

Abstract
We have established a new clonal assay system that can evenly support the development of T and natural killer (NK) cells. With this system, we show that all T cell progenitors in the earliest CD44+CD25−FcγRII/III− fetal thymus (FT) cell population retain NK potential, and that the NK lineage–committed progenitors (p-NK) also exist in this population. T cell lineage–committed progenitors (p-T), which are unable to generate NK cells, first appear at the CD44+CD25− FcγRII/III+ stage in day 12 FT. The proportion of p-T markedly increases during the transition from the CD44+CD25− stage to the CD44+CD25+ stage in day 14 FT. On the other hand, p-NK preferentially increase in number at the CD44+CD25− stage between days 12 and 14 of gestation. The production of p-NK continues up to the CD44+CD25+ stage, but ceases before the rearrangement of T cell receptor β chain genes. It was further shown that the CD44+CD25− CD122+ population of day 14 FT exclusively contains p-NK. These results indicate that the earliest T cell progenitor migrating into the FT is T/NK bipotent, and strongly suggest that the bipotent progenitor continuously produces p-NK and p-T until the CD44+CD25+ stage.
Keywords: T cell, natural killer cell, hematopoietic stem cell, thymus, clonal assay
The T cell system is composed of functionally distinct subsets, including helper T cells, killer T cells, and T cells mediating delayed type hypersensitivity, and these T cells comprise millions of clones expressing the TCR that recognize different antigens. NK cells carry a similar function as killer T cells, in that they discriminate virus-infected cells and some neoplastic cells from normal healthy cells, and kill such cells 1 2. However, NK cells are distinct from T cells since they do not express a TCR 3, and NK cell development is independent of the thymus 4. Nevertheless, experimental evidence has been accumulated proposing that T and NK cells are derived from a common progenitor 5 6 7.
There has been more than a decade of dispute about the characteristics of T cell progenitors migrating into the thymus 8 9 10 11. Early studies made by marking the bone marrow progenitors with chromosome aberrations 12 or bacterial gene introduction 13 14 15 suggested that the progenitors migrating into the thymus are committed to the T cell lineage. On the other hand, it has been shown that the earliest population of thymus cells was able to generate not only T but also B and NK cells when transferred into irradiated recipients 16 17. In the fetus, the earliest population of thymus cells has also been shown to retain myeloid potential 18 19, suggesting that a common progenitor for these lineages exists in the thymus. However, such experiments where cell populations are used as the progenitor source are usually ineffective in discriminating the activity of a mixture of unipotent progenitors for different lineages from that of a common progenitor. To avoid the dilemma of cell populations, we have developed a clonal assay system, the multilineage progenitor (MLP) assay 20, which is capable of determining the developmental potential of single progenitors toward T, B, and myeloid lineages. With the MLP assay, we have shown that the earliest population in fetal thymus (FT) does not contain multipotent progenitors but rather contains progenitors restricted to the T, B, or myeloid lineage 21. Although this study clarified that thymic T cell progenitors are distinct from myeloid and B cell progenitors, it remained unclear whether the T cell progenitors we determined retain the potential to generate NK cells.
In the mouse, it has been suggested that the thymic T cell progenitors are T/NK bipotent. Rodewald et al. 5 showed that the CD4−CD8−TCR−FcγRII/III (FcR)+ population of FT was able to generate both T and NK cells. It was further shown that the c-kit+NK1.1+ subpopulation of CD44+CD25− FT cells has the potential to generate both NK and T cells 7. The intimate relationship between T and NK lineages has also been shown by genetic analyses. Knockout of the common γ chain 22 or the Janus kinase 3 23 gene resulted in depletion of NK cells in addition to T and B cells, and overexpression of the human CD3∈ gene in the mouse brought about complete depletion of both T and NK cells 24. More direct evidence supporting the presence of bipotent progenitors generating T and NK cells (p-T/NK) has been shown in the investigation of human FT progenitors. Sanchez et al. 25 showed that T cells were generated from a pool of cells (∼1,000 cells) from many human FT cell colonies, all of which were able to give rise to NK cells when cultured in the presence of IL-2, by coculturing with a deoxyguanosine (dGuo)-treated murine FT lobe. Moreover, they have shown that thymic NK progenitors can be phenotypically separated from T cell progenitors 26, strongly suggesting that commitment to T and NK cell lineages occurs in the FT. However, it is still unclear at what stage the bipotent progenitor branches to unipotent T and NK progenitors. In this study, we established for the first time a clonal assay system that is effective in determining the developmental potential of individual progenitors toward T and NK cells. With this clonal assay, we verified the presence of p-T/NK not only in the earliest CD44+CD25− population, but also in the CD44+CD25+ population of the FT. The presence of a large number of NK lineage–committed progenitors (p-NK) was also disclosed in the early FT subpopulations. T cell lineage–committed progenitors (p-T) were shown to increase with the progress of the differentiation stage, whereas NK progenitors decreased reciprocally.
Materials and Methods
Mice.
Adult C57BL/6 (B6) mice were purchased from SLC. B6Ly5.1 mice were maintained in our animal facility. Embryos at various stages of gestation were obtained from time-mated pregnant B6Ly5.1 mice.
mAbs.
The following antibodies were used: PE–anti-Thy1.2 (5a-8; Caltag), biotinylated anti-NK1.1 and PE–anti-NK1.1 (PK136; PharMingen), PE–anti-CD45 (30F11.1; PharMingen), PE–anti-CD122 (TM-β1; PharMingen), FITC–anti-CD3∈ (145-2C11; PharMingen), allophycocyanin (APC)–anti-CD3 (CT-CD3; Caltag), FITC–anti-CD25 (PC61; PharMingen), PE–anti-CD44 (IM7.8.1; Caltag), APC–anti-CD8 and FITC–anti-CD8 (YTS169.4; Caltag), APC–anti-CD4 and PE–anti-CD4 (GK1.5; Caltag), FITC anti–TCR-γ/δ (GL-3; Caltag), and PE anti–TCR-α/β (H57-597; Caltag). Anti-FcR (2.4G2) and anti-Ly5.2 (AL1-4A2, donated by Dr. I.L. Weissman, Stanford University, Stanford, CA) were labeled with FITC in our laboratory. Anti-TER119 (established by Dr. T. Kina in our laboratory), anti-CD44 (IM7.8.1), anti-B220 (RA3-6B2), anti–Mac-1 (M1/70), and anti–Gr-1 (RA3-8C5) were biotinylated in our laboratory. Anti–c-kit (ACK-2; donated by Dr. S.-I. Nishikawa, Kyoto University) and anti-Ly5.1 (A20-1.7; donated by Dr. Y. Saga, Banyu Seiyaku, Tokyo, Japan) were conjugated with Cy5 using a labeling kit (Biological Detection Systems). For biotinylated antibodies, Cy5-streptavidin (Caltag) was used as the secondary reagent.
Growth Factors.
Recombinant murine (rm)IL-7 was donated by Dr. T. Sudo (Basic Research Lab, Toray, Kanagawa, Japan). Commercially available recombinant murine stem cell factor (rmSCF), rmIL-2, and rmIL-15 (all from Genzyme) were also used.
Staining and Sorting of Progenitor Cells.
FcR− and FcR+ subpopulations of CD44+CD25− FT cells were obtained from fetuses at 12 d postcoitum (dpc) as described previously 19. To isolate CD44+CD25− and CD44+CD25+ cells in the lineage marker (Lin)−CD3−CD4−CD8− population, 14-dpc FT cells were stained with biotinylated Lin (anti-TER119, anti–Mac-1, anti–Gr-1, anti-NK1.1, and anti-B220), washed, and then stained with a mixture of Cy5-streptavidin, APC–anti-CD4, APC–anti-CD8, APC–anti-CD3, FITC–anti-CD25, and PE–anti-CD44. Cells were subsequently sorted using a FACS Vantage™ (Becton Dickinson). In sorting CD122+ cells, 14-dpc FT cells were stained with the same mAbs as above except that PE–anti-CD122 was used instead of PE–anti-CD44.
Cytokine-supplemented High Oxygen Submersion Culture.
The method for high oxygen submersion (HOS) culture has been described in detail elsewhere 27. Single dGuo-treated lobes were placed into wells of a 96-well V-bottomed plate (Nalge Nunc International), to which progenitors were added. The plates were centrifuged at 150 g for 5 min at room temperature, placed into a plastic bag (Ohmi Oder Air Service), and the air inside was replaced by a gas mixture (70% O2, 25% N2, 5% CO2). The plastic bag was incubated at 37°C. The cultures were maintained in RPMI 1640 medium (GIBCO BRL) supplemented with 10% FCS (M.A. Bioproducts), l-glutamine (2 mM), sodium pyruvate (1 mM), sodium bicarbonate (2 mg/ml), nonessential amino acid solution (0.1 mM; GIBCO BRL), 2-ME (5 × 10−5 M), streptomycin (100 μg/ml), and penicillin (100 U/ml). Culture medium was also supplemented with various cytokines to promote the growth of NK cells (see Results). Medium was replaced by half every 5 d.
After 10 d of culture, cells inside and outside the FT lobe were harvested from each well. One fifth of each sample was stained with FITC–anti-Ly5.2, PE anti–Thy-1, and Cy5–anti-Ly5.1 to be screened for the presence of progenitor type (Ly5.1+) cells and expression of Thy-1 on these cells. The samples containing Ly5.1+ cells were selected for further analysis. The remaining four fifths of cells from the selected samples were divided into four groups. One group was stained with FITC–anti-CD3∈, PE–anti-NK1.1, and Cy5–anti-Ly5.1, the second with FITC–anti-CD8, PE–anti-CD4, and Cy5–anti-Ly5.1, and the third with FITC anti–TCR-γ/δ, PE anti–TCR-α/β, and Cy5–anti-Ly5.1. Surface phenotype was analyzed by a FACS Vantage™. The remaining group was used for the analysis of TCR gene rearrangement.
Cytotoxic Assay.
The cytotoxic assay was carried out as reported previously 28. Yac-1 cells were used as target cells. In brief, target cells were labeled with PKH67 green fluorescence dye (Sigma Chemical Co.) and mixed with various numbers of effector cells. The mixture was incubated at 37°C for 4 h in a V-bottomed 96-well microplate. Culture medium was the same as that used in HOS cultures. Dead cells were stained with propidium iodide red fluorescence dye (Sigma Chemical Co.), and the proportion of propidium iodide–stained cells (dead cells) among PKH67-stained cells was determined by analyzing with a FACS Vantage™.
PCR Analysis of TCR-β and TCR-γ Gene Rearrangement.
One fifth of the cells harvested from each sample (see preceding section) was subjected to PCR analysis. To prepare genomic DNA, 3,000 cells were resuspended in 20 μl of 1× PCR buffer (10 mM Tris-HCl, pH 9.0, 50 mM KCl, and 1.5 mM MgCl2) including 0.45% NP-40, 0.45% Tween 20, and 1.2 μg of Proteinase K (Sigma Chemical Co.), and incubated at 55°C for 1 h, then 95°C for 10 min. Samples of these disrupted cells were used as templates for PCR amplification. Primers were: 5′ of Dβ2, 5′-GCACCTGTGGGGAAGAAACT-3′; 3′ of Jβ2.6, 5′-TGAGAGCTGTCTCCTACTATCGATT-3′; Vγ4, 5′-AGTGTTCAGAAGCCCGATGCA-3′; 3′ of Jγ1, 5′-AGAGGGAATTACTATGAGCT-3′. The reaction volume was 20 μl, containing 5 μl of the cell extract, 1.5 μl of 10× PCR buffer, 0.16 μl of 25 mM dNTPs, 4 pmol of each primer, and 0.6 U of Taq polymerase (Life Technologies). Thermocycling conditions were as follows: 5 min at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 60°C for D-Jβ rearrangement or at 55°C for V-Jγ rearrangement, 2 min at 72°C, and finally 10 min at 72°C. The PCR products were applied to a 1.2% agarose gel, electrophoresed, and stained with ethidium bromide.
Results
Modification of the FT Organ Culture System to Support the Development of T and NK Cells.
We first tried to establish a culture system capable of generating T and NK cells. This was attained by modifying the MLP assay system that was effective in investigating the developmental capability of progenitors toward T, B, and myeloid lineages 20.
Since FT contains a small number of NK cells as well as NK progenitors 29, it is expected that dGuo-treated FT lobes support the differentiation of NK cells. Cytokines IL-2 and IL-15 were used for promoting the growth of NK cells in FT organ culture. FT cells from 12-dpc fetuses (100 cells) of B6Ly5.1 mice, which are exclusively CD44+CD25− (see Fig. 4 A for flow cytometric profiles), were seeded to each well of a V-bottomed 96-well plate, in which a dGuo-treated FT lobe (B6) had been placed. IL-2, IL-15, or a cocktail of these and other cytokines was added, and the plate was incubated under HOS conditions for 10 d. Cells were harvested, counted, and assayed for expression of T and NK cell markers on donor-derived (Ly5.1+) cells. Table indicates that the cell recovery increased slightly by the addition of cytokines, and in all groups >90% of the recovered cells were Thy-1+. Without cytokines, ∼40% were CD3+ NK1.1− (T cells), whereas only 3% were CD3−NK1.1+ cells (NK cells). Addition of IL-2 or IL-15 dramatically increased the proportion of NK cells, while the proportion of T cells declined reciprocally. The effect of IL-2 or IL-15 in supporting the generation or growth of NK cells is compatible with a previous report 30. Addition of IL-7 and SCF does not show any prominent influence on the cell recovery or proportions of T and NK cells. However, these factors are supportive for improving the seeding efficiency in the single progenitor assay (data not shown). In the following experiments, we used a cocktail of IL-2 (25 U/ml), IL-7 (50 U/ml), and SCF (10 ng/ml) as the agent modifying FT organ culture to evenly support the development of NK and T cells.
Figure 4.

Frequency and total number of different types of progenitors in subpopulations of 12- and 14-dpc FT cells. (A) Flow cytometric profiles of 12- and 14-dpc FT cells are shown. Numbers in panels represent percentage of cells. Cells in subpopulations gated with bars or rectangles in A served for the clonal assay (see Fig. 3 A). In all subpopulations, 50 individual cells were investigated. (B and C) The numbers of the different types of progenitors detected among 50 cells are scored (upper scales). In calculating the total number of progenitors in an FT (lower scales), the numbers of cells per 12-dpc FT and 14-dpc FT were regarded as 1,000 and 10,000, respectively.
Table 1.
Effect of Various Cytokines on the Generation of T and NK Cells in FT Organ Cultures
| Cytokine | No. of cells recovered | Percentage | ||
|---|---|---|---|---|
| Thy-1+ | CD3+NK1.1− | CD3−NK1.1+ | ||
| (×103) | ||||
| None | 83 ± 21 | 96 ± 2 | 41 ± 3 | 3 ± 1 |
| IL-2 (25) | 123 ± 5 | 92 ± 3 | 24 ± 3 | 19 ± 3 |
| IL-2 (100) | 110 ± 11 | 92 ± 4 | 18 ± 2 | 36 ± 3 |
| IL-15 (100) | 113 ± 12 | 95 ± 2 | 17 ± 2 | 38 ± 3 |
| IL-2 (50) + IL-15 (50) | 150 ± 30 | 94 ± 3 | 16 ± 2 | 40 ± 3 |
| IL-7 (50) + SCF (10) | 120 ± 13 | 92 ± 2 | 38 ± 2 | 2 ± 1 |
| IL-2 (25) + IL-7 (50) + SCF (10) | 130 ± 20 | 91 ± 3 | 28 ± 2 | 21 ± 3 |
100 12-dpc FT cells (B6Ly5.1) were cultured in each well in the presence of a dGuo-treated lobe (B6) and cytokines for 10 d.
Characterization of NK Cells Generated in the Modified FT Organ Culture.
The time course of T and NK cell generation in FT organ cultures with or without cytokine cocktail was investigated. FT cells from 12-dpc fetuses (100 cells) were cultured with a dGuo-treated lobe in the presence or absence of a cytokine cocktail (IL-2, IL-7, and SCF). At various days after the culture, cells were harvested, counted, and analyzed for their surface phenotypes. Numbers of CD3+ NK1.1− and CD3−NK1.1+ cells were plotted against culture days (Fig. 1a and Fig. b). The results indicate that the generation or growth of NK cells is highly dependent on exogenous cytokines, and that the NK cell generation in cytokine-supplemented cultures preceded T cell generation by ∼1 d. Fig. 1 C shows the representative flow cytometric profiles for expression of NK1.1 versus CD3, CD4 versus CD8, and TCR-α/β versus TCR-γ/δ on cells generated in cultures with or without the cytokine cocktail. These results indicate that the cytokine cocktail used here enhanced the generation of NK cells without modulating T cell generation.
Figure 1.


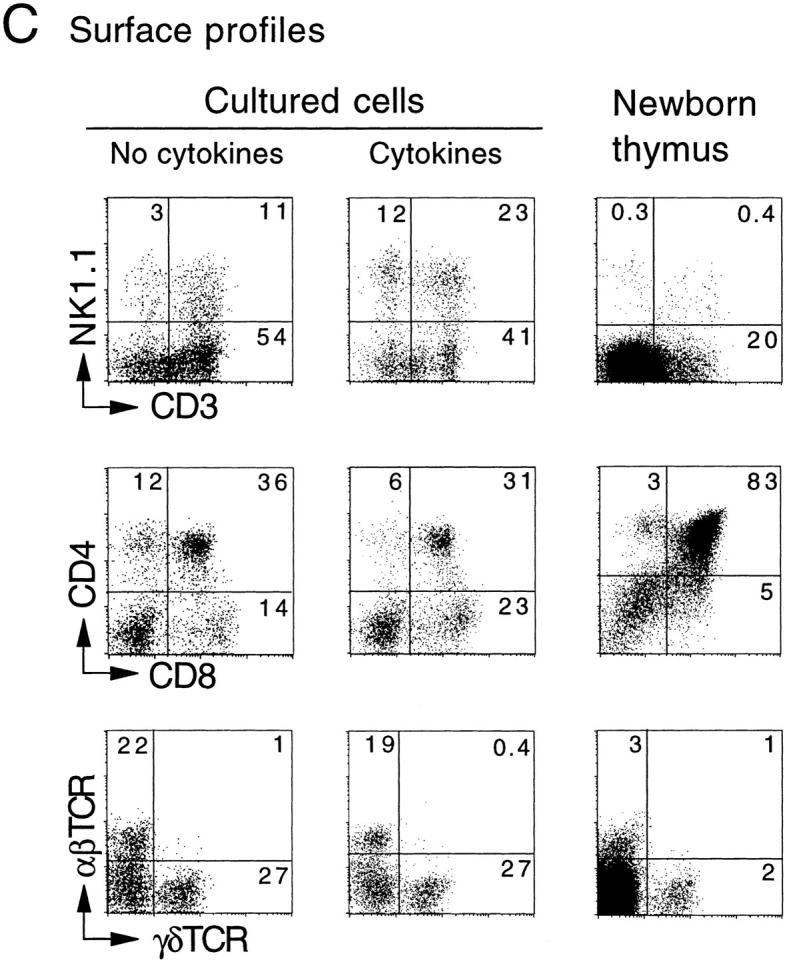
In vitro generation of T and NK cells from FT progenitors. Unfractionated FT cells (102) from 12-dpc B6Ly5.1 fetuses were cultured with a dGuo-treated FT lobe (B6) in the absence or presence of IL-2 (25 U/ml), IL-7 (50 U/ml), and SCF (10 ng/ml). At various days of culture, cells were collected from each well, counted, and analyzed by a flow cytometer. A and B show the kinetics of generation of CD3+NK1.1− and CD3−NK1.1+ cells, respectively. (C) Representative flow cytometric profiles of cells recovered 10 d after culture for expression of NK1.1 vs. CD3, CD4 vs. CD8, and TCR-α/β vs. TCR-γ/δ on Ly5.1+ cells. Profiles of newborn thymus cells are shown for comparison.
We next investigated whether the NK1.1+ cells generated in cytokine-supplemented FT organ cultures were functional as NK cells. Cells were harvested from cytokine-supplemented FT organ cultures seeded with 12-dpc FT cells (100 cells), and CD3−NK1.1+ cells and CD3−NK1.1− cells were isolated with a flow cytometer. These cells were examined for killer activity by culturing with the NK-sensitive cell line Yac-1 at various E/T ratios. Fig. 2 shows that NK1.1+ cells but not NK1.1− cells are cytotoxic, indicating that the NK1.1+ cells generated in the cytokine-supplemented FT organ culture are functional NK cells.
Figure 2.

Cytolytic activity of NK1.1+ cells derived from 12-dpc FT progenitors. Unfractionated FT cells (102) from 12-dpc fetuses were cultured together with a dGuo-treated FT lobe in the presence of a cytokine cocktail (see legend to Fig. 1). After 10 d of culture, cells were recovered and pooled from three wells, then stained in three colors with anti-Ly5.1, anti-CD3, and anti-NK1.1. Ly5.1+CD3−NK1.1+ cells and Ly5.1+CD3− NK1.1− cells were obtained by FACS® sorting, and were assayed for cytolytic activity against the target cell line Yac-1 as detailed in Materials and Methods.
Identification of p-T/NK, p-T, and p-NK in the FT by Clonal Assay.
Individual cells in the CD44+CD25− population from 14-dpc FT (B6Ly5.1) were seeded in the wells in which a dGuo-treated FT (B6) lobe had been placed (Fig. 3 A). The culture medium was supplemented with IL-2, IL-7, and SCF. After 10 d of culture under HOS conditions, cells were harvested from each well for flow cytometric analysis. Expression of T and NK cell markers was examined by gating Ly5.1+ cells. Surface phenotypes of cells derived from three types of progenitors identified in this experiment are shown in Fig. 3 B. The progenitors generating NK1.1+ cells but not CD3+ cells were determined to be p-NK (Fig. 3 B, left panels), whereas those generating only CD3+ cells were determined to be p-T (middle panels). In addition, the presence of p-T/NK was shown unambiguously (right panels). The T cells generated from a p-T as well as from a p-T/NK express CD4 and/or CD8 as well as TCR-α/β or TCR-γ/δ. The generation of NKT-like cells expressing both CD3 and NK1.1 was seen in the case of p-T/NK. Such NKT-like cells were also generated from p-T but not from p-NK when the culture period was extended (data not shown).
Figure 3.
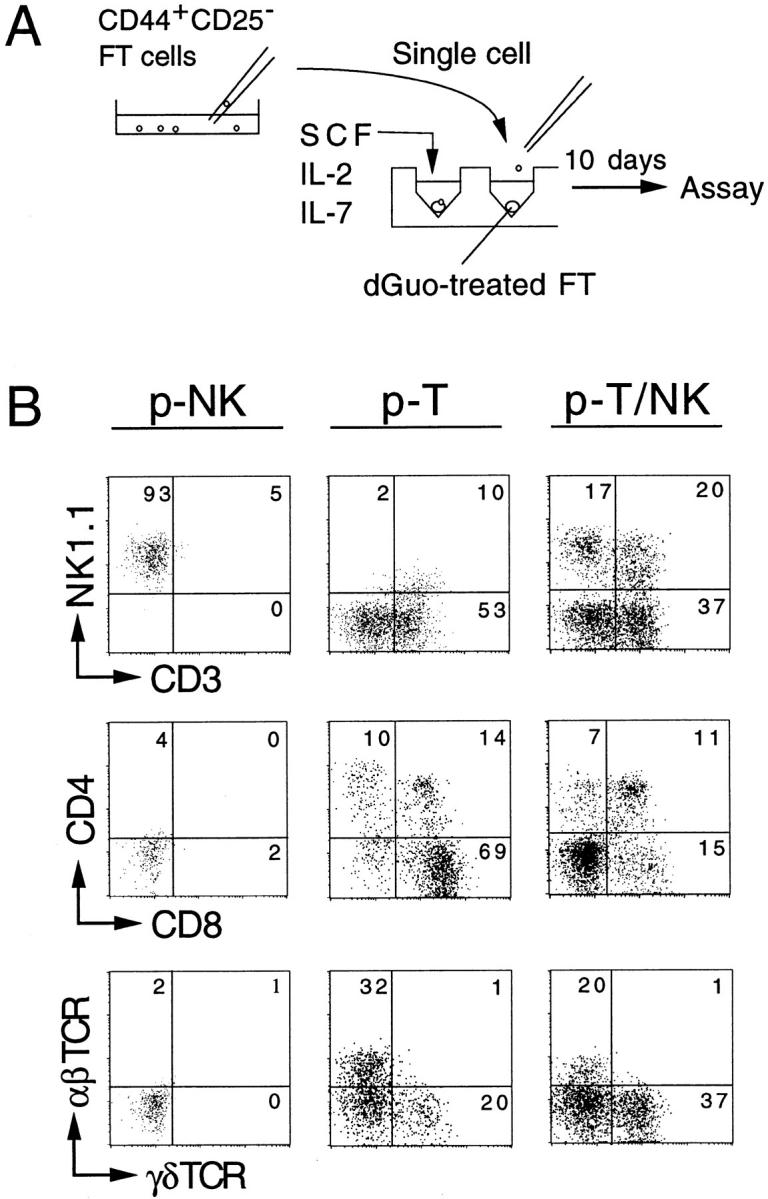
Representative flow cytometric profiles of cells derived from single FT progenitors. (A) Procedure of the clonal assay culture is shown. Single CD44+CD25− FT cells (14 dpc) were picked up under microscopical visualization, then seeded into the wells where a dGuo-treated FT lobe had been placed. The culture medium used was supplemented with a cytokine cocktail (see legend to Fig. 1). Cells were cultured under HOS conditions for 10 d. (B) Examples of the flow cytometric profiles of cells generated from p-NK, p-T, and p-T/NK are shown. Cell recoveries in these clones were 2.4 × 104, 8.0 × 104, and 2.0 × 105, respectively.
In our previous study investigating the developmental potential of thymic progenitors toward T, B, and myeloid lineages, it was indicated that a large majority of the progenitors in FT cells was committed to the T cell lineage 21. Because the culture conditions used in this study were unable to support the generation of NK cells, both p-T/NK and p-T should have been defined as “p-T.”
When Do the p-T and p-NK Split from Bipotent p-T/NK?
Subpopulations of 12- and 14-dpc FT cells (Fig. 4 A) were used as the progenitor source. These subpopulations were sorted, and the single cells (total 50 cells in each group) were cultured for 10 d in cytokine-supplemented FT organ culture as in the preceding section. Cells were recovered from each well, and analyzed with a flow cytometer. Three types of progenitors were found to be present in these subpopulations, and these three types were the same as those seen in Fig. 3 B.
The distribution of different types of progenitors in subpopulations of 12- and 14-dpc FT cells is shown in Fig. 4B and Fig. C, respectively. It was found that all T cell progenitors in the most immature CD44+CD25−FcR− (FcR−) population of 12-dpc FT were p-T/NK, and the frequency of the p-T/NK in this population is equivalent to that determined to be “p-T” in our previous investigation performed with the MLP assay 21. Quite a large number of p-NK also exists in this population. A small number of p-T appears at the next CD44+CD25−FcR+ (FcR+) stage, but p-T/NK and p-NK still predominate in this population. In the CD44+CD25− population of 14-dpc FT, the proportion of p-T becomes higher than in 12-dpc FT. It is also found that p-NK is the largest in number in this population. A prominent increase of p-T was seen at the CD44+CD25+ stage. Of interest is that p-T/NK and p-NK still exist in this stage. At the CD44−CD25+ stage, most of the cells no longer express progenitor activity, although a very small number of p-T can be seen (data not shown). The results shown in Fig. 4 suggest that the NK potential is retained in the T cell progenitor until just before the TCR-β gene rearrangement begins. In contrast, p-NK can split from p-T/NK at any stage of early T cell development in the FT.
NK Lineage Commitment Occurs before the Rearrangement of TCR Genes.
It is well known that NK cells do not express TCR on their surface, nor do they rearrange TCR genes 3. However, because the NK cells generated in the present study are artificially induced from thymic progenitors by culturing them with a thymic lobe in the presence of exogenous cytokines, the possibility could not be ruled out that the progenitors that had already initiated their TCR gene rearrangement were induced to become NK cells. DNA was extracted from NK and T cells derived from single p-T/NK, from NK cells derived from single p-NK, and from T cells derived from single p-T. Rearrangement of TCR-β and TCR-γ genes was examined by PCR using the primer pairs shown in Fig. 5. It is shown that multiple rearrangements had occurred at the Dβ-Jβ and Vγ-Jγ regions in all T cells derived from single progenitors. In marked contrast, TCR gene rearrangement was not observed in NK cells regardless of origin, indicating that the split to the NK progenitors ceases before rearrangement of the TCR-β gene.
Figure 5.

NK cells retain their TCR-β and TCR-γ locus in the germline configuration. The Dβ2-Jβ2 gene rearrangement (A) and Vγ4/Vγ3-Jγ1 gene rearrangement (B) in cells derived from single p-T/NK, p-NK, and p-T were investigated. As a positive control, unfractionated adult thymocytes (AT) were used. The DNA equivalent to 750 cells was PCR amplified in each sample.
Enrichment of p-NK in the IL-2Rβ+ Population of FT.
To ensure the existence of p-NK in the FT, we tried to isolate a population containing exclusively p-NK. For isolation of p-NK, we used an mAb to CD122 (IL-2/15Rβ), because it has been suggested that the NK progenitors express this molecule 6 31.
Lin−CD44+CD25− cells from 14-dpc FT can be separated into CD122− and CD122+ subpopulations (Fig. 6 A). In this experiment, anti-NK1.1 mAb is included in the lineage (Lin) markers. Cells enclosed in the rectangles (right panel of Fig. 6 A) were sorted, and served for the single cell assay to determine NK and T cell generating potential. The results, shown in Fig. 6 B, indicate that the CD122− population contains all three types of progenitors, whereas only p-NK were detected in the CD122+ population. Moreover, p-NK were highly enriched in the CD122+ population: 17 out of 50 cells examined were p-NK. Failure to detect any p-T/NK or p-T indicates that the frequency of these progenitors in this population is <1/50. Such a low frequency of p-T in the CD122+ population was confirmed by culturing single cells with a dGuo-treated lobe in the absence of exogenous IL-2 (data not shown). On the other hand, previous studies in which 103 CD122+ FT cells were cultured with a dGuo-treated FT indicated that this population retains the potential to generate T cells 6 32. Thus, the CD122+ population might contain a very small number of p-T, but the large majority of CD122+ progenitors are NK lineage committed.
Figure 6.

The CD122+ population of FT cells contains exclusively p-NK. (A) Flow cytometric profiles of 14-dpc FT cells. (B) Single CD44+CD25−CD122− or CD44+CD25−CD122+ cells (50 cells each) enclosed with rectangles in the right panel of A were cultured with a dGuo-treated FT lobe in the presence of the cytokine cocktail (see legend to Fig. 3). The numbers of the different types of progenitors detected among 50 CD122− and 50 CD122+ cells are scored.
Discussion
We have previously shown that only progenitors restricted to T, B, or myeloid lineage, but not multipotent progenitors, exist in the FT 21. However, the assay system used in these experiments was not designed to support the generation of NK cells. In this study, we succeeded in establishing a new assay system capable of determining the developmental potential of a single progenitor toward T and/or NK lineages. By examining individual cells with this assay system, the existence of bipotent p-T/NK in FT was unambiguously shown. The unipotent p-T and p-NK detected in this study are also considered to reflect the commitment status in vivo for the following reasons: (a) p-T or p-NK are enriched or purified in subpopulations of FT cells; (b) the addition of IL-2 to the culture medium made p-NK come into view without reducing the frequency of progenitors with T cell potential (our unpublished data), indicating that the progenitors detected as p-NK are distinct from p-T/NK or p-T; (c) detection of p-T/NK but not p-T in the FcR− subpopulation of 12-dpc FT indicated that the NK potential retained by a p-T/NK–type T cell progenitor is very efficiently expressed in this assay; and (d) addition of higher doses of IL-2 (50–100 U/ml) did not increase the frequency of p-NK or p-T/NK (data not shown), strongly suggesting that the progenitors detected as p-T retain no NK potential.
With this clonal assay, we clarified that all of the T cell progenitors previously determined in the CD44+CD25− FcR− population of 12-dpc FT 21 were bipotent p-T/NK (Fig. 4). It has recently been shown that a population capable of generating both T and NK cells is present in fetal spleen and fetal blood of 13–16 dpc fetuses 33 34, and we have found that a large number of p-T/NK exist in 12-dpc fetal liver (FL; our unpublished data). These findings strongly suggest that the p-T/NK that emerged in prethymic tissues have migrated into the FT. This study further showed that the earliest CD44+CD25−FcR− population contained quite a large number of p-NK in addition to p-T/NK. This is in marked contrast to the detection of only a very small number of p-T at early stages (Fig. 4 B). It is likely that the early thymic p-T/NK generate p-NK through uneven cell division, retaining their T/NK bipotent activity. However, the possibility cannot be ruled out that some of these p-NK are of extrathymic origin, as the p-NK have also been detected in FL (our unpublished data).
A rapid increase in the proportion of p-T, which are unable to generate NK cells, between the CD44+CD25− and CD44+CD25+ stages (Fig. 4) may indicate that progenitors are committed to the T cell lineage immediately before TCR-β gene rearrangement. The predominance of p-T at the CD44+CD25+ stage is compatible with previous findings that the CD44+CD25+ population scarcely retains NK potential 9 17. Consistent with this is the finding that the expression of T cell lineage–restricted molecules becomes prominent at this stage 19 35. It remains to be clarified whether the increase of p-T in this population is due to multiple uneven splits of p-T from p-T/NK or to the proliferation of p-T themselves at this stage.
It is well known that NK cell development is independent of the thymus, and NK activity in nu/nu mice is reported to be higher than in normal euthymic mice 4. Thus, a question to be answered is why the progenitors migrating into the thymus retain NK potential. It is possible that the thymus supplies NK cells to peripheral lymphoid tissues during fetal through neonatal ages, since NK cells are generated first in the FT during ontogeny 29 36. An alternative interpretation could be that the NK progenitors or a small number of NK cells generated in the FT play a part in inducing the architecture of the thymic microenvironment. Such an idea is based on recent findings that progenitors expressing surface lymphotoxin, which retain NK potential, participate in forming lymph nodes and Peyer's patches 37 38. However, it seems unlikely that NK progenitors are indispensable for the construction of the thymic environment, since isolated p-T can produce T cells in the dGuo-treated FT lobe (Fig. 3 and Fig. 5). Moreover, thymic T cell development is normal in lymphotoxin knockout mice 39 and Id2 knockout mice 40 that lack lymph nodes and Peyer's patches. There may be no necessity for splitting off NK potential before entering the thymus, since NK potential does not seem to disturb T cell generation in the thymus. On the other hand, it is unlikely that the role of p-NK is to supply NKT cells, as NKT-like cells are generated from p-T but not from p-NK (see text referring to Fig. 3). More detailed investigations are necessary to clarify the role of p-NK or NK cells in the thymus.
Fig. 7 illustrates the stages of commitment of p-T/NK to p-T and p-NK revealed by this study. All T cell progenitors in FL are T/NK bipotent, although p-NK also exist in FL (our unpublished data). The developmental potential toward T and NK lineages is intimately kept together until immediately before TCR-β gene rearrangement begins. Nevertheless, p-NK are generated at a rather broad range of differentiation stages, including prethymic stages. Delivery of p-NK in the FT begins at the earliest CD44+CD25−FcR− stage, and continues up to the CD44+CD25+ stage. In contrast, commitment to the T cell lineage, or the delivery of p-T, occurs mainly at the CD44+CD25+ stage, which is immediately before TCR-β gene rearrangement begins. Such an intimate association of NK potential to the T cell lineage may provide circumstantial evidence for the proposal that T cells have evolved from a prototype of NK cells.
Figure 7.

A model for NK and T cell lineage commitment in the murine FT. The T cell progenitors migrating into the FT are bipotent p-T/NK. Commitment of p-T/NK to p-NK occurs at a broad range of early differentiation stages, whereas commitment to p-T mainly occurs immediately before the TCR-β rearrangement. CD44+CD25− cells in 12-dpc FT comprise FcR− and FcR+ populations (see Fig. 4), whereas a majority of those in 14-dpc FT are FcR+ (reference 19). A portion of p-NK in 14-dpc FT express CD122. The width of the white arrows approximately correlates with the rate of p-T and p-NK generation. It is unclear whether p-NK also immigrate from extrathymic tissues (broken arrow).
To date, no signals, either extracellular or intracellular, mediating commitment of p-T/NK to p-T and p-NK have been determined. It has previously been shown that high doses of IL-2 and IL-15 may interfere with the cell fate choice of progenitors between T and NK lineages 6. Knockout of IL-2/15Rβ 41 or IL-15Rα 42 gene results in the complete absence of NK cells. Targeting of the IFN regulatory factor 1 (IRF-1) gene, which is indispensable for IL-15 production, also brings about deletion of NK cells 43. However, the role of cytokines or cytokine receptors seems to be mostly related to the promotion or inhibition of the growth of precursors and/or mature cells rather than to lineage commitment. Some transcription factors (GATA-3, TCF-1) have been reported to be required for T cell development 44, but it has not yet been clarified whether these molecules are also involved in NK cell development. Molecules that seem to be more directly involved in the branching point between T and NK lineages are natural dominant negative helix-loop-helix factors Id2 and Id3. Transduction of the Id3 gene into CD34+ cells from human FL was found to result in enhancement of NK cell generation at the cost of T cell generation 45. In the Id2-deficient mice, NK cell development was found to be severely depleted without affecting T cell development 40. A transcription factor, Ets-1, was also found to play an important role in the generation of NK but not T cells 46. A combination of these genetic investigations and the single progenitor assay established in this study will contribute significantly to the elucidation of molecular and cellular mechanisms underlying the commitment and differentiation of T and/or NK cells. We are currently setting up such an experimental system.
Acknowledgments
The authors are indebted to Dr. W.T.V. Germeraad (University Hospital Utrecht, Utrecht, The Netherlands) for reading the manuscript, and to Ms. Y. Takaoki for secretarial assistance.
This study was partially supported by grants from the Ministry of Education, Science, Sports and Culture, Japan.
Footnotes
Abbreviations used in this paper: APC, allophycocyanin; B6, C57BL/6; dGuo, deoxyguanosine; dpc, day(s) postcoitum; FcR, FcγRII/III; FL, fetal liver; FT, fetal thymus; HOS, high oxygen submersion; Lin, lineage marker; MLP, multilineage progenitor; p-NK, NK lineage–committed progenitor; p-T, T cell lineage–committed progenitor; p-T/NK, bipotent progenitor generating T and NK cells; SCF, stem cell factor.
References
- Herberman R.B., Reynolds C.W., Ortaldo J.R. Mechanism of cytotoxicity by natural killer (NK) cells. Annu. Rev. Immunol. 1986;4:651–680. doi: 10.1146/annurev.iy.04.040186.003251. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Biology of natural killer cells. Adv. Immunol. 1989;47:187–376. doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier L.L., Spits H., Phillips J.H. The developmental relationship between NK cells and T cells. Immunol. Today. 1992;13:392–395. doi: 10.1016/0167-5699(92)90087-N. [DOI] [PubMed] [Google Scholar]
- Herberman R.B., Nunn M.F., Lavrin D.H. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int. J. Cancer. 1975;16:230–239. doi: 10.1002/ijc.2910160205. [DOI] [PubMed] [Google Scholar]
- Rodewald H.R., Moingeon P., Lucich J.L., Dosiou C., Lopez P., Reinherz E.L. A population of early fetal thymocytes expressing FcγRII/III contains precursors of T lymphocytes and natural killer cells. Cell. 1992;69:139–150. doi: 10.1016/0092-8674(92)90125-v. [DOI] [PubMed] [Google Scholar]
- Leclercq G., Debacker V., Smedt M.D., Plum J. Differential effects of interleukin-15 and interleukin-2 on differentiation of bipotential T/natural killer progenitor cells. J. Exp. Med. 1996;184:325–336. doi: 10.1084/jem.184.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyle J.R., Michie A.M., Furlonger C., Nakano T., Lenardo M.J., Paige C.J., Zuniga-Pflucker J.C. Identification of a novel developmental stage marking lineage commitment of progenitor thymocytes. J. Exp. Med. 1997;186:173–182. doi: 10.1084/jem.186.2.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spits H. Early stages in human and mouse T-cell development. Curr. Opin. Immunol. 1994;6:212–221. doi: 10.1016/0952-7915(94)90094-9. [DOI] [PubMed] [Google Scholar]
- Moore T.A., Zlotnik A. T-cell lineage commitment and cytokine responses of thymic progenitors. Blood. 1995;86:1850–1860. [PubMed] [Google Scholar]
- Rodewald H.R. Pathways from hematopoietic stem cells to thymocytes. Curr. Opin. Immunol. 1995;7:176–187. doi: 10.1016/0952-7915(95)80002-6. [DOI] [PubMed] [Google Scholar]
- Shortman K., Wu L. Early T lymphocyte progenitors. Annu. Rev. Immunol. 1996;14:29–47. doi: 10.1146/annurev.immunol.14.1.29. [DOI] [PubMed] [Google Scholar]
- Abramson S., Miller R.G., Phillip R.A. The identification in adult bone marrow of pluripotent and restricted stem cells of the myeloid and lymphoid systems. J. Exp. Med. 1977;145:1567–1579. doi: 10.1084/jem.145.6.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick J.E., Magli M.C., Huszar D., Phillips R.A., Bernstein A. Introduction of a selectable gene into primitive stem cells capable of long-term reconstitution of the hemopoietic system of W/W vmice. Cell. 1985;42:71–79. doi: 10.1016/s0092-8674(85)80102-1. [DOI] [PubMed] [Google Scholar]
- Keller G., Paige C., Gilboa E., Wagner E.F. Expression of a foreign gene in myeloid and lymphoid cells derived from multipotent haematopoietic precursors. Nature. 1985;318:149–154. doi: 10.1038/318149a0. [DOI] [PubMed] [Google Scholar]
- Lemischka I.R., Raulet D.H., Mulligan R.C. Developmental potential and dynamic behavior of hematopoietic stem cells. Cell. 1986;45:917–927. doi: 10.1016/0092-8674(86)90566-0. [DOI] [PubMed] [Google Scholar]
- Matsuzaki Y., Gyotoku J., Ogawa M., Nishikawa S., Katsura Y., Gachelin G., Nakauchi H. Characterization of c-kit positive intrathymic stem cells that are restricted to lymphoid differentiation. J. Exp. Med. 1993;178:1283–1292. doi: 10.1084/jem.178.4.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga-Pflucker J.C., Jiang D., Lenardo M.J. Requirement for TNF-α and IL-1α in fetal thymocyte commitment and differentiation. Science. 1995;268:1906–1909. doi: 10.1126/science.7541554. [DOI] [PubMed] [Google Scholar]
- Peault B., Khazaal I., Weissman I.L. In vitro development of B cells and macrophages from early mouse fetal thymocytes. Eur. J. Immunol. 1994;24:781–784. doi: 10.1002/eji.1830240345. [DOI] [PubMed] [Google Scholar]
- Hattori N., Kawamoto H., Katsura Y. Isolation of the most immature population of murine fetal thymocytes that includes progenitors capable of generating T, B, and myeloid cells. J. Exp. Med. 1996;184:1901–1908. doi: 10.1084/jem.184.5.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto H., Ohmura K., Katsura Y. Direct evidence for the commitment of hematopoietic stem cells to T, B and myeloid lineages in murine fetal liver. Int. Immunol. 1997;9:1011–1019. doi: 10.1093/intimm/9.7.1011. [DOI] [PubMed] [Google Scholar]
- Kawamoto H., Ohmura K., Katsura Y. Presence of progenitors restricted to T, B, or myeloid lineage, but absence of multipotent stem cells, in the murine fetal thymus. J. Immunol. 1998;161:3799–3802. [PubMed] [Google Scholar]
- DiSanto J.P., Muller W., Guy-Grand D., Fischer A., Rajewsky K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor γ chain. Proc. Natl. Acad. Sci. USA. 1995;92:377–381. doi: 10.1073/pnas.92.2.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.Y., Saijo K., Takahashi T., Osawa M., Arase H., Hirayama N., Miyake K., Nakauchi H., Shirasawa T., Saito T. Developmental defects of lymphoid cells in Jak3 kinase-deficient mice. Immunity. 1995;3:771–782. doi: 10.1016/1074-7613(95)90066-7. [DOI] [PubMed] [Google Scholar]
- Wang B., Biron C., She J., Higgins K., Sunshine M.J., Lacy E., Lonberg N., Terhorst C. A block in both early T lymphocyte and natural killer cell development in transgenic mice with high-copy numbers of the human CD3E gene. Proc. Natl. Acad. Sci. USA. 1994;91:9402–9406. doi: 10.1073/pnas.91.20.9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M.J., Muench M.O., Roncarolo M.G., Lanier L.L., Phillips J.H. Identification of a common T/natural killer cell progenitor in human fetal thymus. J. Exp. Med. 1994;180:569–576. doi: 10.1084/jem.180.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M.J., Spits H., Lanier L.L., Phillips J.H. Human natural killer cell committed thymocytes and their relation to the T cell lineage. J. Exp. Med. 1993;178:1857–1866. doi: 10.1084/jem.178.6.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y., Katsura Y. Development of T cell receptor αβ-bearing T cells in the submersion organ culture of murine fetal thymus at high oxygen concentration. Eur. J. Immunol. 1993;23:200–205. doi: 10.1002/eji.1830230131. [DOI] [PubMed] [Google Scholar]
- Slezak S.E., Horan P.K. Cell-mediated cytotoxity. A highly sensitive and informative flow cytometric assay. J. Immunol. Methods. 1989;117:205–214. doi: 10.1016/0022-1759(89)90142-7. [DOI] [PubMed] [Google Scholar]
- Carlyle J.R., Michie A.M., Cho S.K., Zuniga-Pflucker J.C. Natural killer cell development and function precede αβ T cell differentiation in mouse fetal thymic ontogeny. J. Immunol. 1998;160:744–753. [PubMed] [Google Scholar]
- Aiba Y., Hirayama F., Ogawa M. Clonal proliferation and cytokine requirement of murine progenitors for natural killer cells. Blood. 1997;89:4005–4012. [PubMed] [Google Scholar]
- Williams N.S., Klem J., Puzanov I.J., Sivakumar P.V., Schatzle J.D., Bennett M., Kumar V. Natural killer cell differentiationinsights from knockout and transgenic mouse models and in vitro systems. Immunol. Rev. 1998;165:47–61. doi: 10.1111/j.1600-065x.1998.tb01229.x. [DOI] [PubMed] [Google Scholar]
- Falk I., Levelt C.N., Eichmann K. Lineage relationships of the fetal thymocyte subset that expresses the β chain of the interleukin-2 receptor. Eur. J. Immunol. 1993;23:3373–3376. doi: 10.1002/eji.1830231248. [DOI] [PubMed] [Google Scholar]
- Carlyle J.R., Zuniga-Pflucker J.C. Lineage commitment and differentiation of T and natural killer lymphocytes in the fetal mouse. Immunol. Rev. 1998;165:63–74. doi: 10.1111/j.1600-065x.1998.tb01230.x. [DOI] [PubMed] [Google Scholar]
- Carlyle J.R., Zuniga-Pflucker J.C. Requirement for the thymus in αβ T lymphocyte lineage commitment. Immunity. 1998;9:187–197. doi: 10.1016/s1074-7613(00)80601-9. [DOI] [PubMed] [Google Scholar]
- Wilson A., MacDonald H.R. Expression of genes encoding the pre-TCR and CD3 complex during thymus development. Int. Immunol. 1995;7:1659–1664. doi: 10.1093/intimm/7.10.1659. [DOI] [PubMed] [Google Scholar]
- Koo J.C., Peppard J.R., Hatzfeld A. Ontogeny of Nk-1+ natural killer cells. I. Promotion of Nk-1+ cells in fetal, baby, and old mice. J. Immunol. 1982;129:867–871. [PubMed] [Google Scholar]
- Mebius R.E., Rennert P., Weissman I.L. Developing lymph nodes collect CD4+CD3− LTβ+ cells that can differentiate to APC, NK cells and follicular cells but not T or B cells. Immunity. 1997;7:493–504. doi: 10.1016/s1074-7613(00)80371-4. [DOI] [PubMed] [Google Scholar]
- Yoshida H., Honda K., Shinkura R., Adachi S., Nishikawa S., Maki K., Ikuta K., Nishikawa S.I. IL-7 receptor α+ CD3+ cells in the embryonic intestine induces the organizing center of Peyer's patches. Int. Immunol. 1999;11:643–655. doi: 10.1093/intimm/11.5.643. [DOI] [PubMed] [Google Scholar]
- Togni P.D., Goellner J., Ruddle N.H., Streeter P.R., Flick A., Mariathasan S., Smith S.C., Carlson R., Shornick L.P., Schoenberger J.S. Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science. 1994;264:703–707. doi: 10.1126/science.8171322. [DOI] [PubMed] [Google Scholar]
- Yokota Y., Mansouri A., Mori S., Sugawara S., Adachi S., Nishikawa S., Gruss P. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397:702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]
- Suzuki H., Duncan G.S., Takimoto H., Mak T.W. Abnormal development of intestinal intraepithelial lymphocytes and peripheral natural killer cells in mice lacking the IL-2 receptor β chain. J. Exp. Med. 1997;185:499–505. doi: 10.1084/jem.185.3.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodoice J.P., Boone D.L., Chai S., Swain R.E., Dassopolos T., Trettin S., Ma A. IL-15 receptor maintains lymphoid homeostasis by supporting lymphocyte homing and proliferation. Immunity. 1998;9:669–676. doi: 10.1016/s1074-7613(00)80664-0. [DOI] [PubMed] [Google Scholar]
- Ogasawara K., Hida S., Azimi N., Tagaya Y., Sato T., Yokochi-Fukuda T., Waldmann T.A., Taniguchi T., Taki S. Requirement for IRF-1 in the microenvironment supporting development of natural killer cells. Nature. 1998;391:700–703. doi: 10.1038/35636. [DOI] [PubMed] [Google Scholar]
- Clevers H.C., Grosschedl R. Transcriptional control of lymphoid developmentlessons from gene targeting. Immunol. Today. 1996;17:336–343. doi: 10.1016/0167-5699(96)10019-0. [DOI] [PubMed] [Google Scholar]
- Heemskerk M.H., Blom B., Nolan G., Stegmann A.P., Bakker A.Q., Weijer K., Res P.C., Spits H. Inhibition of T cell and promotion of natural killer cell development by the dominant negative helix loop helix factor Id3. J. Exp. Med. 1997;186:1597–1602. doi: 10.1084/jem.186.9.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton K., Muthusamy N., Fischer C., Ting C.N., Walunas T.L., Lanier L.L., Leiden J.M. The Ets-1 transcription factor is required for the development of natural killer cells in mice. Immunity. 1998;9:555–563. doi: 10.1016/s1074-7613(00)80638-x. [DOI] [PubMed] [Google Scholar]