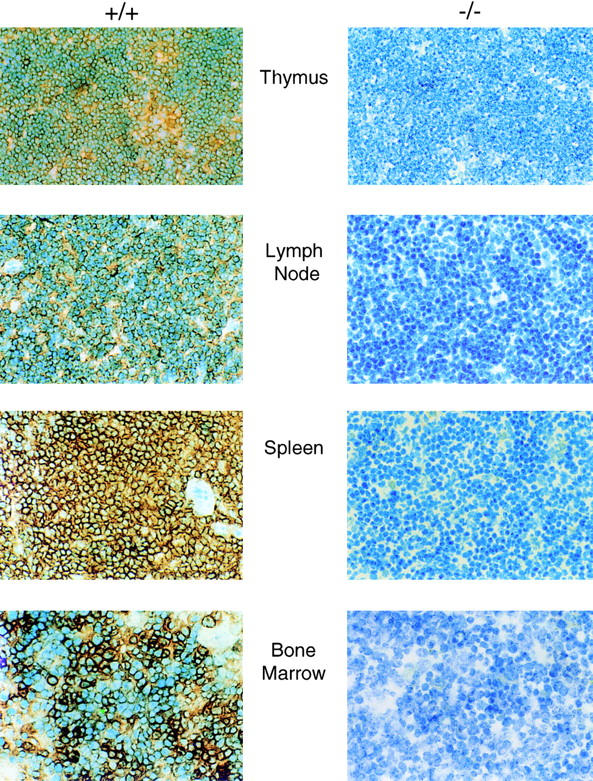
Immunohistochemical analysis of spleen, thymus, lymph node, and bone marrow. Tissue from homozygous null (−/−) and wild-type (+/+) mice is indicated. Fixed tissue sections were treated with normal goat serum followed by rabbit anti–mouse PSGL-1 antibody. Bound anti–mouse PSGL-1 antibody was visualized with biotinylated goat anti–rabbit antibody reacted with peroxidase-conjugated streptavidin. The samples were developed with 3,3′-diaminobenzidine and counterstained with methylene blue.
PSGL-1–deficient Mice Develop Normally.
PSGL-1−/− mice developed normally, and their postnatal growth rate was comparable with that of wild-type mice. Both sexes were fertile. P-selectin and PSGL-1 were previously shown to be cell surface components of the sperm and ovum, respectively, leading to the suggestion that cell adhesion involving P-selectin and PSGL-1 might mediate sperm–ova interaction 69. Our results indicate that the presence of PSGL-1 on the ova, if confirmed, is not required for fertilization. Similar results were obtained for the P-selectin–deficient mouse 6.
Histological analysis of thymus, spleen, and peripheral and mesenteric lymph nodes, where PSGL-1 expression is high, showed no abnormalities, indicating that PSGL-1 is not required for normal lymphoid organ structure. This is in contrast to the abnormal lymph node architecture that characterizes L-selectin–deficient mice 11. Examination of the bone marrow revealed normal erythroid and myeloid maturation; no abnormalities in cell morphology were observed. PSGL-1−/− mice showed no signs of infection up to 12 mo of age, and specifically lacked the ulcerative cutaneous infections exhibited by P- and E-selectin double-deficient mice 12 13.
PSGL-1–deficient Mice Have Mild Neutrophilia.
Leukocytosis is well known to be associated with an inadequate leukocyte adhesion and emigration phenotype. Modest elevation of neutrophil counts is observed in P-selectin–deficient mice 6 and in C2GlnNAc-deficient mice that exhibit partial deficiency of the selectin ligands 70, whereas a remarkable granulocytosis is observed in P- and E-selectin double-deficient mice 12 and fucosyltransferase (Fuc-T) VII–deficient mice that lack ligand activity for all three selectins 71. Therefore, we examined the peripheral blood counts of wild-type and PSGL-1−/− mice. The total leukocyte counts were not significantly different between wild-type and PSGL-1−/− mice, whereas the neutrophil counts in PSGL-1−/− mice were 2.9-fold higher than those in the wild-type mice (Table ). The eosinophil counts tended to be higher in PSGL-1−/− mice (3.5-fold elevation, P = 0.07). The hemoglobin levels and platelet counts in PSGL-1−/− mice are essentially identical to those of the matched wild-type controls. The mild neutrophilia observed in PSGL-1−/− mice is very similar to the 2.4-fold elevation reported with P-selectin–deficient mice 6. The elevation of neutrophil counts in the PSGL-1−/− mice suggests a defect in neutrophil adhesion and migration.
Table 1.
Peripheral Blood Counts
| Blood cell types | Wild-type | PSGL-1−/− |
|---|---|---|
| Total leukocytes (per μl) | 7,060 ± 597 | 7,110 ± 506 |
| Neutrophils | 580 ± 60 | 1,673 ± 121* |
| Lymphocytes | 6,282 ± 586 | 5,106 ± 394 |
| Monocytes | 139 ± 35 | 190 ± 41 |
| Eosinophils | 55 ± 20 | 193 ± 68 |
| Hemoglobin (g/dl) | 15.0 ± 0.2 | 15.4 ± 0.2 |
| Platelets (×103/μl) | 1,038 ± 36 | 1,021 ± 39 |
Blood was collected from the retroorbital plexus of 10 male mice of each genotype. Data are presented as mean ± SEM. *P < 0.001 vs. wild-type.
Early Neutrophil Migration during Experimental Peritonitis Is Impaired in PSGL-1–deficient Mice.
Neutrophil migration in the PSGL-1−/− mice was examined in a thioglycollate-induced peritonitis model. Chemical peritonitis was induced by intraperitoneal thioglycollate injection in both PSGL-1–deficient mice and matched wild-type controls. At 2 h after thioglycollate injection, the number of neutrophils migrating into the peritoneal cavity in PSGL-1−/− mice was 4.3-fold lower than wild-type mice (Fig. 5 A, inset). At the 4-h time point, the difference in neutrophil recruitment between wild-type and PSGL-1−/− mice was reduced to 1.9-fold (Fig. 5 A). At 8 h after thioglycollate injection, the absolute number of neutrophils in the peritoneal cavity of PSGL-1–deficient mice was only slightly lower (1.5-fold) than in the matched wild-type controls. These results demonstrate that the kinetics of neutrophil influx into the peritoneal cavity in PSGL-1−/− mice are characterized by an early defect after the initiation of the inflammatory stimuli. Neutrophil migration approaches normal levels 8 h after exposure to the inflammatory stimuli. To directly compare the results obtained with the PSGL-1–deficient mice to those with P-selectin–deficient mice, we reexamined neutrophil migration in the P-selectin–deficient mice. We observed a 15.7-fold decrease in neutrophil migration at 2 h (Fig. 5 B, inset), a 3.9-fold decrease at 4 h, and a 1.7-fold decrease at 8 h compared with wild-type matched controls (Fig. 5 B), thus confirming earlier results 6. Our observation in PSGL-1–deficient mice of an early defect followed by a significant recovery at later time points is very similar to that of P-selectin–deficient mice, but distinct from that observed in P- and E-selectin double-deficient mice in which neutrophil influx remains low at all time points 12. Although PSGL-1−/− mice and P-selectin−/− mice showed similar kinetics of neutrophil migration in the peritonitis model, PSGL-1−/− mice recruited more neutrophils at each time point than P-selectin–deficient mice when each strain was compared with its wild-type control. It is possible that the difference in the genetic background of the PSGL-1−/− mice (mixed 129Sv/C57BL/6J/SJL) and the P-selectin−/− mice (100% C57BL/6J) may affect the neutrophil extravasation defect in these strains. Alternatively, these results are consistent with the presence of an additional P-selectin ligand.
Figure 5.

Neutrophil migration in thioglycollate-induced peritonitis. PSGL-1–deficient mice with matched controls and P-selectin–deficient mice with matched controls were studied in parallel. Absolute neutrophil counts in the peritoneal exudate were determined at the indicated times after induction of experimental peritonitis with thioglycollate. An expanded comparison at 2 h is presented in the inset. (A) PSGL-1−/− mice (•, and [inset] black bars) and matched wild-type control mice (○, and [inset] white bars). (B) P-selectin−/− mice (▪, and [inset] black bars) and matched wild-type control mice (□, and [inset] white bars). Each data point represents the average of the results of 8–10 mice. Data are presented as mean ± SEM. Differences were statistically significant (*P < 0.001; # P = 0.002) compared with respective wild-type controls.
Leukocyte Rolling In Vivo.
Leukocyte rolling in the postcapillary venules of the cremaster muscle was examined by intravital microscopy in wild-type mice, PSGL-1–deficient mice, and P-selectin–deficient mice to determine the relative role of P-selectin and PSGL-1 in leukocyte trafficking in vivo. This model enables direct examination of granulocytes, which comprise >90% of the adherent leukocytes, with the blood vessel wall 72. In the first series of experiments, leukocyte rolling was assessed at time points <30 min after introduction of the inflammatory stimuli in a model of mild trauma-induced inflammation. A severe early defect in leukocyte rolling was observed in the PSGL-1–deficient animals (Fig. 6). The rolling flux fraction was 1.2% compared with 20.9% in matched wild-type controls (Fig. 6, and Table , top) The microvessel and hemodynamic parameters in the observed vessels were closely matched across all of the genotype groups (Table , top). In agreement with the results of other investigators 6, P-selectin–deficient animals had a pronounced defect in leukocyte rolling, with an average leukocyte rolling flux fraction of 0.4% compared with 18.4% in matched wild-type controls (Fig. 6, and Table , top). These results emphasize that in mild trauma-induced inflammation, leukocyte rolling is markedly decreased but not absent in both PSGL-1–deficient mice and P-selectin–deficient mice within 30 min of introduction of inflammatory stimuli.
Table 2.
Microvessel Parameters
| Genotype | Mice | Vessels | Vessel diameter | Wall shear rate | Rolling flux fraction |
|---|---|---|---|---|---|
| μm | s−1 | % | |||
| Trauma-induced inflammation | |||||
| PSGL-1+/+ | 4 | 23 | 40.1 ± 1.9 | 1,460 ± 158 | 20.9 ± 1.6 |
| PSGL-1−/− | 7 | 36 | 33.2 ± 1.2 | 1,311 ± 148 | 1.2 ± 0.3 |
| P-selectin+/+ | 5 | 31 | 37.3 ± 1.6 | 1,259 ± 143 | 18.4 ± 1.2 |
| P-selectin−/− | 4 | 24 | 33.1 ± 1.7 | 1,034 ± 110 | 0.4 ± 0.2 |
| TNF-α–induced inflammation | |||||
| PSGL-1+/+ | 5 | 25 | 37.5 ± 2.4 | 905 ± 101 | 13.9 ± 1.3 |
| PSGL-1−/− | 4 | 27 | 33.7 ± 1.1 | 857 ± 95 | 5.2 ± 0.7 |
| PSGL-1−/− + 9A9 | 4 | 24 | 36.0 ± 1.7 | 767 ± 97 | 0.6 ± 0.1 |
| P-selectin+/+ | 4 | 36 | 34.7 ± 1.6 | 805 ± 91 | 12.7 ± 1.6 |
| P-selectin−/− | 4 | 28 | 34.1 ± 1.5 | 767 ± 76 | 5.1 ± 0.7 |
To determine the role of PSGL-1 in cytokine-induced inflammation that is thought to be both P-selectin and E-selectin mediated, we compared the rolling behavior of leukocytes in TNF-α–treated cremaster muscles of PSGL-1–deficient mice and matched wild-type controls. Mice were treated with TNF-α, then rolling in the venules of the cremaster muscle was evaluated 2 h later. In contrast to trauma-induced inflammation, PSGL-1–deficient mice had a significant number of rolling leukocytes. A leukocyte rolling flux fraction of 5.2% (Fig. 7, and Table , bottom) was observed. The rolling flux fraction of the TNF-α–treated wild-type mice was 13.9% (Fig. 7, and Table , bottom). TNF-α treatment led to reduced blood flow in the microcirculation of the cremaster muscle and resulted in lower wall shear rate values for TNF-α–treated animals than was observed under conditions of trauma alone (Table , bottom). Within the TNF-α–treated group, the microvessel and hemodynamic parameters were similar in both the wild-type control and PSGL-1 null animals (Table , bottom). Comparable results were obtained with the P-selectin–deficient mice (Fig. 7 D).
To prove that the residual leukocyte rolling in the TNF-α–treated PSGL-1–deficient mice is mediated by E-selectin, the blocking anti–E-selectin antibody 9A9 was infused into the PSGL-1–deficient mouse 8 73. In TNF-α–treated PSGL-1−/− mice, the leukocyte rolling flux decreased to 0.6% in the presence of 9A9 (Fig. 7, and Table , bottom). Leukocytes visible in the still photograph of the cremaster venules from a PSGL-1–deficient mouse treated with both TNF-α and antibody 9A9 (Fig. 7 C) are stationary on the vessel wall and likely adhered via integrins before the infusion of 9A9. In contrast, antibody 9A9 infused into TNF-α–treated wild-type mice had a leukocyte rolling flux of 24.9% (Fig. 7 D). This is likely due to the increased average leukocyte rolling velocity when slow E-selectin–mediated rolling is substituted by faster P-selectin–mediated rolling. In sum, these results indicate that PSGL-1 is not required for E-selectin–mediated leukocyte rolling. The basis for residual rolling after blocking of E-selectin–mediated leukocyte–vessel wall interaction is uncertain, but is consistent with interaction of P-selectin with a ligand on leukocytes other than PSGL-1.
Discussion
The counterreceptors on cell surfaces that bind specifically to the selectin family of adhesion molecules have been difficult to identify, although many candidate selectin ligands have been put forth. As lectins, the selectins bind predominantly oligosaccharide sequences of sialylated fucosylated lactosamine. These sequences are recognized by the selectins in vitro, albeit with low affinity (for a review, see Varki 74). Many carbohydrates, glycoproteins, or glycolipids bearing these oligosaccharide sequences will bind to the selectins in vitro when the receptor or ligand is presented at high density or under static conditions. The issue in understanding the biology of selectin ligands has been to prove specific interaction of candidate ligands with selectins under physiological conditions 47 75.
Although in vitro studies performed with isolated selectins and potential ligands or by cell adhesions assays have been invaluable in defining candidates in the inflammatory pathway, the determination of physiological import of each of these adhesion molecules requires study in animal models. The three selectins contribute to leukocyte rolling on the endothelial cell lining of venules in a cooperative and sequential manner in the early stages of the inflammatory process. Genetic disruption of the P-selectin gene in mice demonstrated that P-selectin is required for neutrophil rolling in postcapillary venules and for neutrophil influx into inflamed peritoneum at early time points 6 7. These studies defined a critical role for P-selectin in early inflammatory events and led to the understanding that P-selectin initiates leukocyte rolling at sites of inflammation and tissue injury. Disruption of the L-selectin gene in mice established that this molecule is also essential for normal leukocyte rolling and emigration into the inflamed peritoneum 7 11. L-selectin mediates leukocyte rolling in inflamed venules sequentially after P-selectin at time points between 40 and 120 min 7. In addition to defects in neutrophil rolling and migration, the L-selectin–deficient mice had significant defects in lymphocyte homing to peripheral lymphoid tissue 11 76. Mice lacking E-selectin initially demonstrated no defects in the inflammatory response 77. However, detailed analysis later showed that slow, stable granulocyte rolling was absent in E-selectin null mice 10 and that E-selectin was required for firm leukocyte attachment 78. Further studies using blocking E-selectin antibodies demonstrated that E-selectin adhesive functions overlap with P-selectin in the later phases (>2 h) of inflammation 79. The cooperative and overlapping function of the selectins is demonstrated in mice that are doubly deficient in both P-selectin and E-selectin. Unlike mice deficient in a single selectin, mice deficient for both P-selectin and E-selectin are characterized by spontaneous skin infections, leukocytosis, and drastic reduction in leukocyte rolling and neutrophil migration into the inflamed peritoneum at both early and late time points 12 13.
Thus, animal studies have demonstrated that P-selectin is required to initiate early contact of leukocytes with the vessel wall, and that after this initial step there is significant cooperativity of function of all three selectins. Using a PSGL-1–deficient mouse, we have explored the role of this glycoprotein in selectin-mediated leukocyte adhesion with the specific goal of determining whether PSGL-1 is the sole ligand for P-selectin and whether PSGL-1 interaction with E-selectin is important for the propagation of the normal inflammatory response.
We have now demonstrated that early neutrophil migration after chemically induced peritonitis is impaired in PSGL-1–deficient mice. Previous reports demonstrated that neutrophil migration into inflamed peritoneum is impaired in mice that lack P-selectin or L-selectin but not E-selectin 6 11 77. P-selectin–deficient mice and L-selectin–deficient mice demonstrate depressed neutrophil migration at the early time points (2–4 h) after chemical insult. In contrast, mice that lack Fuc-T VII or are deficient in both P-selectin and E-selectin show a marked reduction in neutrophil migration at time points up to 8 h 12 71. Thus, in this assay deficiency of multiple selectin functions results in a more severe phenotype. In our chemical peritonitis studies, the PSGL-1–deficient animals demonstrated a defect in neutrophil migration that was marked at 2 h (77% reduction) but was only minimally abnormal at the later time points. This phenotype more closely resembled that of P-selectin–deficient mice than the severe migration defect seen in mice deficient in both P-selectin and E-selectin 12 13. When compared with the P-selectin–deficient mice, the PSGL-1–deficient mice had significantly more neutrophils migrating into the inflamed peritoneum at parallel time points. These findings suggest the presence of additional P-selectin ligands other than PSGL-1, although PSGL-1 nonetheless appears to be an important and dominant P-selectin ligand.
Our leukocyte rolling experiments have shown that early trauma-induced leukocyte–endothelial cell interaction is greatly reduced, whereas cytokine-induced leukocyte rolling is only modestly impacted in the PSGL-1–deficient mice. Trauma-induced neutrophil rolling in the postcapillary venules of P-selectin–deficient and L-selectin–deficient mice is reduced initially but approaches normal levels as inflammation progresses 6 7 11. In contrast, neutrophil rolling is virtually absent in trauma- and TNF-α–induced inflammation in mice deficient in both P-selectin and E-selectin 12 13. In our current work, we have established that PSGL-1 is required for the early, P-selectin–mediated phases of leukocyte rolling at times <30 min after the inflammatory stimulus. Similar to the P-selectin–deficient mice, neutrophil rolling in PSGL-1–deficient mice is reduced by ∼95% compared with wild-type mice. Based on this observation, PSGL-1 appears to be the dominant P-selectin ligand early in trauma-induced inflammatory events.
TNF-α is a cytokine that induces E-selectin biosynthesis in endothelial cells and promotes leukocyte rolling in part mediated by E-selectin. Mice deficient in both P-selectin and E-selectin have profound leukocyte rolling defects in TNF-α–treated venules 12 13. In our studies, the PSGL-1–deficient mice demonstrated a significant amount of leukocyte rolling (5.2% flux) after 2 h of pretreatment with TNF-α. The leukocyte rolling flux for the TNF-α–treated PSGL-1–deficient mice (5.2%) was identical to that of TNF-α–treated P-selectin–deficient mice (5.1%) and is significantly higher than the leukocyte rolling flux reported in TNF-α–primed mice deficient in both P-selectin and E-selectin 12 13. This finding suggests that PSGL-1 functions mainly as a P-selectin ligand in this system; the preserved function of E-selectin and L-selectin with their complementary counterreceptors supports a TNF-α–induced rolling flux in the absence of P-selectin or PSGL-1. Furthermore, the presence of a measurable leukocyte rolling flux in the PSGL-1–deficient mouse raises questions of the importance of leukocyte–leukocyte rolling interactions mediated by L-selectin and PSGL-1.
Leukocytosis, the hallmark of a major defect in host defense, is often associated with recurrent infection or chronic ulceration as a manifestation of blockade of leukocyte activity. Mice deficient in both P-selectin and E-selectin 12 13 and mice deficient in the fucosyltransferase required for synthesis of the selectin ligands 71 exhibit leukocytosis. However, our PSGL-1–deficient mice exhibited only a mild elevation of neutrophils. Since genetic deficiency of P-selectin, E-selectin, L-selectin, or, from these studies, PSGL-1, is not associated with significant leukocytosis, it would appear that the presence of 5% of normal leukocyte rolling and delayed neutrophil migration comprises an adequate host defense. Thus, no single deficiency of a selectin or selectin ligand causes a phenotype of leukocytosis, chronic infection, and early demise. This observation speaks to the importance of redundancy in these host defense systems. It is also consistent with our observation that PSGL-1 is the dominant ligand for P-selectin, but not required for the function of E-selectin within the context of leukocyte rolling.
These experiments using PSGL-1–deficient mice have established that PSGL-1 is a critical P-selectin ligand early in inflammation. Comparison of the phenotype of the PSGL-1–deficient mouse and the mouse deficient in both P-selectin and E-selectin 12 13 provides evidence that PSGL-1 is not a required neutrophil ligand for E-selectin–mediated neutrophil rolling. In contrast to the mice deficient in both P-selectin and E-selectin, the PSGL-1–deficient mice have no leukocytosis, no susceptibility to spontaneous infection, and have detectable leukocyte rolling and adequate neutrophil migration into the inflamed peritoneum after inflammatory stimuli. If PSGL-1 were the sole ligand for both P-selectin and E-selectin, the PSGL-1–deficient mice and the mice deficient in both P-selectin and E-selectin would have the same phenotype. These data provide strong evidence for the existence of additional physiological ligands for E-selectin, perhaps ESL-1 31, and do not support a requirement for PSGL-1 in E-selectin–mediated neutrophil rolling. The relative importance of PSGL-1–E-selectin interaction during T cell homing 40 80 is unresolved, but further studies using PSGL-1–deficient mice will address this important question.
Acknowledgments
We are grateful to Dr. Harold Dvorak for his interpretation of the mouse tissue histology, Dr. Lia Palomba for preparing the antibodies to mouse PSGL-1, Dr. Andrea Bottaro for technical advice in generating the knockout construct, Dr. Barry Wolitzky for supplying antibodies to E-selectin, Dr. Pamela Stanley for the ES cells, and Dr. Klaus Ley for teaching us his in vivo leukocyte rolling assay.
This work was supported by grant HL51926 from the National Institutes of Health.
Footnotes
J. Yang's present address is Department of Medicine, University of Pennsylvania School of Medicine, Philadelphia, PA 19104.
The online version of this article contains supplemental material.
J. Yang, T. Hirata, and K. Croce contributed equally to this work.
Abbreviations used in this paper: ES, embryonic stem; PSGL-1, P-selectin glycoprotein ligand 1.
References
- Springer T.A. Traffic signals for lymphocyte recirculation and leukocyte emigrationthe multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- Tedder T.F., Steeber D.A., Chen A., Engel P. The selectinsvascular adhesion molecules. FASEB J. 1995;9:866–873. [PubMed] [Google Scholar]
- McEver R.P., Moore K.L., Cummings R.D. Leukocyte trafficking mediated by selectin-carbohydrate interactions. J. Biol. Chem. 1995;270:11025–11028. doi: 10.1074/jbc.270.19.11025. [DOI] [PubMed] [Google Scholar]
- Stenberg P.E., McEver R.P., Shuman M.A., Jacques Y.V., Bainton D.F. A platelet alpha-granule membrane protein GMP140 is expressed on the plasma membrane after activation. J. Cell Biol. 1985;101:880–886. doi: 10.1083/jcb.101.3.880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman C.L., Yeo E., Wencel-Drake J.D., Furie B.C., Ginsberg M.H., Furie B. A platelet alpha granule membrane protein that is incorporated into the plasma membrane during activation. Characterization and subcellular localization of PADGEM protein. J. Clin. Invest. 1986;78:130–137. doi: 10.1172/JCI112542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayadas T.N., Johnson R.C., Rayburn H., Hynes R.O., Wagner D.D. Leukocyte rolling and extravasation are severely compromised in P-selectin-deficient mice. Cell. 1993;74:541–554. doi: 10.1016/0092-8674(93)80055-j. [DOI] [PubMed] [Google Scholar]
- Ley K., Bullard D.C., Arbones M.L., Bosse R., Vestweber D., Tedder T.F., Beaudet A.L. Sequential contribution of L- and P-selectin to leukocyte rolling in vivo. J. Exp. Med. 1995;181:669–675. doi: 10.1084/jem.181.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkel E.J., Jung U., Bullard D.C., Norman K.E., Wolitzky B.A., Vestweber D., Beaudet A.L., Ley K. Absence of trauma-induced leukocyte rolling in mice deficient in both P-selectin and intercellular adhesion molecule 1. J. Exp. Med. 1996;183:57–65. doi: 10.1084/jem.183.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua M.P., Stengalin S., Gimbrone M.A., Jr., Seed B. Endothelial leukocyte adhesion molecule 1an inducible receptor for neutrophils related to complement regulatory proteins and lectins. Science. 1989;243:1160–1165. doi: 10.1126/science.2466335. [DOI] [PubMed] [Google Scholar]
- Kunkel E.J., Ley K. Distinct phenotype of E-selectin-deficient mice. E-selectin is required for slow leukocyte rolling in vivo. Circ. Res. 1996;79:1196–1204. doi: 10.1161/01.res.79.6.1196. [DOI] [PubMed] [Google Scholar]
- Arbones M.L., Ord D.C., Ley K., Ratech H., Maynard C.C., Otten G., Capon D.J., Tedder T.F. Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin-deficient mice. Immunity. 1994;1:247–260. doi: 10.1016/1074-7613(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Frenette P.S., Mayadas T.N., Rayburn H., Hynes R.O., Wagner D.D. Susceptibility to infection and altered hematopoiesis in mice deficient in both P- and E-selectins. Cell. 1996;84:563–574. doi: 10.1016/s0092-8674(00)81032-6. [DOI] [PubMed] [Google Scholar]
- Bullard D.C., Kunkel E.J., Kubo H., Hicks M.J., Lorenzo I., Doyle N.A., Doerschuk C.M., Ley K., Beaudet A.L. Infectious susceptibility and severe deficiency of leukocyte rolling and recruitment in E-selectin and P-selectin double mutant mice. J. Exp. Med. 1996;183:2329–2336. doi: 10.1084/jem.183.5.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen E., Palabrica T., Sajer S., Gilbert G.E., Wagner D.D., Furie B.C., Furie B. PADGEM-dependent adhesion of platelets to monocytes and neutrophils is mediated by a lineage-specific carbohydrate, LNF III (CD15) Cell. 1990;63:467–474. doi: 10.1016/0092-8674(90)90443-i. [DOI] [PubMed] [Google Scholar]
- Polley M.J., Phillips M.L., Wayner E., Nudelman E., Singhal A.K., Hakomori S., Paulson J.C. CD62 and endothelial cell-leukocyte adhesion molecule 1 ELAM-1 recognize the same carbohydrate ligand, sialyl-Lewis x. Proc. Natl. Acad. Sci. USA. 1991;88:6224–6228. doi: 10.1073/pnas.88.14.6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxall C., Watson S.R., Dowbenko D., Fennie C., Lasky L.A., Kiso M., Hasegawa A., Asa D., Brandley B.K. The three members of the selectin receptor family recognize a common carbohydrate epitope, the sialyl Lewis(x) oligosaccharide. J. Cell Biol. 1992;117:895–902. doi: 10.1083/jcb.117.4.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picker L.J., Warnock R.A., Burns A.R., Doerschuk C.M., Berg E.L., Butcher E.C. The neutrophil selectin LECAM-1 presents carbohydrate ligands to the vascular selectins ELAM-1 and GMP-140. Cell. 1991;66:921–933. doi: 10.1016/0092-8674(91)90438-5. [DOI] [PubMed] [Google Scholar]
- Lasky L.A., Singer M.S., Dowbenko D., Imai Y., Henzel W.J., Grimley C., Fennie C., Gillett N., Watson S.R., Rosen S.D. An endothelial ligand for L-selectin is a novel mucin-like molecule. Cell. 1992;69:927–938. doi: 10.1016/0092-8674(92)90612-g. [DOI] [PubMed] [Google Scholar]
- Baumheuter S., Singer M.S., Henzel W., Hemmerich S., Renz M., Rosen S.D., Lasky L.A. Binding of L-selectin to the vascular sialomucin CD34. Science. 1993;262:436–438. doi: 10.1126/science.7692600. [DOI] [PubMed] [Google Scholar]
- Baumheuter S., Dybdal N., Kyle C., Lasky L.A. Global vascular expression of murine CD34, a sialomucin-like endothelial ligand for L-selectin. Blood. 1994;84:2554–2565. [PubMed] [Google Scholar]
- Hemmerich S., Butcher E.C., Rosen S.D. Sulfation-dependent recognition of high endothelial venule (HEV)-ligands by L-selectin and MECA 79, and adhesion-blocking monoclonal antibody. J. Exp. Med. 1994;180:2219–2226. doi: 10.1084/jem.180.6.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg E.L., McEvoy L.M., Berlin C., Bargatze R.F., Butcher E.C. L-selectin-mediated lymphocyte rolling on MAdCAM-1. Nature. 1993;366:695–698. doi: 10.1038/366695a0. [DOI] [PubMed] [Google Scholar]
- Guyer D.A., Moore K.L., Lynam E.B., Schammel C.M., Rogelj S., McEver R.P., Sklar L.A. P-selectin glycoprotein ligand-1 (PSGL-1) is a ligand for L-selectin in neutrophil aggregation. Blood. 1996;88:2415–2421. [PubMed] [Google Scholar]
- Spertini O., Cordey A.S., Monai N., Giuffre L., Schapira M. P-selectin glycoprotein ligand 1 is a ligand for L-selectin on neutrophils, monocytes, and CD34+ hematopoietic progenitor cells. J. Cell Biol. 1996;135:523–531. doi: 10.1083/jcb.135.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu L., Chen A., Delahunty M.D., Moore K.L., Watson S.R., McEver R.P., Tedder T.F. L-selectin binds to P-selectin glycoprotein ligand-1 on leukocytesinteractions between the lectin, epidermal growth factor, and consensus repeat domains of the selectins determine ligand binding specificity. J. Immunol. 1996;157:3995–4004. [PubMed] [Google Scholar]
- Alon R., Fuhlbrigge R.C., Finger E.B., Springer T.A. Interactions through L-selectin between leukocytes and adherent leukocytes nucleate rolling adhesions on selectins and VCAM-1 in shear flow. J. Cell Biol. 1996;135:849–865. doi: 10.1083/jcb.135.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargatze R.F., Kurk S., Butcher E.C., Jutila M.A. Neutrophils roll on adherent neutrophils bound to cytokine-induced endothelial cells via L-selectin on the rolling cells. J. Exp. Med. 1994;180:1785–1792. doi: 10.1084/jem.180.5.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jutila M.A., Kurk S. Analysis of bovine gamma delta T cell interactions with E-, P-, and L-selectin. Characterization of lymphocyte on lymphocyte rolling and the effects of O-glycoprotease. J. Immunol. 1996;156:289–296. [PubMed] [Google Scholar]
- Walcheck B., Moore K.L., McEver R.P., Kishimoto T.K. Neutrophil–neutrophil interactions under hydrodynamic shear stress involve L-selectin and PSGL-1. A mechanism that amplifies initial leukocyte accumulation of P-selectin in vitro. J. Clin. Invest. 1996;98:1081–1087. doi: 10.1172/JCI118888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel K.D., McEver R.P. Comparison of tethering and rolling of eosinophils and neutrophils through selectins and P-selectin glycoprotein ligand-1. J. Immunol. 1997;159:4555–4565. [PubMed] [Google Scholar]
- Levinovitz A., Muhlhoff J., Isenmann S., Vestweber D. Identification of a glycoprotein ligand for E-selectin on mouse myeloid cells. J. Cell Biol. 1993;121:449–459. doi: 10.1083/jcb.121.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones W.M., Watts G.M., Robinson M.K., Vestweber D., Jutila M.A. Comparison of E-selectin-binding glycoprotein ligands on human lymphocytes, neutrophils, and bovine gamma delta T cells. J. Immunol. 1997;159:3574–3583. [PubMed] [Google Scholar]
- Zollner O., Lenter M.C., Blanks J.E., Borges E., Steegmaier M., Zerwes H.G., Vestweber D. L-selectin from human, but not from mouse neutrophils binds directly to E-selectin. J. Cell Biol. 1997;136:707–716. doi: 10.1083/jcb.136.3.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako D., Chang X.J., Barone K.M., Vachino G., White H.M., Shaw G., Veldman G.M., Bean K.M., Ahern T.J., Furie B. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell. 1993;75:1179–1186. doi: 10.1016/0092-8674(93)90327-m. [DOI] [PubMed] [Google Scholar]
- Asa D., Raycroft L., Ma L., Aeed P.A., Kaytes P.S., Elhammer A.P., Geng J.G. The P-selectin glycoprotein ligand functions as a common human leukocyte ligand for P- and E-selectins. J. Biol. Chem. 1995;270:11662–11670. doi: 10.1074/jbc.270.19.11662. [DOI] [PubMed] [Google Scholar]
- Goetz D.J., Greif D.M., Ding H., Camphausen R.T., Howes S., Comess K.M., Snapp K.R., Kansas G.S., Luscinskas F.W. Isolated P-selectin glycoprotein ligand-1 dynamic adhesion to P- and E-selectin. J. Cell Biol. 1997;137:509–519. doi: 10.1083/jcb.137.2.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F., Wilkins P.P., Crawley S., Weinstein J., Cummings R.D., McEver R.P. Post-translational modifications of recombinant P-selectin glycoprotein ligand-1 required for binding to P- and E-selectin. J. Biol. Chem. 1996;271:3255–3264. [PubMed] [Google Scholar]
- Moore K.L., Eaton S.F., Lyons D.E., Lichenstein H.S., Cummings R.D., McEver R.P. The P-selectin glycoprotein ligand from human neutrophils displays sialylated, fucosylated, O-linked poly-N-acetyllactosamine. J. Biol. Chem. 1994;269:23318–23327. [PubMed] [Google Scholar]
- Patel K.D., Moore K.L., Nollert M.U., McEver R.P. Neutrophils use both shared and distinct mechanisms to adhere to selectins under static and flow conditions. J. Clin. Invest. 1995;96:1887–1896. doi: 10.1172/JCI118234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges E., Pendl G., Eytner R., Steegmaier M., Zollner O., Vestweber D. The binding of T cell-expressed P-selectin glycoprotein ligand-1 to E- and P-selectin is differentially regulated. J. Biol. Chem. 1997;272:28786–28792. doi: 10.1074/jbc.272.45.28786. [DOI] [PubMed] [Google Scholar]
- Sammar M., Aigner S., Hubbe M., Schirrmacher V., Schachner M., Vestweber D., Altevogt P. Heat-stable antigen CD24 as ligand for mouse P-selectin. Int. Immunol. 1994;6:1027–1036. doi: 10.1093/intimm/6.7.1027. [DOI] [PubMed] [Google Scholar]
- Aigner S., Ramos C.L., Hafezi-Moghadam A., Lawrence M.B., Friederichs J., Altevogt P., Ley K. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J. 1998;12:1241–1251. doi: 10.1096/fasebj.12.12.1241. [DOI] [PubMed] [Google Scholar]
- Aigner S., Sthoeger Z.M., Fogel M., Weber E., Zarn J., Ruppert M., Zeller Y., Vestweber D., Stahel R., Sammar M., Altevogt P. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood. 1997;89:3385–3395. [PubMed] [Google Scholar]
- Aruffo A., Kolanus W., Walz G., Fredman P., Seed B. CD62/P-selection recognition of myeloid and tumor cell sulfatides. Cell. 1991;67:35–44. doi: 10.1016/0092-8674(91)90570-o. [DOI] [PubMed] [Google Scholar]
- Diacovo T.G., Puri K.D., Warnock R.A., Springer T.A., von Andrian U.H. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 1996;273:252–255. doi: 10.1126/science.273.5272.252. [DOI] [PubMed] [Google Scholar]
- Moore K.L., Patel K.D., Bruehl R.E., Li F., Johnson D.A., Lichenstein H.S., Cummings R.D., Bainton D.F., McEver R.P. P-selectin glycoprotein ligand-1 mediates rolling of human neutrophils on P-selectin. J. Cell Biol. 1995;128:661–671. doi: 10.1083/jcb.128.4.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Selectin ligandswill the real ones please stand up? J. Clin. Invest. 1997;99:158–162. doi: 10.1172/JCI119142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman K.E., Moore K.L., McEver R.P., Ley K. Leukocyte rolling in vivo is mediated by P-selectin glycoprotein ligand-1. Blood. 1995;86:4417–4421. [PubMed] [Google Scholar]
- Borges E., Eytner R., Moll T., Steegmaier M., Campbell M.A., Ley K., Mossmann H., Vestweber D. The P-selectin glycoprotein ligand-1 is important for recruitment of neutrophils into inflamed mouse peritoneum. Blood. 1997;90:1934–1942. [PubMed] [Google Scholar]
- Takada M., Nadeau K.C., Shaw G.D., Tilney N.L. Early cellular and molecular changes in ischemia/reperfusion injuryinhibition by a selectin antagonist, P-selectin glycoprotein ligand-1. Transplant. Proc. 1997;29:1324–1325. doi: 10.1016/s0041-1345(96)00577-5. [DOI] [PubMed] [Google Scholar]
- Laszik Z., Jansen P.J., Cummings R.D., Tedder T.F., McEver R.P., Moore K.L. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood. 1996;88:3010–3021. [PubMed] [Google Scholar]
- Vachino G., Chang X.J., Veldman G.M., Kumar R., Sako D., Fouser L.A., Berndt M.C., Cumming D.A. P-selectin glycoprotein ligand-1 is the major counter-receptor for P-selectin on stimulated T cells and is widely distributed in non-functional form on many lymphocytic cells. J. Biol. Chem. 1995;270:21966–21974. doi: 10.1074/jbc.270.37.21966. [DOI] [PubMed] [Google Scholar]
- Fujimoto T.T., Noda M., Takafuta T., Shimomura T., Fujimura K., Kuramoto A. Expression and functional characterization of the P-selectin glycoprotein ligand-1 in various cells. Int. J. Hematol. 1996;64:231–239. doi: 10.1016/0925-5710(96)00474-4. [DOI] [PubMed] [Google Scholar]
- Diacovo T.G., Roth S.J., Morita C.T., Rosat J.P., Brenner M.B., Springer T.A. Interactions of human α/β and γ/δ T lymphocyte subsets in shear flow with E-selectin and P-selectin. J. Exp. Med. 1996;183:1193–1203. doi: 10.1084/jem.183.3.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J., Galipeau J., Kozak C.A., Furie B.C., Furie B. Mouse P-selectin glycoprotein ligand-1molecular cloning, chromosomal localization, and expression of a functional P-selectin receptor. Blood. 1996;87:4176–4186. [PubMed] [Google Scholar]
- Snapp K.R., Craig R., Herron M., Nelson R.D., Stoolman L.M., Kansas G.S. Dimerization of P-selectin glycoprotein ligand-1 (PSGL-1) required for optimal recognition of P-selectin. J. Cell Biol. 1998;142:263–270. doi: 10.1083/jcb.142.1.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce K., Freedman S.J., Furie B.C., Furie B. Interaction between soluble P-selectin and soluble P-selectin glycoprotein ligand 1equilibrium binding analysis. Biochemistry. 1998;37:16472–16480. doi: 10.1021/bi981341g. [DOI] [PubMed] [Google Scholar]
- Sako D., Comess K.M., Barone K.M., Camphausen R.T., Cumming D.A., Shaw G.D. A sulfated peptide segment at the amino terminus of PSGL-1 is critical for P-selectin binding. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]
- Pouyani T., Seed B. PSGL-1 recognition of P-selectin is controlled by a tyrosine sulfation consensus at the PSGL-1 amino terminus. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]
- Wilkins P.P., Moore K.L., McEver R.P., Cummings R.D. Tyrosine sulfation of P-selectin glycoprotein ligand-1 is required for high affinity binding to P-selectin. J. Biol. Chem. 1995;270:22677–22680. doi: 10.1074/jbc.270.39.22677. [DOI] [PubMed] [Google Scholar]
- Symon F.A., Lawrence M.B., Williamson M.L., Walsh G.M., Watson S.R., Wardlaw A.J. Functional and structural characterization of the eosinophil P-selectin ligand. J. Immunol. 1996;157:1711–1719. [PubMed] [Google Scholar]
- Veldman G.M., Bean K.M., Cumming D.A., Eddy R.L., Sait S.N., Shows T.B. Genomic organization and chromosomal localization of the gene encoding human P-selectin glycoprotein ligand. J. Biol. Chem. 1995;270:16470–16475. doi: 10.1074/jbc.270.27.16470. [DOI] [PubMed] [Google Scholar]
- Li F., Erickson H.P., James J.A., Moore K.L., Cummings R.D., McEver R.P. Visualization of P-selectin glycoprotein ligand-1 as a highly extended molecule and mapping of protein epitopes for monoclonal antibodies. J. Biol. Chem. 1996;271:6342–6348. doi: 10.1074/jbc.271.11.6342. [DOI] [PubMed] [Google Scholar]
- Liu W., Ramachandran V., Kang J., Kishimoto T.K., Cummings R.D., McEver R.P. Identification of N-terminal residues on P-selectin glycoprotein ligand-1 required for binding to P-selectin. J. Biol. Chem. 1998;273:7078–7087. doi: 10.1074/jbc.273.12.7078. [DOI] [PubMed] [Google Scholar]
- Ioffe E., Liu Y., Bhaumik M., Poirier F., Factor S.M., Stanley P. WW6an embryonic stem cell line with an inert genetic marker that can be traced in chimeras. Proc. Natl. Acad. Sci. USA. 1995;92:7357–7361. doi: 10.1073/pnas.92.16.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky H.H., Zweifach B.W. Application of the “two-slit” photometric technique to the measurement of microvascular volumetric flow rates. Microvasc. Res. 1978;15:93–101. doi: 10.1016/0026-2862(78)90009-2. [DOI] [PubMed] [Google Scholar]
- Krayev A.S., Markusheva T.V., Kramerov D.A., Ryskov A.P., Skyabin K.G., Bayev A.A., Georgiev G.P. Ubiquitous transposon-like repeats B1 and B2 of the mouse genomeB2 sequencing. Nucleic Acids Res. 1982;10:7461–7475. doi: 10.1093/nar/10.23.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Volpe A., Simeone A., D'Esposito M., Scotto L., Fidanza V., de Falco A., Boncinelli E. Molecular analysis of the heterogeneity region of the human ribosomal spacer. J. Mol. Biol. 1985;183:213–223. doi: 10.1016/0022-2836(85)90214-1. [DOI] [PubMed] [Google Scholar]
- Geng J.G., Raub T.J., Baker C.A., Sawada G.A., Ma L., Elhammer A.P. Expression of a P-selectin ligand in zona pellucida of porcine oocytes and P-selectin on acrosomal membrane of porcine sperm cells. Potential implications for their involvement in sperm–egg interactions. J. Cell Biol. 1997;137:743–754. doi: 10.1083/jcb.137.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellies L.G., Tsuboi S., Petryniak B., Lowe J.B., Fukuda M., Marth J.D. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- Maly P., Thall A., Petryniak B., Rogers C.E., Smith P.L., Marks R.M., Kelly R.J., Gersten K.M., Cheng G., Saunders T.L. The alpha1,3fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–653. doi: 10.1016/s0092-8674(00)80137-3. [DOI] [PubMed] [Google Scholar]
- Jung U., Bullard D.C., Tedder T.F., Ley K. Velocity differences between L- and P-selectin-dependent neutrophil rolling in venules of mouse cremaster muscle in vivo. Am. J. Physiol. 1996;271:H2740–H2747. doi: 10.1152/ajpheart.1996.271.6.H2740. [DOI] [PubMed] [Google Scholar]
- Norton C.R., Rumberger J.M., Burns D.K., Wolitzky B.A. Characterization of murine E-selectin expression in vitro using novel anti-mouse E-selectin monoclonal antibodies. Biochem. Biophys. Res. Commun. 1993;195:250–258. doi: 10.1006/bbrc.1993.2037. [DOI] [PubMed] [Google Scholar]
- Varki A. Selectin ligands. Proc. Natl. Acad. Sci. USA. 1994;91:7390–7397. doi: 10.1073/pnas.91.16.7390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestweber D. Ligand-specificity of the selectins. J. Cell. Biochem. 1996;61:585–591. doi: 10.1002/(SICI)1097-4644(19960616)61:4%3C585::AID-JCB12%3E3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Steeber D.A., Green N.E., Sato S., Tedder T.F. Lymphocyte migration in L-selectin-deficient mice. Altered subset migration and aging of the immune system. J. Immunol. 1996;157:1096–1106. [PubMed] [Google Scholar]
- Labow M.A., Norton C.R., Rumberger J.M., Lombard G.K., Shuster D.J., Hubbard J., Bertko R., Knaack P.A., Terry R.W., Harbison M.L. Characterization of E-selectin-deficient micedemonstration of overlapping function of the endothelial selectins. Immunity. 1994;1:709–720. doi: 10.1016/1074-7613(94)90041-8. [DOI] [PubMed] [Google Scholar]
- Ley K., Allietta M., Bullard D.C., Morgan S. Importance of E-selectin for firm leukocyte adhesion in vivo. Circ. Res. 1998;83:287–294. doi: 10.1161/01.res.83.3.287. [DOI] [PubMed] [Google Scholar]
- Ramos C.L., Kunkel E.J., Lawrence M.B., Jung U., Vestweber D., Bosse R., McIntyre K.W., Gillooly K.M., Norton C.R., Wolitzky B.A., Ley K. Differential effect of E-selectin antibodies on neutrophil rolling and recruitment to inflammatory sites. Blood. 1997;89:3009–3018. [PubMed] [Google Scholar]
- Fuhlbrigge R.C., Kieffer J.D., Armerding D., Kupper T.S. Cutaneous lymphocyte antigen is a specialized form of PSGL-1 expressed on skin-homing T cells. Nature. 1997;389:978–981. doi: 10.1038/40166. [DOI] [PubMed] [Google Scholar]