

Abstract
The protozoan parasite Toxoplasma gondii actively penetrates its host cell by squeezing through a moving junction that forms between the host cell plasma membrane and the parasite. During invasion, this junction selectively controls internalization of host cell plasma membrane components into the parasite-containing vacuole. Membrane lipids flowed past the junction, as shown by the presence of the glycosphingolipid GM1 and the cationic lipid label 1.1′-dihexadecyl-3-3′-3-3′-tetramethylindocarbocyanine (DiIC16). Glycosylphosphatidylinositol (GPI)-anchored surface proteins, such as Sca-1 and CD55, were also readily incorporated into the parasitophorous vacuole (PV). In contrast, host cell transmembrane proteins, including CD44, Na+/K+ ATPase, and β1-integrin, were excluded from the vacuole. To eliminate potential differences in sorting due to the extracellular domains, parasite invasion was examined in host cells transfected with recombinant forms of intercellular adhesion molecule 1 (ICAM-1, CD54) that differed in their mechanism of membrane anchoring. Wild-type ICAM-1, which contains a transmembrane domain, was excluded from the PV, whereas both GPI-anchored ICAM-1 and a mutant of ICAM-1 missing the cytoplasmic tail (ICAM-1–Cyt−) were readily incorporated into the PV membrane. Our results demonstrate that during host cell invasion, Toxoplasma selectively excludes host cell transmembrane proteins at the moving junction by a mechanism that depends on their anchoring in the membrane, thereby creating a nonfusigenic compartment.
Keywords: membrane sorting, lipid domains, phagocytosis, moving junction, invasion
Toxoplasma gondii is a member of the Apicomplexa phylum, a diverse group of obligate intracellular protozoan parasites, including Plasmodium, the causative agent of human malaria, that are unified by a common set of apical structures involved in cell invasion 1. Toxoplasma has the broadest host range among this diverse group of parasites, and is able to invade and infect virtually any nucleated cell from a warm-blooded vertebrate 2. Using a mechanism that is likely to be shared by other members of this phylum, Toxoplasma enters both phagocytic and nonphagocytic cells by active penetration, a rapid process (20–30 s) that does not rely on the host cell endocytic machinery for uptake 3 4. A direct result of this active invasion process is the formation of a specialized compartment called the parasitophorous vacuole (PV), which resists fusion with all levels of the endocytic network 5 6 7 8. The parasite remains within this segregated compartment throughout its intracellular lifecycle, dividing within the vacuole and eventually lysing the host cell. Defining how this unique intracellular lifestyle is established is important for an understanding of host defense in the context of antigen presentation and activation of cells for destruction of intracellular microbes.
Recent electrophysiological studies 9 confirm an earlier model that the Toxoplasma-containing PV forms primarily by invagination of the host cell plasma membrane 10. During invasion, the parasite squeezes through a tight constriction formed at the junction of the host cell and parasite plasma membrane that has been called the moving junction 10 11. Freeze–fracture analysis during Toxoplasma 12 or Plasmodium 13 invasion has shown that the nascent PV is devoid of intramembranous particles, implying that the majority of host cell membrane proteins are absent from the PV. Such a process may be accomplished by the moving junction; however, the mechanism of this sorting remains a mystery.
To determine the selectivity and mechanism of sorting during formation of the PV, we have analyzed the partitioning of host cell surface membrane lipids versus proteins that are differentially tethered in the plasma membrane. These studies reveal a novel mechanism of sorting that restricts access to the PV of host cell plasma membrane components based on their anchoring in the membrane.
Materials and Methods
Antibodies and Reagents.
Chemicals were obtained from Sigma Chemical Co., and tissue culture reagents from GIBCO BRL. Antibodies to host proteins were obtained from the following sources: American Type Culture Collection, mAb IM7.8.1 (CD44) and mAb E13 161-7 (Sca-1); Michael Kashgarian (Yale University, New Haven, CT), mAb c464.6 (alpha subunit Na+/K+ ATPase); Francis Brodsky, University of California at San Francisco, San Francisco, CA), mAb AIIB2 (β1-integrin); Chemicon International Inc., mAb RR1/1 (CD54); PharMingen, mAb IA10 (CD55); and Transduction Laboratories, mAb C34120 (caveolin-1). Biotinylated cholera toxin B (CTB) was obtained from List Biological Laboratories, Inc. Rabbit anti-SAG1 was provided by Lloyd Kasper (Dartmouth Medical School, Hanover, NH), and rabbit anti-Toxoplasma ACT1 was produced commercially (Cocalico Biologicals, Inc.). Secondary antibodies were obtained from Jackson ImmunoResearch Labs or Molecular Probes, Inc.
Parasite and Cell Culture.
Toxoplasma tachyzoites of the RH strain were propagated by serial passage in monolayers of human fibroblasts (HFs) as described previously 4. For invasion assays, host cells on LabTek chamber slides (Fisher Scientific) were challenged with either parasites or collagen-coated zymosan in DMEM/3% FCS for 5 min at 37°C 5. The cytochalasin-resistant clone CydR-1 of Toxoplasma was compared with its wild-type parental line, PLK, using similar invasion protocols supplemented with 0.5 μM cytochalasin D as described previously 4. Monolayers were either fixed immediately after challenge or washed in PBS, and returned to culture at 37°C in DMEM/10% FCS for defined intervals. All cell cultures were free of mycoplasma contamination as verified by testing with the Gen-Probe rapid detection kit.
DiIC16 Labeling of Host Cells.
Monolayers of HF cells were surface-labeled with 1.1′-dihexadecyl-3-3′-3-3′-tetramethylindocarbocyanine (DiIC16; Molecular Probes, Inc.) before challenge with parasites or zymosan as described previously 5. In experiments that required permeabilization of cells, monolayers were labeled with CM-DiI (Molecular Probes, Inc.), an analogue of DiIC16 that is retained in cells throughout permeabilization. To isolate PVs, infected monolayers were disrupted by gentle scraping and passage through a 23-g needle in PBS containing 3% FCS. This procedure released a mixture of intact PVs and free parasites that were distinguished by phase–contrast and examined by fluorescence microscopy for the presence of DiIC16. The percentage of vacuoles or parasites that were stained with DiIC16 was determined by examining 50 or more vacuoles in each of 2 or more experiments, and results are presented as the mean ± SE.
Labeling of Host Cell Surface GM1 with CTB.
Monolayers of 3T3 fibroblasts were incubated with 10 μg/ml of biotinylated CTB in DMEM/10% FCS for 5 min at 10°C, rinsed, and challenged with parasites or zymosan as described previously 5. Biotinylated CTB was detected with Oregon green–conjugated streptavidin (Molecular Probes, Inc.) and examined by epifluorescence or confocal microscopy.
Biotinylation of Host Cell Surface Membrane Proteins.
Monolayers of HF cells were rinsed with PBS (pH 7.8), then incubated with 1 mg/ml sulfo-NHS-LC-LC biotin (Pierce Chemical Co.) in PBS (pH 7.8) for 20 min at 10°C. After permeabilization, biotinylated proteins were detected with Oregon green–conjugated streptavidin (Molecular Probes, Inc.) and viewed by epifluorescence or confocal microscopy.
Immunofluorescence and Confocal Microscopy.
For immunofluorescence (IF) and confocal microscopy, monolayers were fixed and processed for IF as described previously 6. Slides were rinsed in PBS and mounted in ProLong® Antifade (Molecular Probes, Inc.), and examined using Zeiss Axioplan or Bio-Rad Confocal 1024 microscopes. To confirm that cell-associated parasites were internalized, monolayers were incubated with rabbit anti-SAG1 before permeabilization, then with secondary antibodies conjugated to Texas red. For quantitative analysis, cells were examined microscopically and vacuoles were scored as positive or negative based on a prominent, continuous rim of fluorescence around the vacuole. Percentages represent the mean and SD from 3 separate counts of 25 PVs each, unless otherwise stated.
We have shown previously that collagen-coated zymosan is taken up into compartments that resemble phagosomes in HFs 5. Here, we used this system to study the early kinetics of phagosome formation. After a 5-min pulse and extensive washing, fibroblast monolayers were examined by phase–contrast microscopy to distinguish internalized zymosan by their phase dark outline. The percentage of zymosan-containing phagosomes that were positive for labeled host proteins or lipids, as determined by a rim of fluorescence staining, was determined by counting 25 vacuoles. Each experiment was performed at least twice.
Immunoelectron Microscopy.
Monolayers of host cells were grown on tissue culture plates and challenged with freshly isolated parasites at a multiplicity of 20:1. After incubation for 2 min, cells were washed in cold PBS, then removed by trypsinization and centrifuged at 200 g for 10 min. Cells were fixed and processed as described previously 5. For quantification, the density of gold label was determined from 25 separate negatives (magnification ×20,000) from 2 or more experiments, and expressed as density of gold particles per vacuole or per micron of membrane length.
Transient Intercellular Adhesion Molecule 1 Expression.
Baby hamster kidney (BHK) cells were transfected with human intercellular adhesion molecule 1 (ICAM-1) cDNA subcloned into the CDM8 expression vector (wild-type ICAM-1; Invitrogen), or with a construct that replaced the transmembrane and cytoplasmic domain with the glycosylphosphatidylinositol (GPI) signal sequence from CD58 (ICAM-1–GPI), or with a construct that had the cytoplasmic domain deleted (ICAM-1–Cyt− [14]). Transfections were performed using Lipofectamine (GIBCO BRL) as instructed by the manufacturer. Cells were plated on chamber slides 24 h after transfection and cultured for an additional 24 h before being challenged with parasites for 5 min. Cells were fixed, permeabilized, and then stained with an antibody against human ICAM-1 and an anti-Toxoplasma SAG1 antibody, and examined by confocal microscopy. For quantitative analysis, cells were examined by confocal microscopy, and PVs were scored as positive or negative based on a prominent, continuous rim of fluorescence around the PV within a 0.5-μm section. Percentages represent the mean ± SD from two separate experiments in which >20 PVs were counted per experiment. For densitometry analysis, images were converted to 256 gray levels, and plots were obtained using the linear transect feature of NIH Image v1.61 (available at http://rsb.info.nih.gov/nih-image/).
Results
Distribution of Host Cell Plasma Membrane Proteins and Lipids in PVs Versus Phagosomes.
Previous studies have demonstrated that Toxoplasma invasion is completed within 20–30 s of initial contact with the host cell 3. To examine the formation of the PV from the host cell plasma membrane, we relied on recently developed protocols for pulse invasion of Toxoplasma 5 15. In brief, host cells were challenged with a high multiplicity of infection (ratio 50:1) for 2.5–5 min, washed extensively, and either directly fixed (T0) or returned to culture for chase intervals of 5, 10, 15, 30, or 60 min before fixation and examination by confocal, IF microscopy, or immunoelectron microscopy (immuno EM). Staining with rabbit anti-SAG1 antibodies before permeabilization revealed that >95% of cell-associated parasites were internalized during this protocol (data not shown).
Internalization of host cell plasma membrane proteins was examined by surface biotinylation, followed by challenge with Toxoplasma or zymosan particles. Vacuoles were classified as positive for the host surface proteins based on a prominent rim of fluorescence staining surrounding the PV or zymosan particle as detected with fluorescent streptavidin. Biotinylated surface proteins were abundantly detected on the cell surface, but were absent or very faintly present in PVs containing Toxoplasma (Fig. 1a and Fig. b). The short pulse used for infection (5 min) implies that these compartments were negative because of exclusion of the majority of surface proteins at the time of formation of the vacuole. In contrast, surface biotinylated proteins were readily internalized during phagocytosis of zymosan particles (Fig. 1a and Fig. b). Host cell plasma membrane lipids were simultaneously followed using the fluorescent label DiIC16. The long-chain carbocyanine dye DiIC16 was loaded into the outer leaflet of the cell, where it is stably maintained 16 17. DiIC16-lipids were detected in 85% of Toxoplasma-containing PVs and zymosan-containing vacuoles. The presence of DiIC16-lipids in the PV was not due to direct transfer of the DiIC16 to the parasite membrane, since liberated Toxoplasma parasites were invariably negative for DiIC16 (0.8 ± 0.6%), whereas isolated PVs remained positive (85.4 ± 5.5%; Fig. 1 C). Over time, the percentage of Toxoplasma PVs that was positive for DiIC16 decreased, whereas it remained relatively constant for zymosan (Fig. 1 B).
Figure 1.



Confocal localization of cell surface DiIC16-lipids versus biotinylated proteins within Toxoplasma- versus zymosan-containing vacuoles in HF cells (0.5-μm section). (A) Biotinylated surface proteins (green channel) were efficiently excluded from PVs (top), but were found in phagosomes containing zymosan (bottom). In contrast, surface DiIC16-lipids (red channel) were internalized into both Toxoplasma- and zymosan-containing vacuoles. HF cells were surface labeled with DiIC16 and sulfo-biotin, exposed to zymosan or parasites for 5 min, fixed, and stained. Parasites and zymosan were visualized with rabbit anti-p30 or antizymosan, respectively, followed by Cy5-conjugated goat anti–rabbit IgG. Biotinylated proteins were detected with Oregon green conjugated to streptavidin. Arrowheads mark the position of Toxoplasma- or zymosan-containing vacuole. (B) Kinetic analysis of the presence of host cell lipids versus proteins within Toxoplasma-containing PVs versus zymosan-containing phagosomes. Values represent mean and SD from a representative experiment. (C) Fluorescence localization of DiIC16-lipids in mechanically isolated PVs (top) versus liberated parasites (Tg, bottom). DiIC16 was present in intact PVs, but not in liberated parasites.
Incorporation of Plasma Membrane Lipids into PVs Versus Phagosomes.
To examine the internalization of endogenous host plasma membrane lipids, the distribution of the surface glycolipid GM1 was monitored by selective staining with biotinylated CTB 18 before parasite invasion. CTB bound to GM1 was detected in the PV membrane, and was also incorporated into zymosan-containing phagosomes (Fig. 2a and Fig. b). Over time, the percentage of vacuoles containing CTB-GM1 decreased in Toxoplasma PVs and remained relatively constant in zymosan vacuoles.
Figure 2.


Confocal localization of surface CTB bound to GM1 ganglioside versus CD44 protein within Toxoplasma- versus zymosan-containing vacuoles in 3T3 cells (0.5-μm section). (A) Surface CD44 (red channel) was efficiently excluded from PVs (top), but was incorporated into phagosomes containing zymosan (bottom). In contrast, GM1 (green channel) was internalized into both Toxoplasma- and zymosan-containing vacuoles. 3T3 cells were prelabeled with biotinylated CTB, incubated with zymosan or parasites for 5 min, and fixed. Biotinylated CTB was detected with Oregon green streptavidin. CD44 was stained with the mAb IM7.8.1, followed by Texas red goat anti–rat IgG. Parasites and zymosan were visualized with rabbit anti-p30 or antizymosan, respectively, followed by Cy5-conjugated goat anti–rabbit IgG. Arrowheads mark the position of Toxoplasma- or zymosan-containing vacuole. (B) Kinetic analysis of CTB-GM1 versus CD44 in Toxoplasma- versus zymosan-containing vacuoles. Values represent mean and SD from a representative experiment.
Exclusion of Plasma Membrane Proteins from Toxoplasma-containing PVs.
We also examined the internalization of specific host cell surface proteins into Toxoplasma-containing PVs, since surface biotinylation may underestimate the distribution of individual proteins. Simultaneously, the internalization of host cell surface GM1 was monitored by prelabeling with biotinylated CTB. Although the surface membrane lipid GM1 was incorporated into the PV, the cell surface protein CD44 was efficiently excluded from the vacuole (Fig. 2a and Fig. b). When fibroblasts were infected with Toxoplasma in the absence of CTB, CD44 and two additional plasma membrane proteins, Na+/K+ ATPase (100-kD subunit) and β1-integrin (130 kD), were also excluded from the PV (data not shown). In marked contrast, CD44 (Fig. 2a and Fig. b) and β1-integrin (data not shown) were readily internalized into zymosan-containing phagosomes. Consistent with the normal remodeling that accompanies phagosome maturation, CD44 was gradually removed from zymosan-containing phagosomes after initial internalization (Fig. 2 B).
IF staining may misrepresent the distribution of a protein because of insensitivity of detection or low spatial resolution, making it difficult to resolve subcellular localizations precisely. Therefore, the distribution of CD44 in newly infected monolayers of 3T3 cells (2-min pulse) was also examined by cryoimmunoEM. Immunogold staining revealed that CD44 was present along the outer surface of the plasma membrane, but not within the PV membrane (Fig. 3 A). Quantitative analysis of the distribution of CD44 demonstrated that the majority (>75%) of PVs were negative, with a few vacuoles containing limited staining. In contrast, the density of plasma membrane labeling ranged from two to seven particles per micron of membrane (Fig. 3 B). Collectively, these findings indicate that transmembrane proteins are efficiently excluded from the PV during its formation.
Figure 3.


CryoimmunoEM localization of CD44 in 3T3 cells infected with Toxoplasma. (A) CD44 was found along the host cell plasma membrane (arrowheads), but not within newly formed PVs. T, Toxoplasma cell. (B) Quantitative analysis indicated an absence of CD44 within a majority of PVs while the plasma membrane was uniformly labeled (T = 2-min pulse, no chase). Results are shown as the percentage of cells or vacuoles versus the density of immunogold staining.
GPI-anchored Membrane Proteins Are Incorporated into the PV.
We reasoned that the observed exclusion of host transmembrane proteins from the PV could be a property of the size of the extracellular domain or a consequence of their membrane anchoring. GPI-anchored proteins exhibit increased lateral mobility in the membrane 19; therefore, we examined internalization of cell surface GPI-anchored proteins into the PV. When 3T3 cells were challenged with Toxoplasma, fixed, and stained for Sca-1, this 18-kD GPI-anchored protein was readily internalized from the host cell plasma membrane into 95–100% of PVs (Fig. 4a [top] and B). To determine if larger GPI-anchored proteins were also capable of entering the PV, we examined CD55, a 75-kD GPI-anchored protein on the surface of HeLa cells, and found it was also internalized into 85% of PVs during parasite invasion (Fig. 4a [middle] and B). Consequently, independent of the size of the extracellular domain, GPI-anchored proteins are internalized from the plasma membrane into the PV. Taken together with our finding that surface biotinylated proteins were not detected within the PV, these data suggest that GPI-anchored proteins represent a minor population on the surface of fibroblast cells.
Figure 4.


Confocal localization of GPI-linked proteins Sca-1 and CD55 versus caveolin-1 in Toxoplasma PVs (0.5-μm section). (A) Sca-1 (top, green) and CD55 (middle, green) were incorporated into PVs, whereas caveolin-1 (bottom, green) was excluded (5-min pulse infection). Parasites were detected with rabbit anti-p30 (top and middle) or rabbit anti-ACT1 (bottom), followed by Texas red–conjugated goat anti–rabbit IgG. Sca-1 was detected with mAb E13 161-7, followed by bodipy-conjugated goat anti–rat IgG. CD55 and caveolin-1 were detected with mAb IA10 and mAb C37120, respectively, followed by Oregon green–conjugated goat anti–mouse IgG. Sca-1 was detected in 3T3 cells, CD55 in HeLa cells, and caveolin-1 in HF cells. Arrowheads mark the position of PVs. (B) The percentage of PVs that internalized Sca-1, CD55, and caveolin-1 after a 5-min challenge with parasites. Values represent the mean and SE from two experiments.
Exclusion of Caveolin-1 Protein from the PV.
The finding that the PV selectively incorporates plasma membrane lipids and GPI-anchored proteins suggests that invasion could occur selectively through specialized lipid-rich microdomains, possibly including such specialized structures as caveoli 20. To determine if parasite invasion resulted in the incorporation of caveolar proteins into the PV, we monitored caveolin-1 distribution during parasite invasion. Unlike GM1, caveolin-1 was not detected in the PV (Fig. 4a [bottom] and B), indicating that the parasite does not interact substantially with caveoli or vesicles derived from them.
Protein Exclusion from the PV Is Dependent on the Mechanism of Membrane Anchoring.
The observation that GPI-anchored proteins are incorporated into the PV suggests that membrane anchoring determines internalization. However, to eliminate potential differences due to the size and configuration of the extracellular domain, parasite invasion was examined in BHK cells transiently transfected with recombinant forms of ICAM-1 (CD54) that were anchored into the host cell plasma membrane by different means, as diagrammed in Fig. 5 C. Wild-type ICAM-1 (CD54) is a 90-kD molecule that possesses a single transmembrane segment and short cytoplasmic domain 21. We compared the wild-type ICAM-1 to a lipid-anchored form (ICAM-1–GPI) and a cytoplasmic deletion form (ICAM-1–Cyt−) of the protein 22 23. Like other transmembrane proteins, wild-type ICAM-1 was excluded from the PV, whereas ICAM-1–GPI and ICAM-1–Cyt− were both incorporated into PVs. All three forms of ICAM were abundantly expressed and visualized with the same antibody to the extracellular domain, ruling out possible differences due to detection. Exclusion was also not due to size constraints, since the molecular masses of wild-type ICAM-1 and ICAM-1–Cyt− only differ by 3 kD (90 and 87 kD, respectively [22]). To quantify the distribution of the three forms of ICAM-1 in the PV membrane, confocal images were scaled to 256 gray levels and analyzed using the line transect feature of NIH Image. The relative intensity of a line extending through the plasma membrane and across the PV was determined for each of the three forms (Fig. 5 C). These analyses revealed that ICAM-1–GPI and ICAM-1–Cyt− were enriched by two- to threefold in the PV membrane relative to the plasma membrane, whereas wild-type ICAM-1 was found at background levels (Fig. 5 A).
Figure 5.


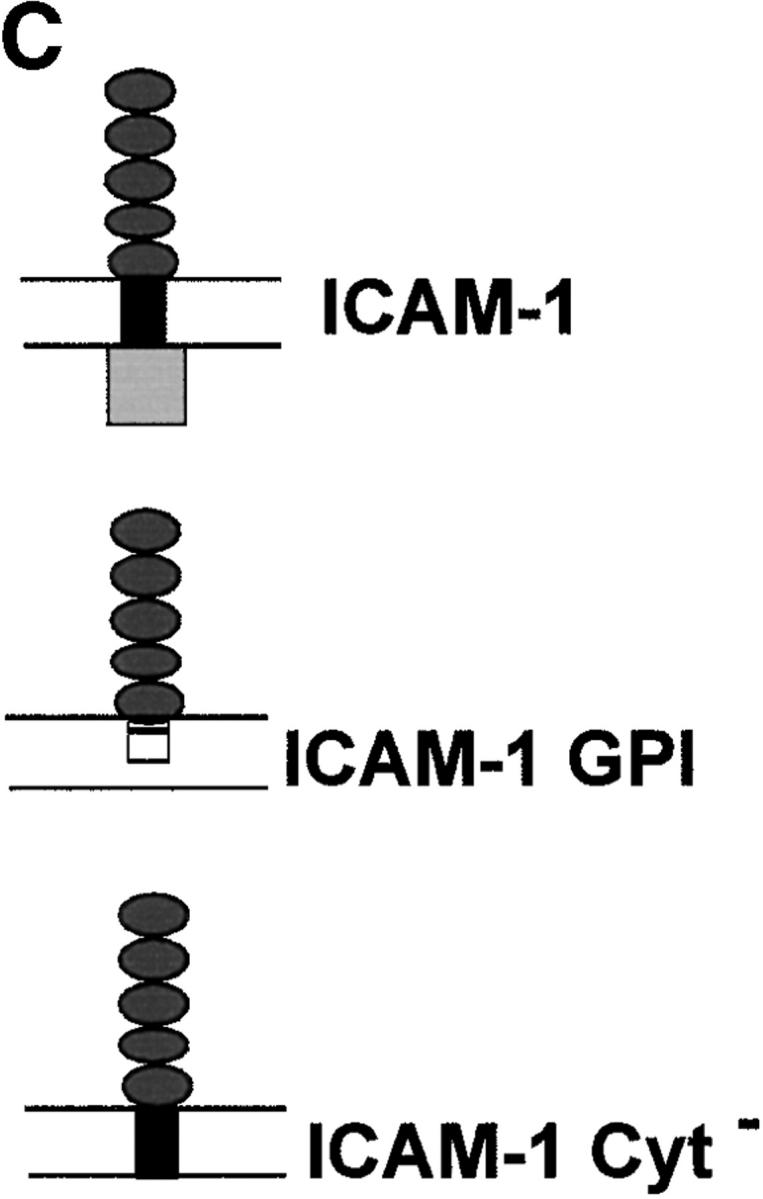
Confocal localization of wild-type ICAM-1 versus ICAM-1–GPI, and ICAM-1–Cyt− in BHK cells infected with Toxoplasma (5-min pulse; 0.5-μm section). (A) Wild type ICAM-1 (top, green) was excluded from PVs, whereas ICAM-1–GPI (middle, green) and ICAM-1–Cyt− (bottom, green) were internalized into PVs. Parasites were detected with rabbit anti-p30, followed by Texas red–conjugated goat anti–rabbit IgG. ICAM-1 was detected with mAb RR1/1, followed by bodipy-conjugated goat anti–mouse IgG. Arrowheads mark the position of PVs. Red lines indicate the transects used for densitometry analysis as plotted to the right. The location of the PV along the line is shown by a cartoon of the parasite above the plot. (B) Confocal quantitative analysis of the percentage of PVs that internalized various forms of ICAM-1 after a 5-min challenge with parasites. Results shown are the mean and SE from two experiments. (C) Diagram of the ICAM-1, ICAM-1–GPI, and ICAM-1–Cyt− constructs anchored in the membrane.
The Host Cell Cytoskeleton and Restricted Access to the PV.
The internalization of the ICAM-1–Cyt− form suggested that restricted access to the PV is mediated by interactions with the host cell cytoskeleton. To test this hypothesis, we examined the invasion of a cytochalasin-resistant mutant of Toxoplasma into HF cells in the presence of sufficient concentrations of the drug to disrupt actin microfilaments. During invasion of the CydR-1 mutant, the host cell surface protein CD44 was excluded from 94% of vacuoles formed in the absence of cytochalasin D and from 87% of the vacuoles formed in the presence of the drug. Moreover, the prominent constriction seen at the junction during invasion 3 was still observed in the presence of cytochalasin D (data not shown).
Discussion
We show here that the PV is formed by invagination of the host cell plasma membrane, while simultaneously excluding transmembrane proteins. The formation of a moving junction at the interface of the host cell plasma membrane and the parasite appears to mediate this differential sorting process. In contrast, during uptake of zymosan, both plasma membrane lipids and transmembrane proteins are internalized into phagocytic vacuoles. Despite the efficient exclusion of transmembrane proteins at the moving junction, the presence of a GPI anchor or the absence of a cytoplasmic tail is sufficient to allow internalization of membrane proteins into the PV. Thus, sorting occurs by a mechanism that acts within the membrane spanning region or on the cytoplasmic region of the protein, and which is largely independent of their extracellular domains.
Several observations indicate that, during parasite invasion, the bulk of the PV is derived from internalization of the host cell plasma membrane. First, electrophysiological studies have shown that during Toxoplasma invasion there is no net change in capacitance across the host cell plasma membrane, implying that the host cell surface area remains constant until the PV pinches off from the host cell plasma membrane 9. Second, our results demonstrate that the PV membrane is formed by invagination of lipids from the host cell plasma membrane. For example, when the fluorescent lipophilic dye DiIC16 16 24 was inserted into the host cell plasma membrane before parasite invasion, it was readily internalized into the PV membrane. Endogenous plasma membrane lipids are also incorporated into the PV, as shown by the internalization of surface GM1 sphingolipid. The presence of these labels in the PV was necessarily due to internalization at the time of entry, as neither one is prone to rapid diffusion, and we have shown previously that there is no vesicular lipid traffic to preformed PVs 5. However, in both cases the percentage of PVs stained with these labels decreased over time, which may be a result of diffusion, transfer to the parasite, or vesicular traffic from an unidentified source that results in dilution of the label.
Although the vacuole surrounding Toxoplasma is derived from the host cell plasma membrane, the transmembrane proteins Na+/K+ ATPase, β1-integrin, and CD44 were excluded from the PV, even though they were readily incorporated during formation of zymosan-containing phagosomes. CryoimmunoEM examination of newly formed PVs revealed that the vacuolar membrane contained at least 20-fold lower levels of host cell proteins than the plasma membrane. Similar to our observations on Toxoplasma, when Plasmodium invades red blood cells, host cell plasma membrane lipids are incorporated into the parasite-containing vacuole, whereas transmembrane proteins in the red cell membrane, including glycophorins and band 3, are excluded 25.
Sorting of plasma membrane proteins during formation of phagosomes has been ascribed previously to their involvement as receptors in particle uptake 26. In contrast, our results indicate that protein exclusion from Toxoplasma-containing PVs is a property of the membrane-anchoring and cytoplasmic extension of proteins, and is independent of the extracellular domain. Initially, this conclusion was supported by examining a variety of different surface proteins that were anchored by either a transmembrane domain (CD44, β1-integrin, Na+/K+ ATPase) or a GPI moiety (Sca-1, CD55). Although there was a perfect correlation between mechanism of anchoring and exclusion versus inclusion, these findings may also be influenced by variability in the size and configuration of the extracellular domains of the proteins examined.
To eliminate potential differences due to the size of the extracellular domain, we compared the internalization of recombinant ICAM-1 that was anchored in the membrane by different means. ICAM-1 is a heavily glycosylated 90-kD protein containing a single transmembrane segment and five tandem Ig-like extracellular domains 21 22 27 28. Removal of the cytoplasmic tail from ICAM-1 eliminates the molecule's association with the actin cytoskeleton 14, and replacement of ICAM-1's transmembrane region with a GPI anchor is likely to increase the molecule's lateral mobility 19 20. Either the presence of a GPI anchor or the deletion of the cytoplasmic region was sufficient to allow ICAM-1 internalization into Toxoplasma-containing PVs. These results conclusively demonstrate that internalization is largely independent of the extracellular domain, and instead support the conclusion that sorting is based on the lateral mobility of the protein within the membrane.
Exclusion of transmembrane proteins from the PV is likely due to establishment of a barrier to protein diffusion that operates at the moving junction, and which may be due to interactions between the cytoplasmic domain and proteins in the cytosol. Lateral diffusion of transmembrane proteins is limited by their direct attachment to the cytoskeleton (tethering) and indirect constraints imposed by the cytoskeleton 29 30 31. Despite the observation that treatment with cytochalasin D did not allow access of CD44, a transmembrane-anchored protein, past the moving junction, we cannot rule out the possibility that short actin filaments remaining after this treatment were still sufficient to retard its mobility. Alternatively, the colloidal nature of the cytosol may be sufficient to retard the mobility of proteins with cytoplasmic extensions such that they have restricted access to the vacuole. In the case of ICAM-1, the lateral mobility of wild-type ICAM-1 is restricted by its direct association with the cytoskeleton and by general constraints due to its cytoplasmic tail. Based on analogous mutations that have been analyzed for other transmembrane proteins 19 29 30 32, ICAM-1–Cyt− and ICAM-1–GPI are expected to display increased lateral mobility that evidently allows them to pass through the moving junction and gain access to the Toxoplasma-containing PV.
Free diffusion of plasma membrane lipids and restricted passage of transmembrane proteins past the moving junction indicate that this unique structure acts as a molecular sieve. Classical tight junctions act as a diffusion barrier in the outer leaflet of the plasma membrane, but allow diffusion in the inner bilayer 33 34. In contrast, our results indicate that GPI-linked proteins anchored in the outer leaflet of the plasma membrane freely enter the PV, yet proteins that extend beyond the membrane bilayer are excluded. Freeze–fracture EM analysis during Plasmodium invasion of erythrocytes has shown a band of rhomboidally arrayed particles at this junction that may represent aggregates of proteins that are prevented from entering the vacuole 13. The molecular basis of this junction is not understood, and identification of host or parasite proteins that are exclusively localized to this structure is an important area for future research.
The selective enrichment of GPI-anchored proteins within the Toxoplasma PV indicates that they are specifically recruited or retained within the vacuole. Enrichment could occur if the parasite binds to one or more GPI-anchored proteins, thereby assuring their inclusion in the vacuole. Such a process occurs during internalization of Escherichia coli into macrophages through FimH-mediated attachment to CD48 35. However, our data do not support this model for Toxoplasma invasion for two reasons: (a) the wide range of different GPI-anchored proteins that are internalized is inconsistent with use of a specific receptor; and (b) the extracellular domain of ICAM-1 was specifically excluded when attached to a transmembrane domain, yet it readily gained access to the vacuole when GPI anchored or when the cytoplasmic tail was truncated.
There are two alternative explanations for the enrichment of GPI-anchored proteins in the PV: (a) exclusion of transmembrane proteins from this membrane allows GPI-anchored proteins to be more densely packed than in the plasma membrane; and (b) GPI-anchored proteins are enriched in the PV because of a selective alteration in membrane lipids. Although the first alternative does not necessitate a reorganization of membrane lipids, the second model suggests that lipid microdomains are also selectively recruited into the PV during invasion. Coalescence of lipid microdomains, or rafts, to form the PV would result in the enrichment of GPI proteins and lipids commonly associated with them (i.e., cholesterol and sphingolipids [20]). Recent evidence indicates that lipid microdomains exist as small patches in the plasma membrane where GPI-anchored proteins are clustered in association with cholesterol and sphingolipids, and that these structures become enlarged by cross-linking 36 37. How such domains, which are estimated to be ∼50 nm in size, might be reorganized during formation of the PV is uncertain, but a similar process of selective lipid recruitment has been described during influenza viral budding from mammalian cells 38.
Our findings are best explained by a model based on parasite-induced invagination of the plasma membrane in combination with tethering of transmembrane proteins at the moving junction. Plasma membrane lipids, GPI-anchored proteins, and proteins without cytoplasmic domains readily diffuse past the moving junction and gain entry into the nascent PV, whereas transmembrane proteins are efficiently excluded. A similar process of protein and lipid sorting has been proposed for erythrocyte membrane vesiculation, caused by mechanical disruption, that results in the restriction of cytoskeleton and cytoskeletal-associated proteins to the cell body while lipids and GPI-anchored proteins are enriched in the vesicle 39. Toxoplasma appears to generate such a process in reverse by distending the host cell plasma membrane inward to form the PV, a process that is driven by the active penetration by the parasite 4. The restricted access of host cell proteins to the PV is likely to underlie its subsequent segregation from the exocytic and endocytic networks, which allows for the parasite's unique and highly successful intracellular lifestyle.
Acknowledgments
The authors thank Olivia Giddings and Marilyn Levy for expert technical assistance, Tom Steinberg and Dan Goldberg for critical review of the manuscript, Gary Ward and Kasturi Haldar for helpful comments, and individuals listed in Materials and Methods for generous supplies of antibodies.
This work was supported by grants from the National Institutes of Health to L.D. Sibley (AI34036) and M. Dustin (AI44931).
Footnotes
Abbreviations used in this paper: BHK, baby hamster kidney; CM-DiI, chloromethyl-benzamido DiIC16; CTB, cholera toxin B; DiIC16, 1.1′-dihexadecyl-3-3′-3-3′-tetramethylindocarbocyanine; EM, electron microscopy; GPI, glycosylphosphatidylinositol; HF, human fibroblast; ICAM-1, intercellular adhesion molecule 1; IF, immunofluorescence; immunoEM, immunoelectron microscopy; PV, parasitophorous vacuole.
References
- Aikawa M., Sterling C.R. Intracellular Parasitic Protozoa 1974. Academic Press, Inc; New York: pp. 76 [Google Scholar]
- Dubey J.P., Beattie C.P. Toxoplasmosis of Animals and Man 1988. CRC Press; Boca Raton, FL: pp. 220 [Google Scholar]
- Morisaki J.H., Heuser J.E., Sibley L.D. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J. Cell Sci. 1995;108:2457–2464. doi: 10.1242/jcs.108.6.2457. [DOI] [PubMed] [Google Scholar]
- Dobrowolski J.M., Sibley L.D. Toxoplasma invasion of mammalian cells is powered by the actin cytoskeleton of the parasite. Cell. 1996;84:933–939. doi: 10.1016/s0092-8674(00)81071-5. [DOI] [PubMed] [Google Scholar]
- Mordue D.G., Håkansson S., Niesman I., Sibley L.D. Toxoplasma gondii resides in a vacuole that avoids fusion with host cell endocytic and exocytic vesicular trafficking pathways. Exp. Parasitol. 1999;92:87–99. doi: 10.1006/expr.1999.4412. [DOI] [PubMed] [Google Scholar]
- Mordue D.G., Sibley L.D. Intracellular fate of vacuoles containing Toxoplasma gondii is determined at the time of formation and depends on the mechanism of entry. J. Immunol. 1997;159:4452–4459. [PubMed] [Google Scholar]
- Joiner K.A., Furhman S.A., Miettinen H.M., Kasper L.H., Mellman I. Toxoplasma gondiifusion competence of parasitophorous vacuoles in Fc receptor-transfected fibroblasts. Science. 1990;249:641–646. doi: 10.1126/science.2200126. [DOI] [PubMed] [Google Scholar]
- Endo T., Pelster B., Piekarski G. Infection of murine peritoneal macrophages with Toxoplasma gondii exposed to ultraviolet light. Z. Parasitenkd. 1981;65:121–129. doi: 10.1007/BF00929177. [DOI] [PubMed] [Google Scholar]
- Suss-Toby E., Zimmerberg J., Ward G.E. Toxoplasma invasionthe parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fusion pore. Proc. Natl. Acad. Sci. USA. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aikawa M., Komata Y., Asai T., Midorikawa O. Transmission and scanning electron microscopy of host cell entry by Toxoplasma gondii . Am. J. Pathol. 1977;87:285–295. [PMC free article] [PubMed] [Google Scholar]
- Nichols B.A., O'Connor R.G. Penetration of mouse peritoneal macrophages by the protozoan Toxoplasma gondii . Lab. Invest. 1981;44:324–335. [PubMed] [Google Scholar]
- Porchet-Hennere E., Torpier G. Relations entre Toxoplasma et sa cellule-hote. Protisologica. 1983;19:357–370. [Google Scholar]
- Aikawa M., Miller L.H., Rabbege J.R., Epstein N. Freeze–fracture study on the erythrocyte membrane during malaria invasion. J. Cell Biol. 1981;91:55–62. doi: 10.1083/jcb.91.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpen O., Pallai P., Staunton D.E., Springer T.A. Association of intercellular adhesion molecule-1 (ICAM-1) with actin-containing cytoskeleton and alpha-actinin. J. Cell Biol. 1992;118:1223–1234. doi: 10.1083/jcb.118.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers V.B., Sibley L.D. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 1997;73:114–123. [PubMed] [Google Scholar]
- Wolf D.E. Determination of the sidedness of carbocyanine dye labeling of membranes. Biochemistry. 1985;24:582–586. doi: 10.1021/bi00324a006. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J., Blumenthal R., Sarkar D.P., Curran M., Morris S.J. Restricted movement of lipid and aqueous dyes through pores formed by influenza hemagglutinin during cell fusion. J. Cell Biol. 1994;127:1885–1894. doi: 10.1083/jcb.127.6.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley D.R., Streuli C.H., Kellie S., Ansell S., Patel B. Characterization of the cholera toxin receptor on Balb/c 3T3 cells as a ganglioside similar to, or identical with, ganglioside GM1 . Biochem. J. 1982;204:209–219. doi: 10.1042/bj2040209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M., Kuo S.C., Sheetz M.P. Lateral movements of membrane glycoproteins restricted by dynamic cytoplasmic barriers. Science. 1991;254:1379–1382. doi: 10.1126/science.1835798. [DOI] [PubMed] [Google Scholar]
- Simons K., Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- Staunton D.E., Marlin S.D., Stratowa C., Dustin M.L., Springer T.A. Primary structure of intercellular adhesion molecule 1 (ICAM-1) demonstrates interaction between members of the immunoglobulin and integrin superfamilies. Cell. 1988;52:925–933. doi: 10.1016/0092-8674(88)90434-5. [DOI] [PubMed] [Google Scholar]
- Staunton D.E., Dustin M.L., Erickson H.P., Springer T.A. The arrangement of the immunoglobulin-like domains of ICAM-1 and the binding sites for LFA-1 and rhinovirus. Cell. 1990;61:243–254. doi: 10.1016/0092-8674(90)90805-o. [DOI] [PubMed] [Google Scholar]
- Staunton D.E., Guar A., Chan P.Y., Springer T.A. Internalization of a major group human rhinovirus does not require cytoplasmic or transmembrane domains of ICAM-1. J. Immunol. 1992;148:3271–3274. [PubMed] [Google Scholar]
- Struck D.K., Pagano R.E. Insertion of fluorescent phospholipids into the plasma membrane of a mammalian cell. J. Biol. Chem. 1980;255:5404–5410. [PubMed] [Google Scholar]
- Ward G.E., Miller L.H., Dvorak J.A. The origin of parasitophorous vacuole membrane lipids in malaria-infected erythrocytes. J. Cell Sci. 1993;106:237–248. doi: 10.1242/jcs.106.1.237. [DOI] [PubMed] [Google Scholar]
- Brown E.J. Phagocytosis. Bioessays. 1995;17:109–117. doi: 10.1002/bies.950170206. [DOI] [PubMed] [Google Scholar]
- Simmons D., Makgoba M.W., Seed B. ICAM, an adhesion ligand of LFA-1, is homologous to the neural cell adhesion molecule NCAM. Nature. 1988;331:624–627. doi: 10.1038/331624a0. [DOI] [PubMed] [Google Scholar]
- Dustin M.L., Rothlein R., Bhan A.K., Dinarello C.A., Springer T.A. Induction by IL-1 and interferon, tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1) J. Immunol. 1986;137:245–254. [PubMed] [Google Scholar]
- Edidin M., Zuniga M.C., Sheetz M.P. Truncation mutants define and locate cytoplasmic barriers to lateral mobility of membrane glycoproteins. Proc. Natl. Acad. Sci. USA. 1994;91:3378–3382. doi: 10.1073/pnas.91.8.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomishige M., Sako Y., Kusumi A. Regulation mechanism of the lateral diffusion of band 3 in erythrocyte membranes by the membrane skeleton. J. Cell Biol. 1998;142:989–1000. doi: 10.1083/jcb.142.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y., Nagafuchi A., Tsukita S., Takeichi M., Kusumi A. Cytoplasmic regulation of the movement of E-cadherin on the free cell surface as studied by optical tweezers and single particle trackingcorralling and tethering by the membrane skeleton. J. Cell Biol. 1998;140:1227–1240. doi: 10.1083/jcb.140.5.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sako Y., Kusumi A. Barriers for lateral diffusion of transferrin receptor in the plasma membrane as characterized by receptor dragging by lazer tweezersfence versus tether. J. Cell Biol. 1995;129:1559–1574. doi: 10.1083/jcb.129.6.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragsten P., Blumenthal R., Handler J.S. Membrane asymmetry in epitheliais the tight junction a barrier to diffusion in the plasma membrane? Nature. 1981;294:718–722. doi: 10.1038/294718a0. [DOI] [PubMed] [Google Scholar]
- van Meer G., Simons K. The function of tight junctions in maintaining differences in lipid composition between the apical and the basolateral cell surface domains of MDCK cells. EMBO (Eur. Mol. Biol. Organ.) J. 1986;5:1455–1464. doi: 10.1002/j.1460-2075.1986.tb04382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baorto D.M., Gao Z., Malaviya R., Dustin M.L., van der Merwe A., Lublin D.M., Abraham S.A. Survival of FimH-expressing enterobacteria in macrophages relies on glycolipid traffic. Nature. 1997;389:636–639. doi: 10.1038/39376. [DOI] [PubMed] [Google Scholar]
- Friedrichson T., Kurzchalia T.V. Microdomains of GPI-anchored proteins in living cells revealed by crosslinking. Nature. 1998;394:802–805. doi: 10.1038/29570. [DOI] [PubMed] [Google Scholar]
- Varma R., Mayor S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature. 1998;394:798–801. doi: 10.1038/29563. [DOI] [PubMed] [Google Scholar]
- Scheiffele P., Rietveld A., Wilk T., Simons K. Influenza viruses select ordered lipid domains during budding from the plasma membrane. J. Biol. Chem. 1999;274:2038–2044. doi: 10.1074/jbc.274.4.2038. [DOI] [PubMed] [Google Scholar]
- Knowles D.W., Tilley L., Mohandas N., Chasis J.A. Erythrocyte membrane vesiculationmodel for the molecular mechanism of protein sorting. Proc. Natl. Acad. Sci. USA. 1997;94:12969–12974. doi: 10.1073/pnas.94.24.12969. [DOI] [PMC free article] [PubMed] [Google Scholar]