

Abstract
Immune evasion is critical for survival of viruses that establish persistent or recurrent infections. However, at the molecular level, little is known about how viruses evade immune attack in vivo. Herpes simplex virus (HSV)-1 glycoprotein gC has two domains that are involved in modulating complement activation; one binds C3, and the other is required for blocking C5 and properdin (P) binding to C3. To evaluate the importance of these regions in vivo, HSV-1 gC mutant viruses were constructed that lacked one or both gC domains and studied in a murine model of infection. Each gC region of complement regulation contributed to virulence; however, the C3 binding domain was far more important, as virus lacking this domain was much less virulent than virus lacking the C5/P inhibitory domain and was as attenuated as virus lacking both domains. Studies in C3 knockout mice and mice reconstituted with C3 confirmed that the gC domains are inhibitors of complement activation, accounting for a 50-fold difference in virulence between mutant and wild-type viruses. We conclude that the C3 binding domain on gC is a major contributor to immune evasion and that this site explains at a molecular level why wild-type virus resists complement attack.
Keywords: immunology, pathogenesis, innate immunity, disease, C3
Herpes simplex virus has developed many strategies for survival in the human host. One of the most successful is its ability to establish latent infection in neurons without expressing viral proteins that serve as targets for immune attack 1 2. However, during latency, virus cannot spread person to person; therefore, the virus must reactivate to successfully infect another host. HSV has the ability to replicate for days after reactivation in the immune host, which may be related to viral immune evasion strategies. HSV-1 immune evasion molecules block activities of antibodies 3 4 5 6 7 and complement 8 9 10 and prevent antigen presentation by the MHC class I complex 11 12. Molecules that perform similar functions have been identified in adenoviruses, vaccinia virus, and other members of the herpes virus family 13 14 15 16 17 18.
HSV-1 glycoprotein gC is among the best characterized of the viral immune evasion molecules. gC binds complement components C3, C3b, iC3b, and C3c, accelerates the decay of the alternative pathway C3 convertase, and blocks the interaction of C5 and properdin (P)1 with C3 9 19 20. The C5 and P blocking domain is near the NH2 terminus, whereas the C3 binding domain is in the central region of the molecule 9 21. gC inhibits complement activation 9 20, protects cell free virus from complement-mediated neutralization 10 22 23, inhibits complement-mediated lysis of HSV-infected cells 24 25, and protects the virus against complement attack in vivo 26.
Vaccines incorporating molecules required for virus entry, including glycoproteins gB and gD, failed to prevent infection and had little effect on recurrence rates 27 28 29. A better understanding of HSV immune evasion strategies may be required to design more effective vaccines. With this goal in mind, we evaluated virulence of gC mutant viruses deleted in the C3 binding domain, the C5/P blocking domain, or both to define the contributions of each domain to immune evasion. We chose to study gC–complement interactions in mice because C3 knockout mice are available to establish the importance of C3. We selected the murine flank model, as we previously showed that complement significantly contributes to defense against HSV-1 in this model 24. We used C3 reconstitution 30 to confirm that gC is highly effective in protecting wild-type virus from complement attack and to define an impressive role for the C3 binding domain in pathogenesis.
Materials and Methods
Construction of gC Mutant Viruses.
HSV-1 strain NS, a low passage clinical isolate, was the parental strain for the mutant viruses. NS-gCnull virus has the entire gC protein coding sequence replaced by a lacZ expression cassette under the control of the HSV-1 infected cell protein 6 early promoter, as previously described 23. NS-gCΔC5/P virus has a deletion of the gC domain involved in blocking C5 and P binding to C3 9 21. This deletion was constructed from the NS BamH1 “I” fragment (reference 31; nucleotides 91,602–98,254; sequence available from EMBL/GenBank/DDBJ under accession numbers X14122, D00317, D00374, S40493, and XD2138) by cutting with Nsi1 and EcoR1 and cloning this fragment (nucleotides 94,911–96,751) into pGEM7Z (Promega Corp.) to create plasmid pLW1. An Nhe1–EcoR1 fragment (nucleotides 96,276–96,751) was excised from pLW1 and cut with Apa1 to delete 91 gC amino acids (amino acids 33–123; nucleotides 96,407–96,679). The Nhe1–EcoR1 fragment deleted of gC amino acids 33–123 was inserted back into pLW1, and then the Nsi1–EcoR1 fragment from pLW1 was cloned back into the BamH1 I fragment to create a flanking sequence vector with a deletion of the gC C5/P domain.
gCΔC3 has a deletion of 92 amino acids (gC amino acids 275–367), prepared by cutting plasmids B29 and H71 32 with BglII, religating the 5′ portion of B29 with the 3′ portion of H71, and then cloning this fragment into the EcoR1–EcoRV site of the BamH1 I fragment. The construction adds a 12-mer BglII linker after gC amino acid 275 and creates a flanking sequence vector that has a deletion of two gC C3 binding regions, resulting in a net loss of 88 amino acids.
A flanking sequence vector to construct gCΔC5/P,C3 was prepared by replacing the BamH1 I EcoR1–EcoRV fragment in the gCΔC5/P flanking sequence vector with the EcoR1–EcoRV fragment of the gCΔC3 flanking sequence vector. Recombinant viruses were prepared in Vero cells by cotransfection using NS-gCnull virus DNA and the various flanking sequence vectors. Recombinant viruses were identified by anti-gC immunoperoxidase staining of infected cells 21. Virus pools were prepared after three rounds of plaque purification.
Sucrose Gradient Purification of Virus.
Vero cells were infected at an MOI of 5 for 20 h at 37°C, and then virus from supernatant fluids was purified on a 5–65% sucrose gradient 23. The visible virus band was collected, dialyzed against PBS at 4°C, and stored at −70°C. Virus titers were determined by plaque assay on Vero cells.
One-Step Growth Curves.
Vero cells were inoculated at an MOI of 5, and at 1, 4, 8, 12, 20, and 24 h after infection, cells plus supernatant fluids were harvested, cells were lysed by sonication, and viral titers were determined by plaque assay.
Southern Blot Analysis.
DNA was digested with BamH1 and HincII, electrophoresed in 1.2% agarose gels, transferred to Immobilon-S membranes (Millipore Corp.), and UV cross-linked. A biotin-labeled gC-1 fragment spanning the entire protein coding sequence (nucleotides 96,276–97,945) was used as probe, and bands were detected by chemiluminescence (New England Biolabs) 6.
Western Blot Analysis.
Purified virus was run on 10% SDS-PAGE, transferred to Immobilon-P membranes (Millipore Corp.), and visualized using polyclonal rabbit anti-gC antibody and horseradish peroxidase–conjugated goat anti–rabbit IgG (Amersham Pharmacia Biotech) 23.
Antibody-independent Complement Neutralization.
Neutralization assays were performed by incubating 104–105 PFU of purified virus with HSV-1 and -2–seronegative human serum as the source of complement for 1 h at 37°C. As a control, complement was inactivated by heat treatment at 56°C for 30 min. Virus titers were then determined by plaque assay on Vero cells. Complement neutralization was calculated as the difference in titers between virus incubated with active or heat-inactivated serum 23.
C3b Rosetting Assays.
Vero cells were infected at an MOI of 2 for 20 h, and then cells were removed using cell dissociation buffer (GIBCO BRL), incubated with C3b-coated erythrocytes for 1 h at 37°C, and viewed by microscopy for rosettes. Cells with four or more bound erythrocytes were considered positive 23.
C3 Purification.
Human C3 was purified according to Hammer et al. 33 with modifications. 20 parts human plasma were treated with 1 part inhibitor containing 1 M KH2PO4, 0.2 M Na4EDTA, 0.2 M benzamidine, and 1 mM PMSF. C3 was precipitated from plasma first with 4.5% polyethylene glycol (PEG) and then with 12% PEG at 0°C. The pellet was dissolved in buffer A (20 mM Na2HPO4, pH 7.4) and loaded onto a DEAE 40HR column (5 × 6.3 cm; Waters) preequilibrated with buffer A. The bound proteins were eluted using a 0–0.5 M NaCl linear salt gradient. Fractions containing C3 were detected by SDS-PAGE and immunodiffusion, dialyzed against buffer A, loaded onto a Mono Q HR 10/10 column (Amersham Pharmacia Biotech), and eluted with a linear salt gradient to 0.5 M NaCl. Homogeneous C3 fractions were identified by SDS-PAGE and immunodiffusion, pooled, and dialyzed against PBS (10 mM Na2HPO4 and 145 mM NaCl, pH 7.4). The purified C3 contained 80% native C3 and 20% C3(H2O), as determined by analyzing the protein sample on a Mono S column (Amersham Pharmacia Biotech) 34.
Murine Flank Model.
The flank model and the C3 knockout mice derived from C57BL/6 mice have been described previously 26 35. In brief, purified virus was scratched onto the flank that was shaved and chemically denuded. Disease at the inoculation site first appeared on day 3 and was scored as follows: erythema or swelling with no vesicles was assigned 0.5 points; individual vesicles were scored as 1 point each with a total maximum daily score of 5. If lesions coalesced, up to 5 points were assigned based on the size of the lesions. Zosteriform disease first appeared on day 4 or 5 and was scored similarly, except that the maximum daily score was 10 because more lesions could be counted over the larger skin area involved.
C3 reconstitution experiments were performed by intraperitoneal injection of C3 knockout mice using 3 mg of human C3 per mouse. C3 knockout mouse serum was assayed for total hemolytic complement activity before and after reconstitution with human C3 as described 36.
Results
Characterization of gC Mutant Viruses.
Features of wild-type gC and three gC mutants are shown in Fig. 1. Wild-type gC (Fig. 1, top) has four C3 binding regions (light gray), each of which is required to bind C3 32. The gC domain that interferes with C5 and P binding to C3 is located at the NH2-terminal region between residues 33 and 123 9 21. gCΔC5/P mutant (Fig. 1, second from top) contains a deletion of gC amino acids 33–123. gCΔC3 mutant (Fig. 1, third from top) has a deletion of amino acids 275–367 that includes C3 binding regions II and III. This deletion removes a disulfide-bonded cysteine pair 37 to minimize the effect of the deletion on gC conformation. gCΔC5/P,C3 mutant (Fig. 1, bottom) has both domains deleted that interact with complement.
Figure 1.

Stick diagram of wild-type and mutant gC proteins. The top stick figure shows wild-type gC with a signal sequence (SIG), transmembrane domain (TM), the C5/P blocking domain, and four regions (labeled I–IV) shown as shaded ovals that are required for C3 binding. Black balloons show the positions of N-linked glycosylation sites. Cysteines are indicated (C), and lines joining two Cs reflect disulfide bonding pattern. The second stick figure shows the deletion from amino acids 33–123 that eliminates the C5/P blocking domain. The third stick figure shows the deletion from amino acids 275–367 that eliminates C3 binding domain regions II and III. The fourth (bottom) stick figure shows the double deletion that eliminates the C5/P blocking and the C3 binding domain.
The mutant gC proteins were recombined into virus, and the size of the gC DNA was evaluated by Southern blot analysis. HSV-1 was cut with BamH1 and HincII to cut once within the gC gene and probed with sequences spanning the entire gC protein coding sequence. Fig. 2 A (left blot) shows two gC bands for wild-type NS and smaller upper bands for gC mutant viruses NS-gCΔC5/P, C3 and NS-gCΔC3; the right blot shows the smaller upper band for gC mutant NS-gCΔC5/P. Bands were of the expected size for each of the gC mutant viruses. Western blots were performed on purified virus to demonstrate that mutant gC glycoproteins are incorporated into the virion and are of the expected size (Fig. 2 B). The double mutant virus NS-gCΔC5/P,C3 makes the smallest of the three mutant proteins. NS-gCΔC5/P has a deletion of 91 amino acids and on SDS-PAGE runs smaller than NS-gCΔC3, which has a deletion of 88 amino acids. However, the difference in migration of the two glycoproteins was greater than expected based on amino acid size. A likely explanation is that five N-linked glycosylation sites and numerous predicted O-linked glycosylation sites lie within the deleted domain of NS-gCΔC5/P, whereas one N-linked and few O-linked glycosylation sites lie within the deleted fragment of NS-gCΔC3.
Figure 2.

Southern blot (A) and Western blot (B) of wild-type and gC mutant viruses. (A) Left panel: DNA from wild-type virus, NS, and gC mutant viruses NS-gCΔC5/P,C3 and NS-gCΔC3 was cut with BamH1 and HincII to generate two gC fragments of different sizes consistent with the deletion mutations introduced. Right panel: DNA from NS and gC mutant virus NS-gCΔC5/P was cut with the same restriction enzymes as above. (B) Western blot of purified viruses showing wild-type (NS) gC protein and the smaller proteins made by each of the mutant viruses NS-gCΔC5/P, NS-gCΔC3, and NS-gCΔC5/P,C3. No gC protein is detected in the gCnull virus lane.
The kinetics of virus replication and peak titers achieved for each gC mutant virus were similar to those of wild-type virus when evaluated by single-step growth curves (result not shown). gC protein expression at the infected cell surface was confirmed for each mutant virus by immunoperoxidase staining, and the ability of gC to bind C3b was determined by rosetting assays. Mutant viruses NS-gCΔC3, NS-gCΔC5/P,C3, and NS-gCnull lack the C3 binding domain and fail to form C3b rosettes, whereas NS and NS-gCΔC5/P mutant viruses have the C3 binding domain and form rosettes (Fig. 3). The results suggest that the gC mutations have not drastically altered protein conformation, as mutant virus NS-gCΔC5/P formed rosettes, and each mutant virus incorporated gC into the envelope and expressed gC at the infected cell surface.
Figure 3.

C3b binding was assessed by a rosetting assay. Wild-type virus, NS, and NS-gCΔC5/P (C5/P) form rosettes, whereas NS-gCΔC3 (C3), NS-gCΔC5/P,C3 (C5/P,C3), and NS-gCnull (gCnull) viruses do not form rosettes.
gC Domains that Interact with Complement Protect the Virus from Complement Neutralization.
Previously, we reported that HSV-1 lacking the entire gC protein is rapidly neutralized by human complement, showing 50% neutralization within 2 min and 100–5,000-fold neutralization by 1 h, the higher number occurring if fresh complement was added during the hour-long incubation. In contrast, wild-type virus is resistant to complement (less than or equal to twofold neutralization; reference 23). The panel of gC mutant viruses was incubated with HSV-1 and -2 antibody–negative human serum as source of complement for 1 h or complement-inactivated serum as a control, and viral titers were determined. A glycoprotein gE mutant virus derived from NS 6 served as a control for the effects of removing a glycoprotein from the virion envelope. gE lacks complement interacting domains. Complement has a small effect on NS and NS-gEnull viruses (0.3 log10 and 0.35 log10, respectively; Fig. 4). gC mutant virus lacking the C5/P blocking domain was relatively resistant to complement (0.43 log10, P = 0.22 or 0.76, comparing NS-gCΔC5/P with NS or NS-gEnull, respectively). The gC mutant virus lacking the C3 binding domain was somewhat more susceptible to complement (0.66 log10, P = 0.01 or 0.1, comparing NS-gCΔC3 with NS or NS-gEnull, respectively); however, gC mutant virus lacking both domains was even more susceptible to complement (1.58 log10, P ≤ 0.001, comparing NS-gCΔC5/P,C3 with NS or gE null virus) and significantly more susceptible than either single gC mutant virus (NS-gCΔC5/P,C3 versus NS-gCΔC5/P or NS-gCΔC3, P ≤ 0.003; Fig. 4). We conclude that both gC domains are important in complement regulation, as the gC double mutant virus was more susceptible to complement than either single mutant virus alone.
Figure 4.

Antibody-independent complement neutralization of wild-type virus, gEnull mutant virus, and a panel of gC mutant viruses. 104–105 PFU of purified virus was incubated with HSV-1 and -2 antibody–negative human serum as a source of complement or heat-inactivated serum as control. After a 1-h incubation at 37°C, virus titers were determined by plaque assay. Results are expressed as the differences in titers (log10) ± SEM comparing virus incubated with inactivated complement or active complement. NS, wild-type HSV-1; gEnull, gE deletion virus NS-gEnull 6 derived from NS; C5/P, NS-gCΔC5/P; C3, NS-gCΔC3; C5/P,C3, NS-gCΔC5/P,C3; gCnull, NS-gCnull. Experiments were performed three to five times, as indicated by the numbers above the bars.
gC Domains that Interact with Complement Are Virulence Factors In Vivo.
Studies were performed to determine if the complement-interacting domains are virulence factors in vivo using the murine flank model 26 35. Fig. 5A and Fig. B show inoculation site and zosteriform disease scores, respectively, for complement-intact mice infected with 5 × 104 PFU of NS or gC mutant viruses. Disease scores at the inoculation and zosteriform sites are lower for mutant virus NS-gCΔC5/P than for NS (P ≤ 0.02 for days 4–8 at the inoculation site, and P < 0.001 for day 5 at the zosteriform site), supporting the conclusion that the C5/P domain is a virulence factor. Even greater differences were detected comparing NS-gCΔC3 or double mutant virus NS-gCΔC5/P,C3 with NS (P < 0.001 for days 5–8 at the inoculation and zosteriform sites). Notably, NS-gCΔC3 virus caused significantly less disease than NS-gCΔC5/P virus (P < 0.001 for days 6–8 at the inoculation and zosteriform sites). We conclude that both gC domains have a significant effect on disease severity; however, the C3 domain is more important because the mutant virus lacking this domain is significantly less virulent than the C5/P mutant virus and is as impaired as the double mutant virus.
Figure 5.

Lesion scores in mice infected with wild-type or gC mutant viruses. The left panels (A, C, and E) show disease scores ± SEM at the inoculation site, and the right panels (B, D, and F) show disease scores ± SEM at the zosteriform spread site. A and B demonstrate disease scores in complement-intact mice (normal mice), C and D in C3 knockout mice, and E and F in C3 knockout mice reconstituted with human C3. WT, virus NS; C5, NS-gCΔC5/P; C3, NS-gCΔC3; C5,C3, gC double mutant virus NS-gCΔC5/P,C3. The numbers of animals evaluated in each group are indicated in the legends at right.
Experiments were performed in C3 knockout mice to determine if interactions between gC and complement account for the lowered virulence of gC mutant viruses. Virulence of gC mutant viruses should be similar to wild-type virus in C3 knockout mice, as C5, P, or C3 cannot become activated. Fig. 5C and Fig. D show that virulence for each of the mutant viruses is comparable to wild-type virus in C3 knockout mice.
As further evidence for the importance of the interaction between gC and complement, C3 knockout mice were reconstituted with C3. Total hemolytic complement activity was undetectable in serum from C3 knockout mice; however, after injection of human C3, total hemolytic complement activity was restored to levels detected in complement-intact animals (result not shown). Fig. 5E and Fig. F show that the gC double mutant virus NS-gCΔC5/P,C3 is markedly less virulent in C3 knockout mice reconstituted with C3, whereas the virulence of wild-type virus is unchanged (comparing NS and gC double mutant virus: inoculation site lesions, P < 0.05 on day 3, P < 0.01 on day 4, and P < 0.001 on days 5–8, whereas for zosteriform lesions, P < 0.001 on days 5–8).
Fig. 6 shows photographs of flank lesions on day 6 after infection. The left panels show NS, and the right panels show NS-gCΔC5/P,C3, demonstrating that lesions caused by NS are comparable in the presence or absence of complement, whereas lesions produced by the gC mutant virus are greatly affected by complement. These experiments illustrate the important role of gC–complement interactions in pathogenesis.
Figure 6.
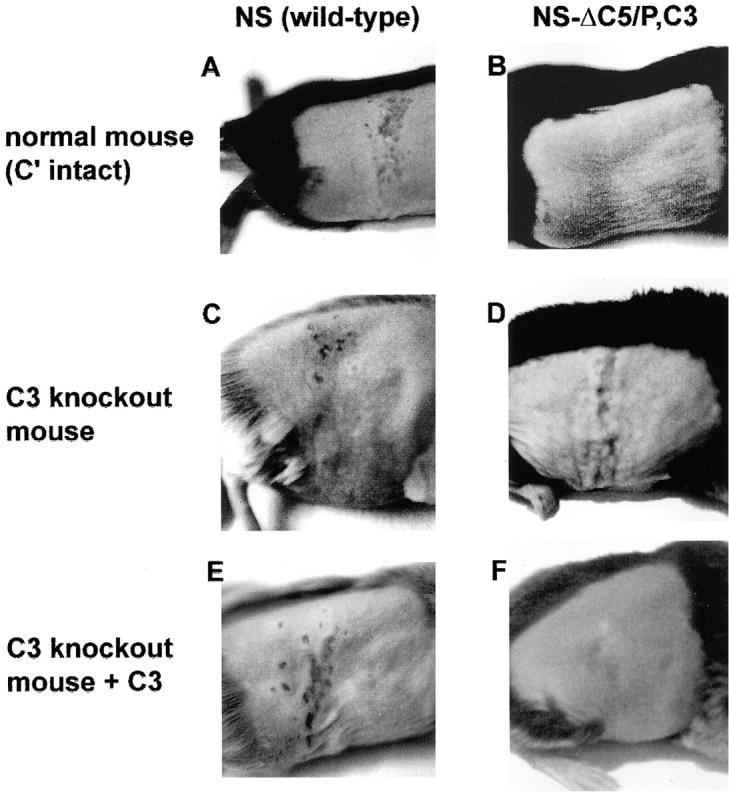
Photographs of denuded flanks of mice 6 d after infection. (A) NS in a complement-intact C57BL/6 mouse showing >10 lesions at the zosteriform site. (B) NS-gCΔC5/P,C3 in a complement-intact C57BL/6 mouse showing no lesions. (C) NS in a C3 knockout mouse showing >10 zosteriform lesions. (D) NS-gCΔC5/P,C3 in a C3 knockout mouse showing >10 zosteriform lesions. (E) NS in a C3 knockout mouse reconstituted with human C3 showing >10 zosteriform lesions. (F) NS-gCΔC5/P,C3 in a C3 knockout mouse reconstituted with human C3 showing no lesions.
Defining the Magnitude of the gC–Complement Effect on Virulence.
Experiments were performed using NS and gC double mutant virus NS-gCΔC5/P,C3 to compare disease scores over a range of inocula. In Fig. 7, the disease scores are expressed as the average cumulative disease scores from days 3–8 after infection 26. In complement-intact mice, infection with 5 × 105 PFU of the gC double mutant virus produced a disease score of 19.3 ± 1.9 at the inoculation site (Fig. 7 A). Producing a comparable disease score required 50-fold less wild-type virus. Infection with 5 × 104 PFU of gC double mutant virus resulted in a disease score of 8.9 ± 0.9, which also correlated with the disease caused by 50-fold less wild-type virus. Similarly, ∼50-fold less NS than gC mutant virus was required to produce comparable zosteriform disease (Fig. 7 B). In marked contrast, in C3 knockout mice, no significant differences were noted comparing wild-type and gC mutant viruses (Fig. 7C and Fig. D). We conclude that domains on gC that interact with complement account for a 50-fold effect on virulence in complement-intact animals, whereas in C3 knockout animals, the domains have no effect. Notably, NS disease scores are virtually identical in complement-intact and C3 knockout mice (comparing the scores for wild-type virus in Fig. 7A with C or B with D), which indicates that gC complement–interacting domains are remarkably effective in protecting wild-type virus from complement attack.
Figure 7.

Disease scores in complement-intact and C3 knockout mice infected with NS or NS-gCΔC5/P,C3 at 5 × 102–5 × 105 PFU. Disease scores are plotted as the average cumulative scores ± SEM for days 3–8 after infection at the inoculation site and for days 4–8 at the zosteriform site. A and B represent disease scores at the inoculation and zosteriform sites, respectively, in complement-intact mice. The numbers of NS (WT) animals evaluated at each dose were n = 4 at 5 × 105 PFU, n = 8 at 5 × 104 PFU, n = 5 at 5 × 103 PFU, and n = 4 at 5 × 102 PFU. C5/P,C3 represents gC double mutant virus NS-gCΔC5/P,C3. The numbers of gC double mutant virus-infected mice evaluated were n = 4 at 5 × 105 PFU, n = 14 at 5 × 104 PFU, n = 3 at 5 × 103 PFU, and n = 4 at 5 × 102 PFU. C and D represent disease scores at the inoculation and zosteriform sites, respectively, in C3 knockout mice. For wild-type virus, the number of animals evaluated was n = 4 except at 5 × 105 PFU, where n = 3. For NS-gCΔC5/P,C3 the numbers of animals evaluated were n = 4 at 5 × 105 PFU, n = 10 at 5 × 104 PFU, n = 4 at 5 × 103 PFU, and n = 3 at 5 × 102 PFU.
Discussion
We have previously defined a role for gC in binding C3 8 9 19, accelerating the decay of the alternative pathway C3 convertase 20 and interfering with the interaction of C5 and P with C3b 9 21. We showed that gCnull virus is highly susceptible to complement neutralization even in the absence of antibodies and that gCnull virus is less virulent in mice and guinea pigs 23 26. We now address the contribution to pathogenesis of two gC domains that interact with complement by constructing gC mutant viruses lacking the C5/P blocking domain, the C3 binding domain, or both domains.
The murine studies indicate that each of the single mutant viruses is significantly less virulent than wild-type virus; however, the C3 binding domain mutant virus is clearly the more important, as this mutant virus was as attenuated as the gC double mutant virus. Fig. 8 A presents a model of complement inhibition mediated by gC domains. In vivo, NS-gCΔC5/P mutant virus is only moderately less virulent than wild-type virus, suggesting that gC affects more than C5 and P binding to C3b, such as accelerating the decay of the alternative pathway C3 convertase, C3bBb 20, or perhaps other C3-mediated functions yet to be determined (Fig. 8 B). The deletion of the C3 binding domain eliminates gC binding to C3b (Fig. 8 C), rendering the C5/P domain ineffective because it lacks proximity to C3b and cannot hinder C5 and P binding. Therefore, a C3 binding domain mutant virus should be as impaired as a double mutant virus (Fig. 8 D). The model explains our in vivo results, but it fails to explain why the C3 binding mutant virus was not as impaired as the double mutant virus in neutralization assays performed in vitro. A possible explanation is that the main effect of gC in vivo may be on a function other than neutralization, such as preventing complement lysis of infected cells 24.
Figure 8.
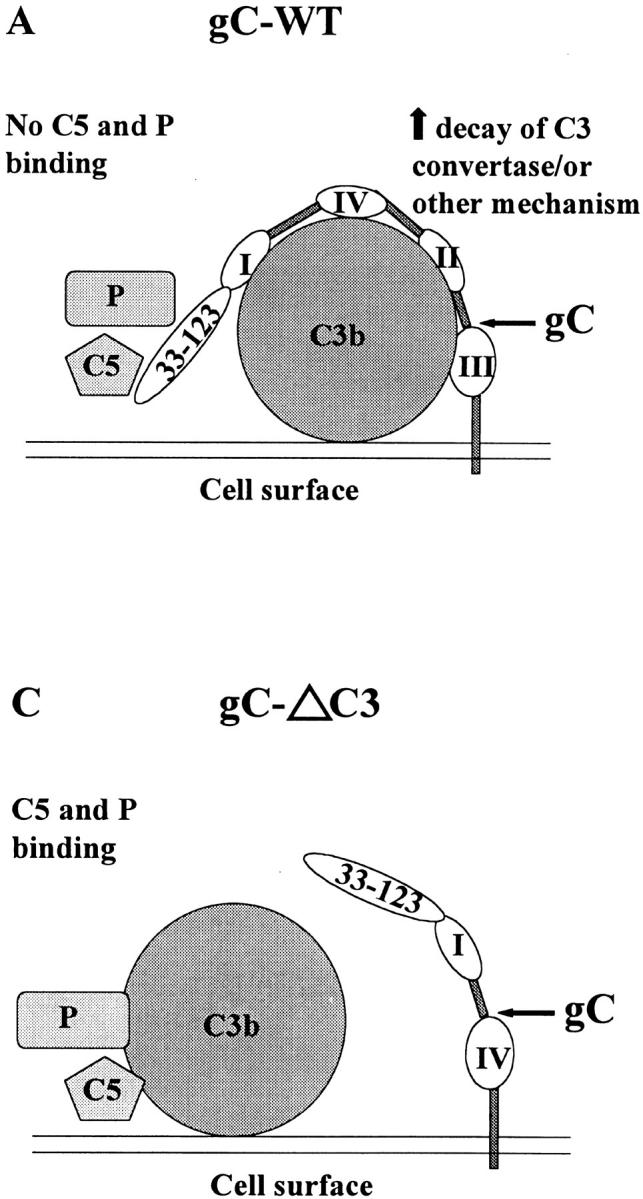
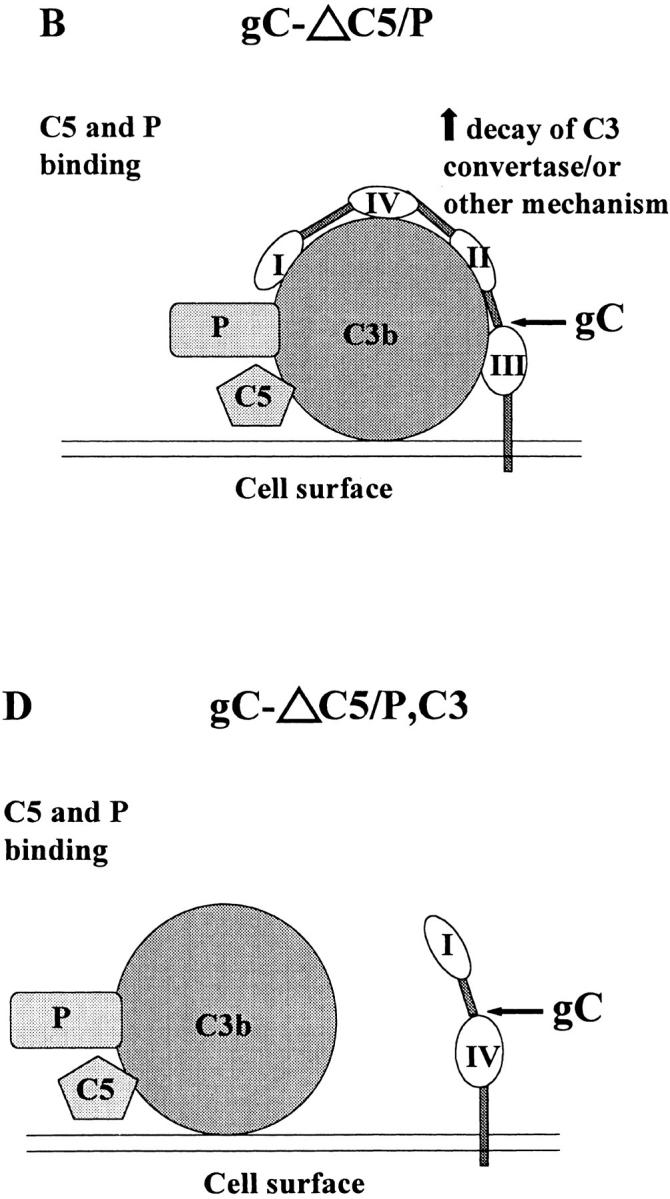
Models showing how gC domains that interact with complement block complement activation. (A) Wild-type gC: the cell surface activates the complement cascade, resulting in C3b fixation on the cell surface. gC regions I–IV bind to C3b, leading to accelerated decay of C3 convertase 20, or inhibit other C3-mediated activities, and the NH2 terminus C5/P blocking domain prevents the interaction of C5 and P with C3b 9 21. (B) gCΔC5/P: gC regions I–IV bind to C3b and inhibit complement activation as in A, whereas the absence of the C5/P blocking domain permits C5 and P binding to C3b, leading to increased complement activation. The net effect is partial inhibition of complement activation. (C) gCΔC3: gC does not bind to C3b; therefore, C3 convertase or other C3-mediated activities are not affected. The C5/P blocking domain is ineffective because it lacks proximity to C3b and cannot block C5 and P binding. The result is that gCΔC3 is as susceptible to complement as gCΔC5/P,C3, shown in D.
Our results definitively demonstrate that gC is a virulence factor because it regulates complement activation. This conclusion is based on observations that gC mutant viruses are significantly less virulent than wild-type virus in complement-intact mice, whereas virulence is comparable to that of wild-type virus in C3 knockout mice. Proof for the requirement for C3 was shown by C3 reconstitution experiments in which C3 reduced virulence of the gC double mutant virus but had no effect on wild-type virus. The magnitude of the gC effect was addressed by evaluating lesion scores using inoculation titers ranging from 5 × 105 to 5 × 102 PFU. In complement-intact mice, 50-fold more gC double mutant virus was required to cause disease comparable to that caused by wild-type virus.
In vitro studies indicate that gC is also involved in the initial stage of virus attachment to cells by binding to heparan sulfate 38 39. In the absence of gC, gB can mediate this effect 38, but a role for gC in attachment has not been established in vivo. Our results indicate that the domains deleted from the gC mutant viruses do not play an important role in attachment, because in C3 knockout mice, the mutant viruses are as virulent as wild-type virus. Of particular interest is the C5/P deletion mutant virus. This mutant virus lacks gC amino acids 33–123, identified as an important domain for gC-mediated attachment to heparan sulfate in vitro 40. Yet this virus shows no difference in virulence from wild-type virus in C3 knockout mice, suggesting that this domain is not important for virus attachment in vivo.
Disease at the inoculation site develops by day 3 after infection, and zosteriform disease generally appears on day 5 6 26. We scored inoculation site and zosteriform disease separately so that we could better evaluate early stages of infection. Interestingly, disease scores at the inoculation site of the C3 single and double mutant viruses did not differ from wild-type virus until day 5. This result suggests that the complement effect on gC mutant virus cannot be detected until rather late after infection. Several explanations are possible. If complement lyses infected cells rather than inhibiting cell free virus, it may take several days until enough cells are infected to create an observable effect. Another possible explanation is that complement concentrations may be too low at the inoculation site early in infection, whereas at later time points, virus-induced tissue injury leads to an increase in vascular permeability and higher levels of complement proteins. Additional considerations include that complement may be mediating its effects by stimulating chemotaxis or other proinflammatory events that may take several days to develop, or that antibodies may be required to augment the effects of complement. This last explanation seems unlikely in view of in vitro results that demonstrate that antibodies are not required for complement-mediated neutralization or lysis of infected cells 23 24.
We have previously described a humoral immune evasion domain on glycoprotein gE that inhibits IgG Fc-mediated functions 4 5 6. We showed that gE blocks complement-enhanced antibody neutralization, reducing the effectiveness of antibody 100-fold in vitro and offering highly significant protection to the virus or virus-infected cell against antibodies in vivo 6. The results of this study indicate that complement has little effect on wild-type virus, as disease scores of wild-type virus are virtually identical in C3 knockout and complement-intact mice. Immune evasion mediated by gC and gE may underlie the long held view that humoral immunity is relatively unimportant in HSV-1 pathogenesis 41. These immune evasion molecules render the Fc domain of antibody and the complement cascade far less effective in host defense. These observations have important implications for vaccines and chemotherapy. They explain why antibodies and complement are poorly protective against HSV infection and provide potential targets for therapies to block HSV-1 immune evasion and render the virus more susceptible to innate and acquired immunity.
Acknowledgments
The authors thank Dr. Ronald Collman for help with graphics.
This work was supported by National Institutes of Health grant RO1 HL 28220.
Footnotes
P, properdin
References
- Stevens J.G., Cook M.L. Latent herpes simplex virus in spinal ganglia of mice. Science. 1971;173:843–845. doi: 10.1126/science.173.3999.843. [DOI] [PubMed] [Google Scholar]
- Kramer M.F., Chen S.-H., Knipe D.M., Coen D.M. Accumulation of viral transcripts and DNA during establishment of latency by herpes simplex virus. J. Virol. 1998;72:1177–1185. doi: 10.1128/jvi.72.2.1177-1185.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucke R.B., Spear P.G. Membrane proteins specified by herpes simplex viruses. V. Identification of an Fc-binding glycoprotein. J. Virol. 1979;32:779–789. doi: 10.1128/jvi.32.3.779-789.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank I., Friedman H.M. A novel function of the herpes simplex virus type 1 Fc receptor, participation in antibody bipolar bridging of antiviral IgG. J. Virol. 1989;63:4479–4488. doi: 10.1128/jvi.63.11.4479-4488.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin G., Socolof E., Frank I., Friedman H.M. The herpes simplex virus Fc receptor protects infected cells from antibody dependent cellular cytotoxicity. J. Virol. 1991;65:7046–7050. doi: 10.1128/jvi.65.12.7046-7050.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashunmugam T., Lubinski J., Wang L., Goldstein L.T., Weeks B.S., Sundaresan P., Kang E.H., Dubin G., Friedman H.M. In vivo immune evasion mediated by the herpes simplex virus type 1 immunoglobulin G Fc receptor. J. Virol. 1998;5351–5359 doi: 10.1128/jvi.72.7.5351-5359.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.C., Frame M.J.C., Ligas M.W., Cross A.M., Stow N.D. Herpes simplex virus immunoglobulin G Fc receptor activity depends on a complex of two viral glycoproteins, gE and gI. J. Virol. 1988;62:1347–1354. doi: 10.1128/jvi.62.4.1347-1354.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman H.M., Cohen G.H., Eisenberg R.J., Seidel C.A., Cines D.B. Glycoprotein C of herpes simplex virus 1 acts as a receptor for the C3b complement component on infected cells. Nature. 1984;309:633–635. doi: 10.1038/309633a0. [DOI] [PubMed] [Google Scholar]
- Kostavasili I., Sahu A., Friedman H.M., Eisenberg R.J., Cohen G.H., Lambris J.D. Mechanism of complement inactivation by glycoprotein C of herpes simplex virus. J. Immunol. 1997;158:1763–1771. [PubMed] [Google Scholar]
- McNearney T.A., Odell C., Holers V.M., Spear P.G., Atkinson J.P. Herpes simplex virus glycoproteins gC-1 and gC-2 bind to the third component of complement and provide protection against complement-mediated neutralization of viral infectivity. J. Exp. Med. 1987;166:1525–1535. doi: 10.1084/jem.166.5.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruh K., Ahn K., Djaballah H., Sempe P., van Endert P.M., Tampe R., Peterson P.A., Yang Y. A viral inhibitor of peptide transporters for antigen presentation. Nature. 1995;375:415–418. doi: 10.1038/375415a0. [DOI] [PubMed] [Google Scholar]
- Hill A., Jugovic P., York I., Russ G., Bennick J., Yewdell J., Ploegh H., Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Fodor W.L., Rollins S.A., Bianco-Caron S., Rother R.P., Guilmette E.R., Burton W.V., Albrecht J.-C., Fleckenstein B., Squinto S.P. The complement control protein homolog of herpesvirus saimiri regulates serum complement by inhibiting C3 convertase activity. J. Virol. 1995;69:3889–3892. doi: 10.1128/jvi.69.6.3889-3892.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huemer H.P., Larcher C., van Drunen Littel-van den Hurk S., Babiuk L.A. Species selective interaction of Alphaherpesvirinae with the “unspecific” immune system of the host. Arch. Virol. 1993;130:353–364. doi: 10.1007/BF01309666. [DOI] [PubMed] [Google Scholar]
- Isaacs S.N., Kotwal G.J., Moss B. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Proc. Natl. Acad. Sci. USA. 1992;89:628–632. doi: 10.1073/pnas.89.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mold C., Bradt B.M., Nemerow G.R., Cooper N.R. Epstein-Barr virus regulates activation and processing of the third component of complement. J. Exp. Med. 1988;168:949–969. doi: 10.1084/jem.168.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother R.P., Rollins S.A., Fodor W.L., Albrecht J.-C., Setter E., Fleckenstein B., Squinto S.P. Inhibition of complement-mediated cytolysis by the terminal complement inhibitor of herpesvirus saimiri. J. Virol. 1994;68:730–737. doi: 10.1128/jvi.68.2.730-737.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ploegh H.L. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- Tal-Singer R., Seidel-Dugan C., Fries L., Huemer H.P., Eisenberg R.J., Cohen G.H., Friedman H.M. Herpes simplex virus glycoprotein C is a receptor for complement component iC3b. J. Infect. Dis. 1991;164:750–753. doi: 10.1093/infdis/164.4.750. [DOI] [PubMed] [Google Scholar]
- Fries L.F., Friedman H.M., Cohen G.H., Eisenberg R.J., Hammer C.H., Frank M.M. Glycoprotein C of herpes simplex virus 1 is an inhibitor of the complement cascade. J. Immunol. 1986;137:1636–1641. [PubMed] [Google Scholar]
- Hung S.-L., Peng C., Kostavasili I., Friedman H.M., Lambris J.D., Eisenberg R.J., Cohen G.H. The interaction of glycoprotein C of herpes simplex virus types 1 and 2 with the alternative complement pathway. Virology. 1994;203:299–312. doi: 10.1006/viro.1994.1488. [DOI] [PubMed] [Google Scholar]
- Gerber S.I., Belval B.J., Herold B.C. Differences in the role of glycoprotein C of HSV-1 and HSV-2 in viral binding may contribute to serotype differences in cell tropism. Virology. 1995;214:29–39. doi: 10.1006/viro.1995.9957. [DOI] [PubMed] [Google Scholar]
- Friedman H.M., Wang L., Fishman N.O., Lambris J.D., Eisenberg R.J., Cohen G.H., Lubinski J. Immune evasion properties of herpes simplex virus type 1 glycoprotein gC. J. Virol. 1996;70:4253–4260. doi: 10.1128/jvi.70.7.4253-4260.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S.L., Frank I., Yee A., Cohen G.H., Eisenberg R.J., Friedman H.M. Glycoprotein C of herpes simplex virus type 1 prevents complement-mediated cell lysis and virus neutralization. J. Infect. Dis. 1990;162:331–337. doi: 10.1093/infdis/162.2.331. [DOI] [PubMed] [Google Scholar]
- Hidaka Y., Sakai Y., Yasushi T., Mori R. Glycoprotein gC of herpes simplex virus type 1 is essential for the virus to evade antibody-independent complement-mediated virus inactivation and lysis of virus-infected cells. J. Gen. Virol. 1991;72:915–921. doi: 10.1099/0022-1317-72-4-915. [DOI] [PubMed] [Google Scholar]
- Lubinski J.M., Wang L., Soulika A.M., Burger R., Wetsel R.A., Colten H., Cohen G.H., Eisenberg R.J., Lambris J.D., Friedman H.M. Herpes simplex virus type 1 glycoprotein gC mediates immune evasion in vivo. J. Virol. 1998;72:8257–8263. doi: 10.1128/jvi.72.10.8257-8263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straus S.E., Wald A., Kost R.G., McKenzie R., Langenberg A.G.M., Hohman P., Lekstrom J., Cox E., Nakamura M., Sekulovich R. Immunotherapy of recurrent genital herpes with recombinant herpes simplex virus type 2 glycoproteins D and Bresults of a placebo-controlled vaccine trial. J. Infect. Dis. 1997;176:129–134. doi: 10.1086/514103. [DOI] [PubMed] [Google Scholar]
- Mertz G.J., Ashley R., Burke R.L., Benedetti J., Critchlow C., Jones C.C., Corey L. Double-blind placebo-controlled trial of a herpes simplex virus type 2 glycoprotein vaccine in persons at high risk for genital herpes infection. J. Infect. Dis. 1990;161:653–660. doi: 10.1093/infdis/161.4.653. [DOI] [PubMed] [Google Scholar]
- Corey L., Langenberg A.G.M., Ashley R., Sekulovich R.E., Izu A.E., Douglas J.M., Handsfield H.H., Warren T., Marr L., Tyring S. Recombinant glycoprotein vaccine for the prevention of genital HSV-2 infection. Two randomized trials. JAMA (J. Am. Med. Assoc.). 1999;282:331–340. doi: 10.1001/jama.282.4.331. [DOI] [PubMed] [Google Scholar]
- Prodeus A.P., Zhou X., Maurer M., Galli S.J., Carroll M.C. Impaired mast cell-dependent natural immunity in complement C3-deficient mice. Nature. 1997;390:172–175. doi: 10.1038/36586. [DOI] [PubMed] [Google Scholar]
- Friedman H.M., Yee A., Diggelmann H., Hastings J.C., Tal-Singer R., Seidel-Dugan C.A., Eisenberg R.J., Cohen G.H. Use of a glucocorticoid-inducible promoter for expression of herpes simplex virus type 1 glycoprotein gC1, a cytotoxic protein in mammalian cells. Mol. Cell. Biol. 1989;9:2303–2314. doi: 10.1128/mcb.9.6.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung S.-L., Srinivasan S., Friedman H.M., Eisenberg R.J., Cohen G.H. Structural basis of C3b binding by glycoprotein C of herpes simplex virus. J. Virol. 1992;66:4013–4027. doi: 10.1128/jvi.66.7.4013-4027.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer C.H., Wirtz G.H., Renfer L., Gresham H.D., Tack B.F. Large scale isolation of functionally active components of the human complement system. J. Biol. Chem. 1981;256:3995–4006. [PubMed] [Google Scholar]
- Pangburn M.K. A fluorimetric assay for native C3. The hemolytically active form of the third component of human complement. J. Immunol. Methods. 1987;102:7–14. doi: 10.1016/s0022-1759(87)80003-0. [DOI] [PubMed] [Google Scholar]
- Simmons A., Nash A.A. Zosteriform spread of herpes simplex as a model of recrudescence and its use to investigate the role of immune cells in prevention of recurrent disease. J. Virol. 1984;52:816–821. doi: 10.1128/jvi.52.3.816-821.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M.B., Prodeus A.P., Nicholson-Weller A., Ma M., Murrow J., Reid R.R., Warren H.B., Lage A.L., Moore F.D., Jr., Rosen F.S. Increased susceptibility to endotoxin shock in complement C3- and C4-deficient mice is corrected by C1 inhibitor replacement. J. Immunol. 1997;159:976–982. [PubMed] [Google Scholar]
- Rux A.H., Moore W.T., Lambris J.D., Abrams W.R., Peng C., Friedman H.M., Cohen G.H., Eisenberg R.J. Disulfide bond structure determination and biochemical analysis of glycoprotein C from herpes simplex virus. J. Virol. 1996;70:5455–5465. doi: 10.1128/jvi.70.8.5455-5465.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold B.C., WuDunn D., Soltys N., Spear P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WuDunn D., Spear P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989;63:52–58. doi: 10.1128/jvi.63.1.52-58.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal-Singer R., Peng C., de Leon M.P., Abrams W.R., Banfield B.W., Tufaro F., Cohen G.H., Eisenberg R.J. Interaction of herpes simplex virus glycoprotein gC with mammalian cell surface molecules. J. Virol. 1995;69:4471–4483. doi: 10.1128/jvi.69.7.4471-4483.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola J.R. Herpes simplex virus vaccines—why don't antibodies protect? JAMA (J. Am. Med. Assoc.). 1999;282:379–380. doi: 10.1001/jama.282.4.379. [DOI] [PubMed] [Google Scholar]