

Abstract
To assess the potency of low-affinity anti–red blood cell (RBC) autoantibodies in the induction of anemia, we generated an immunoglobulin (Ig)G2a class-switch variant of a 4C8 IgM anti–mouse RBC autoantibody, and compared its pathogenic potential with that of its IgM isotype and a high-affinity 34-3C IgG2a autoantibody. The RBC-binding activity of the 4C8 IgG2a variant was barely detectable, at least 1,000 times lower than that of its IgM isotype, having a high-binding avidity, and that of the 34-3C IgG2a monoclonal antibody (mAb). This low-affinity feature of the 4C8 mAb was consistent with the lack of detection of opsonized RBCs in the circulating blood from the 4C8 IgG2a–injected mice. However, the 4C8 IgG2a variant was highly pathogenic, as potent as its IgM isotype and the 34-3C IgG2a mAb, due to its capacity to interact with Fc receptors involved in erythrophagocytosis. In addition, our results indicated that the pentameric form of the low-affinity IgM isotype, by promoting the binding and agglutination of RBCs, is critical for its pathogenic activity. Demonstration of the remarkably high pathogenic potency of low-affinity autoantibodies, if combined with appropriate heavy chain effector functions, highlights the critical role of the Ig heavy chain constant regions, but the relatively minor role of autoantigen-binding affinities, in autoimmune hemolytic anemia.
Keywords: autoantibody, autoimmune hemolytic anemia, Fc receptor, Kupffer cell, knockout mouse
The pathogenesis of autoantibody-mediated cellular and tissue lesions in autoimmune diseases is most straightforwardly attributable to the combined action of self-antigen binding properties and effector functions associated with the Fc regions of the different Ig isotypes, such as the capacity to bind Fc receptors for IgG (FcγR), activate complement, and cause multivalence-induced agglutination. This is best exemplified by remarkable differences among a panel of Coombs' anti-RBC monoclonal autoantibodies, established from lupus-prone NZB mice, in their ability to induce autoimmune hemolytic anemia 1 2 3 4 5.
It has been believed that the high-affinity binding of autoantibodies to self-antigens is likely to be critical for the expression of their pathogenic activities in vivo. This notion has been supported by the demonstration that the Ig genes in a subset of autoantibody-producing B cells, which are clonally expanded, are modified by somatic mutations, especially in their CDR3 6 7, and that the affinity maturation of autoantibodies in association with IgM to IgG class switching coincides with progression of the clinical development of autoimmune diseases, most notably SLE 8 9. However, the affinity maturation of autoantibodies in parallel to Ig class switch and somatic hypermutations is not a consistent observation. Analysis of clonally related anti-IgG2a rheumatoid factors isolated from a lupus-prone MRL-lpr/lpr mouse has demonstrated only limited affinity maturation, despite Ig isotype switching with extensive somatic mutations 10. Most significantly, studies on a panel of anti-IgG2a rheumatoid factor monoclonal autoantibodies disclosed that even low-affinity autoantibodies are able to induce immune complex–mediated vasculitis, with a cryoglobulin activity uniquely associated with murine IgG3 isotype 11 12.
Recent studies on anti–mouse RBC monoclonal autoantibodies revealed a major role of FcγR-mediated erythrophagocytosis in the development of anemia induced by IgG anti-RBC autoantibodies 13 14. Thus, it can be speculated that low-affinity anti-RBC autoantibodies of IgG isotypes could become highly pathogenic, if combined with the capacity to interact with FcγR expressed on phagocytic effector cells. To explore this possibility, we have generated an IgG2a class-switch variant from an NZB-derived 4C8 IgM anti–mouse RBC monoclonal autoantibody 2. Since it has been generally thought that the IgM antibody has a low affinity, we should be able to study the pathogenic activity of low-affinity anti-RBC autoantibody of IgG2a isotype capable of interacting with phagocytic FcγR 13 14. In the present work, comparative analysis of the 4C8 IgG2a variant with its IgM isotype and a high-affinity 34-3C IgG2a anti–mouse RBC monoclonal autoantibody, established from NZB mice 4, demonstrates that despite a low binding affinity, both 4C8 IgM and IgG2a isotypes are remarkably pathogenic, because of the high-avidity binding capacity of polyvalent IgM isotype or because of a high-affinity interaction of IgG2a isotype with FcγR involved in erythrophagocytosis.
Materials and Methods
Mice.
BALB/c mice were purchased from Bomholtgard. Mice deficient in FcR γ chains (FcRγ), which lack functional expression of both FcγR type I (FcγRI) and type III (FcγRIII), and wild-type littermates were developed as described previously 15.
DNA Construction.
The VDJH4C8-Cγ2a plasmid containing the complete 4C8 IgG heavy chain gene of the IgG2a isotype was constructed using the following DNA fragments: the rearranged VDJ region isolated from cDNA encoding the variable region of the heavy chain of the 4C8 mAb 16, the promoter region isolated from pSV-Vμ1 17, the heavy chain enhancer region isolated from pSVE2-neo 18, and the Cγ2a region derived from the genomic clone, pIgH10 19.
mAbs.
Hybridomas secreting the 4C8 IgM and 34-3C IgG2a anti–mouse RBC mAbs were derived from unmanipulated NZB mice 2 4. The 4C8 IgG2a class-switch variant was obtained by transfecting 4C8 heavy chain loss mutant cells by electroporation with the VDJH4C8-Cγ2a plasmid together with a pSVE2-neo plasmid containing the neomycin-resistant gene. After selection for resistance to neomycin and secretion of IgG antibodies, stable transfected cells secreting the 4C8 IgG2a variant were cloned by limiting dilutions. IgG2a anti-TNP (Hy1.2) and IgM anti-IgG2a 6 7 8 9 10 11 12 13 14 15 16 17 18 19 mAbs were used as control. Rat anti–mouse κ chain mAb (H139.52.1.5) was provided by Dr. M. Pierres (Centre d'Immunologie de Marseille-Luminy, Marseille, France 20). IgG mAbs were purified from culture supernatants by protein A column chromatography. IgM mAbs were purified by euglobulin precipitation from culture supernatants concentrated by 50% saturated ammonium sulfate precipitation, according to the method of Garcia-Gonzales et al. 21. The purity of IgG and IgM was >90% as documented by SDS-PAGE.
Reverse Transcriptase PCR and cDNA Sequencing.
RNA was prepared from 4C8 IgG2a–transfected cells by RNeasy Mini Kit (Qiagen). The first strand of cDNA was synthesized with an oligo(dT) primer and 5 μg of total RNA. For amplification with Pfu DNA polymerase (Stratagene), the following primers were used: 5′-untranslated VH primer (5′-CAGTTCTCTCTACAGTTA-3′) and Cγ2a-CH1 primer (5′-GCCAGTGGATAGAC-3′) for the 4C8 heavy chain; 5′-untranslated Vκ primer (5′-CAGGGGAAGCAAGATGG-3′) and Cκ primer (5′-TGGATGGTGGGAAGATG-3′) for the 4C8 light chain. The nucleotide sequence corresponding to the variable region of the 4C8 heavy or light chain was determined by the dideoxynucleotide chain terminating method 22.
Anti–Mouse RBC Assays.
A flow cytometric assay was used to detect anti–mouse RBC activities of purified mAbs. 50 μl of various concentrations of purified mAbs was incubated with 20 μl of 1% RBC suspension, prepared from BALB/c mice, in 1% BSA-PBS for 30 min at 4°C. After washing three times with 1% BSA-PBS, the RBCs were incubated with biotinylated rat anti–mouse κ chain mAb (H139.52.1.5), followed by PE-conjugated streptavidin R (Caltag), and analyzed with a FACScan™ (Becton Dickinson). The presence of opsonized RBCs in mice injected with anti–mouse RBC mAb was detected by a similar flow cytometric assay, using biotinylated rat anti–mouse κ chain mAb. For in vitro and in vivo bindings of anti–mouse RBC mAb on biotinylated mouse RBCs, bound antibodies were revealed with PE-labeled F(ab′)2 goat anti–mouse Ig conjugates (Jackson ImmunoResearch Labs). An indirect radioimmunoassay was also used to detect anti–mouse RBC activities of mAbs, as described previously 4. In brief, 100 μl of various concentrations of purified mAbs was incubated with 10 μl of 25% RBC suspension in 1% BSA-PBS for 30 min at 4°C. After washing three times with 1% BSA-PBS, the RBCs were incubated with 125I-labeled goat anti–mouse Ig in 1% BSA-PBS. Results are expressed as the radioactivity (in cpm) bound to RBCs. Complement-mediated lysis and hemagglutination assays were as described previously 4.
Experimental Autoimmune Hemolytic Anemia.
Autoimmune hemolytic anemia was induced by a single intraperitoneal injection of purified anti-RBC mAb into 2–3-mo-old mice. Blood samples were collected into heparinized microhematocrit tubes and centrifuged in a hematocentrifuge, and hematocrits (Ht) were directly determined after centrifugation, as described previously 4.
In Vivo Biotinylation of Mouse RBCs.
Mouse RBCs were biotinylated in vivo by an intravenous infusion of N-hydroxysuccinimide biotin (NHS-biotin; Boehringer Mannheim), according to the method described by Dale and Daniels 23. In brief, 2 mg NHS-biotin was dissolved in 0.12 ml N,N-dimethylacetamide (Sigma Chemical Co.), incubated for 2 min in the dark at 37°C, and added to 3.2 ml sterile saline. 500 μg of NHS-biotin was then injected intravenously into mice. This in vivo biotinylation procedure resulted in essentially all the RBCs being biotinylated, as judged by staining with streptavidin Red 670 (GIBCO BRL) 24 h after the infusion of NHS-biotin.
Histological Studies.
Livers and spleens were obtained at autopsy, processed for histological examination, and stained with hematoxylin and eosin (HE). The extent of in vivo RBC destruction by Kupffer cell–mediated phagocytosis was revealed by coloration of liver sections with Perls iron staining.
Results
Low-Affinity Binding to Mouse RBCs by the 4C8 IgG2a Isotype-Switch Variant.
The strategy for generating the recombinant IgG2a isotype-switch variant was to clone the heavy chain variable region (VH) gene from the 4C8 IgM anti–mouse RBC hybridoma and to join this gene to the already cloned mouse IgG2a heavy chain constant region gene. The recombinant IgG2a isotype-switch variant was then expressed in a heavy chain loss mutant of the 4C8 IgM hybridoma. Notably, the VH4C8 and Vκ4C8 sequences of the 4C8 heavy and light chain cDNA derived from a reverse transcription PCR amplification of mRNA isolated from the cells secreting 4C8 IgG2a switch variant were identical to the originally published sequence 16.
The in vitro binding activity of the 4C8 IgG2a switch variant, compared with its IgM isotype and the high-affinity 34-3C IgG2a anti–mouse RBC mAb, was investigated by flow cytometric analysis using a biotinylated rat anti–mouse κ chain mAb, followed by PE-conjugated streptavidin. Incubation of mouse RBCs with 0.1 ng of the 4C8 IgM or 34-3C IgG2a mAb resulted in substantial binding, whereas >250 ng of the IgG2a switch variant was required to show significant binding, with a maximal binding at 1 μg (Fig. 1). Thus, mouse RBC-binding activity of the 4C8 IgG2a variant was at least 1,000 times less than that of its IgM isotype and the 34-3C IgG2a mAb. It should also be mentioned that the staining intensity obtained by the 4C8 IgG2a variant was far lower than that observed with the 4C8 IgM or the 34-3C IgG2a mAb. Marginal RBC-binding activities by the 4C8 IgG2a variant were confirmed with a radioimmunoassay (Fig. 2). These results indicated that the 4C8 mAb has a low binding affinity, while its binding activity was markedly promoted by the pentameric high-avidity IgM isotype. Moreover, hemagglutination activity was only detectable with the 4C8 IgM mAb, and neither IgM nor IgG2a 4C8 mAb exhibited complement-mediated hemolysis in vitro in the presence of guinea pig, rabbit, or mouse serum (data not shown).
Figure 1.

Flow cytometric analysis of mouse RBCs stained with different amounts of the 4C8 (IgM and IgG2a) and 34-3C (IgG2a) anti–mouse RBC mAbs. RBCs freshly prepared from BALB/c mice were first incubated with different amounts of various mAbs, and then incubated with biotinylated anti–mouse κ chain mAb, followed by PE-conjugated streptavidin. Shaded histograms indicate the background staining with anti–mouse κ chain mAb alone. As controls, the results obtained with isotype-matched control mAbs are shown.
Figure 2.

Radioimmunoassay for the detection of anti–mouse RBC activities by the 4C8 (IgM and IgG2a) and 34-3C (IgG2a) anti–mouse RBC mAbs. RBCs freshly prepared from BALB/c mice were first incubated with different concentrations of various mAbs, and then incubated with radiolabeled goat anti–mouse Ig. The results are expressed as the radioactivity bound to mouse RBCs in the presence of 4C8 IgM (•), 4C8 IgG2a (□), and 34-3C IgG2a (○). As controls, the results obtained with isotype-matched control mAbs are shown: IgM anti-IgG2a (▪) and IgG2a anti-TNP (▵).
Lack of Detection of Opsonized RBCs, but Efficient Elimination of Circulating RBCs in Mice Injected with the Low-Affinity 4C8 IgG2a Variant.
Because of its markedly limited in vitro binding to mouse RBCs, we determined whether the 4C8 IgG2a variant was capable of binding to circulating RBCs in vivo. When analyzed by a flow cytometric assay 24, 48, and 72 h after a single intraperitoneal injection of 1 mg of the 4C8 IgG2a variant into BALB/c mice, opsonized RBCs were undetectable in the circulating blood (Fig. 3 A). This was in marked contrast to the presence of bound antibodies on essentially all the circulating mouse RBCs between 24 and 72 h after the administration of 50 μg of the 4C8 IgM or 34-3C IgG2a mAb. Notably, the staining pattern obtained by in vitro incubation with the 4C8 IgG2a variant was indistinguishable between RBCs from mice injected with the 4C8 IgG2a and those from control mice, excluding a rapid and selective elimination of a subpopulation of circulating RBCs by the low-affinity 4C8 IgG2a (data not shown). Thus, the lack of detection of opsonized RBCs in the circulating blood further confirmed a low-affinity feature of the 4C8 IgG2a switch variant.
Figure 3.

Failure to detect opsonized circulating RBCs, but development of anemia accompanied by efficient clearance of circulating RBCs after injection of the 4C8 IgG2a variant in BALB/c mice. (A) Mouse RBCs were obtained 24 h after an intraperitoneal injection of 4C8 IgG2a (1 mg), 4C8 IgM (50 μg), or 34-3C IgG2a (50 μg) anti–mouse RBC mAb, and then stained with biotinylated anti–mouse κ chain mAb, followed by PE-conjugated streptavidin. Shaded histograms indicate the background staining with PE-conjugated streptavidin. Essentially identical results were obtained with mouse RBCs obtained 48 and 72 h after injection of anti–mouse RBC mAb (data not shown). (B and C) Mouse RBCs were biotinylated in vivo by an intravenous infusion of NHS-biotin 24 h before the intraperitoneal injection of 1 mg of purified 4C8 IgG2a (•) or control IgG2a Hy1.2 anti-TNP mAb (○) on day 0. The clearance of biotinylated RBCs was followed by the staining with streptavidin Red 670. Results are expressed as percent biotinylated RBCs (Biotin-RBC) of three individual mice, measured 2, 4, and 8 d after the mAb injection (B). The development of anemia was determined by measuring Ht values (C).
Strikingly, mice injected with 1 mg of the 4C8 IgG2a variant developed a severe anemia with a decrease in Ht values down to 21% 4 d after the injection (see below). To verify the elimination of circulating RBCs by the 4C8 IgG2a variant, RBCs were in vivo biotinylated with an intravenous injection of NHS-biotin, and their clearance was then determined after the injection of 1 mg of 4C8 IgG2a or Hy1.2 IgG2a anti-TNP mAb. As shown in Fig. 3 B, biotinylated RBCs were gradually replaced by newly generated RBCs in control mice injected with Hy1.2 anti-TNP mAb, while the injection of the 4C8 IgG2a variant markedly accelerated this process. Although Ht values began to recover ∼5 d after the 4C8 IgG2a mAb injection and returned to almost normal levels by 8 d (Fig. 3 C), the levels of biotinylated RBCs were further decreased. 8 d after 4C8 IgG2a mAb injection, biotinylated RBCs were hardly detectable (<1%), while ∼70% of circulating RBC were still biotinylated in control mice. This indicated that the 4C8 IgG2a variant indeed bound to circulating RBCs in vivo and efficiently eliminated them. Notably, the immunoreactivity of biotinylated RBCs, obtained from 24 to 72 h after in vivo biotinylation, to the 4C8 mAb was comparable to that of control RBCs, as judged by in vitro and in vivo binding analyses, and the kinetics and extent of anemia induced by the 4C8 mAb in biotin-treated mice were essentially identical to those of untreated mice (Fig. 3 C and Fig. 4).
Figure 4.

Development of anemia by the 4C8 and 34-3C anti–mouse RBC mAbs in BALB/c mice. Mice were intraperitoneally injected with different amounts of purified mAbs on day 0: 25 μg, 50 μg, 200 μg, or 1 mg of 4C8 IgG2a mAb; 25, 50, 100, or 250 μg of 4C8 IgM mAb; 12.5, 25, 50, or 100 μg of 34-3C IgG2a mAb. Results are expressed as mean Ht values of three to five mice.
High Pathogenic Activity of the Low-Affinity 4C8 IgG2a Variant Promoted through FcγR-dependent Erythrophagocytosis.
To compare the pathogenic activity of the 4C8 IgG2a variant with its IgM isotype and the high-affinity 34-3C IgG2a mAb, the development of anemia was analyzed by a single intraperitoneal injection of various amounts of purified mAb into BALB/c mice. Despite low-affinity binding to mouse RBCs, the 4C8 IgG2a mAb was highly pathogenic, and 50 μg was sufficient to cause significant anemia (mean Ht values of five mice 4 d after the injection: 35 ± 3%; Fig. 4). This dose was comparable to that required for the induction of anemia by its IgM isotype and the high-affinity 34-3C IgG2a mAb.
Histological examinations showed that erythrophagocytosis by Kupffer cells was the most remarkable pathological change associated with anemia induced by the 4C8 IgG2a isotype (Fig. 5 A), as observed in mice developing anemia after the injection of the 34-3C IgG2a mAb 4. Destruction of RBCs by phagocytosis was further documented by extensive iron deposits in Kupffer cells from mice injected with the 4C8 IgG2a variant (Fig. 5 C). In contrast, the injection of 4C8 IgM mAb at a dose of 250 μg resulted in an enormous accumulation of agglutinated RBCs in the spleen, and in hepatic sinusoids accompanied by occasional necrosis of hepatic parenchymal cells (Fig. 5 B), as shown previously 4.
Figure 5.
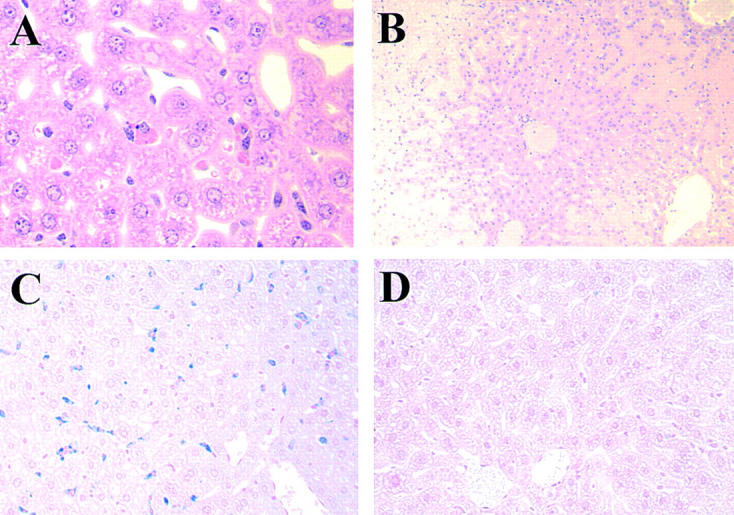
(A) Representative histological appearance of liver 4 d after the injection of 1 mg of the 4C8 IgG2a variant. Note the presence of marked erythrophagocytosis by Kupffer cells (HE; original magnification: ×400). (B) Representative histological appearance of liver from BALB/c mice that died of anemia 2 d after the injection of 250 μg of the 4C8 IgM mAb. Note an enormous accumulation of agglutinated RBCs in sinusoids of liver accompanied by necrosis of hepatic parenchymal cells (HE; original magnification: ×100). (C and D) Representative histological appearance of iron deposits in Kupffer cells from BALB/c and FcRγ-deficient mice after the injection of 1 mg of the 4C8 IgG2a variant. Mice injected with the 4C8 IgG2a variant were killed at day 7. The extent of in vivo RBC destruction by phagocytosis was revealed by coloration of liver sections with Perls iron staining. Note extensive iron deposits in Kupffer cells from BALB/c mice injected with the 4C8 IgG2a mAb (C), in marked contrast to the complete absence of iron deposits in livers from FcRγ-deficient mice injected with the 4C8 IgG2a mAb (D) (original magnification: ×200).
To determine the contribution of FcγR-mediated erythrophagocytosis to the pathogenesis of the 4C8 IgG2a–induced anemia, the development of anemia was assessed in FcRγ-deficient mice lacking functional expression of both FcγRI and FcγRIII involved in phagocytosis of IgG2a-opsonized RBCs 13 14. As shown in Fig. 6, FcRγ-deficient mice were completely resistant to the pathogenic effect of 1 mg of the 4C8 IgG2a mAb, and failed to exhibit erythrophagocytosis, as documented by the lack of iron deposits in their Kupffer cells (Fig. 5 D).
Figure 6.

Development of anemia in FcRγ-deficient (•) and wild-type (○) mice after the injection of 1 mg of the 4C8 IgG2a variant. Mean Ht values (± 1 SD) of three mice are shown.
Discussion
In this study, we have demonstrated that the 4C8 anti–mouse RBC autoantibody, derived from lupus-prone NZB mice, has a low binding affinity to its corresponding self-antigen, yet its IgM and IgG2a isotypes exhibit a remarkably high pathogenic potency. Our results indicate that the pentameric form of a low-affinity IgM isotype, by promoting the binding and agglutination of RBCs, is critical for its pathogenic activity, and that the capacity of the IgG2a isotype to interact with FcγR involved in erythrophagocytosis is responsible, despite its barely detectable RBC-binding activity, for its hemolytic activity. Demonstration of the high pathogenic potency of low-affinity autoantibodies highlights a remarkable role of Ig heavy chain effector functions, as opposed to a relatively minor role of autoantigen-binding affinities, in the pathogenesis of autoimmune hemolytic anemia.
A striking feature of our analysis is that despite the lack of detection of significantly opsonized RBCs in the circulating blood, the 4C8 IgG2a variant with low binding affinity is able to induce a severe anemia as a result of peripheral destruction of circulating RBCs. This was documented by an efficient clearance of in vivo–biotinylated circulating RBCs and by Kupffer cell–mediated erythrophagocytosis. To our surprise, the amount of the IgG2a isotype-switch variant required to induce anemia was comparable to that of the high-avidity IgM isotype exhibiting >1,000 times stronger RBC-binding activity. This indicates that the pathogenic activity of low-affinity IgG anti-RBC autoantibodies is dramatically enhanced when provided with an appropriate Ig heavy chain effector function, namely the capacity to interact with FcγR involved in erythrophagocytosis. Thus, a rapid FcγR-mediated uptake by Kupffer cells of even very poorly opsonized RBCs present in hepatic microcirculation appears to be an extremely efficient mechanism for the development of anemia induced by IgG anti-RBC autoantibodies.
An additional, unexpected observation was that the pathogenic potential of the low-affinity 4C8 IgG2a autoantibody was also comparable to the high-affinity 34-3C IgG2a mAb 4. However, this comparison is only tentative in its interpretation because of possible differences in the specificity of the 4C8 and 34-3C mAbs 24. Only experiments comparing IgG2a mAbs of the same specificity, but with different affinities, would provide definitive conclusions on this issue. Nevertheless, our results strongly suggest that RBC-binding affinities of Coombs' autoantibodies apparently play a relatively minor role for in vivo hemolytic activities of the IgG2a isotype capable of interacting efficiently with two different classes of FcγR (FcγRI and FcγRIII) involved in erythrophagocytosis 13 14 25 26. It may be worth noting that although the pathogenicity of the 4C8 and 34-3C IgG2a mAbs was almost comparable at lower doses, it appears that the 34-3C mAb at higher doses is more pathogenic than the 4C8 IgG2a (Fig. 4). Although we do not have a straightforward explanation for this phenomenon at present, it can be speculated that the IgG2a isotype of anti-RBC autoantibodies could mediate erythrophagocytosis through their binding to the high-affinity FcγRI, followed by their subsequent interaction with circulating RBCs. A significant binding of free IgG2a anti-RBC to FcγRI on phagocytic cells is likely to occur only when higher doses of IgG2a mAb are injected, because of the competition of circulating monomeric IgG2a without anti-RBC activity. Owing to its high-affinity RBC-binding capacity, even limited amounts of the cell-bound 34-3C IgG2a may efficiently capture circulating RBCs, thereby mediating erythrophagocytosis. In contrast, the ability to capture circulating RBCs by the low-affinity 4C8 IgG2a bound on Kupffer cells may be too poor to cause erythrophagocytosis by such a mechanism. Consistent with this hypothesis, our recent analysis on different FcγR-deficient mice has shown that the erythrophagocytosis caused by lower doses of the 34-3C and 4C8 IgG2a mAbs is primarily mediated by the low-affinity FcγRIII, while FcγRI is only involved in the development of anemia induced by higher doses of these mAbs (our unpublished results).
In addition to the remarkable contribution of IgG Fc region mediating FcγR interaction to pathogenic activities of the low-affinity 4C8 IgG2a variant, these studies also suggest that the strong pathogenic activity of the IgM 4C8 isotype is dependent on its pentameric form, which promotes the binding and agglutination of RBCs. Consequently, the IgM isotype of the 4C8 mAb induces a different form of severe anemia, resulting from massive agglutination of RBCs in spleen and liver, which does not involve FcγR-mediated phagocytosis and complement activation 4 13. Although the present study does not formally prove that the pentameric form of the 4C8 IgM antibody is crucial for the in vivo agglutination of RBCs, we have recently observed that the IgG3 variant of the 4C8 mAb exhibits neither FcγR-dependent erythrophagocytosis nor agglutination of RBCs in vivo (our unpublished results), supporting the importance of the IgM pentameric form for the in vivo agglutination of RBCs by this mAb. However, it should be stressed that we have previously observed that two of four IgM anti-RBC mAbs failed to induce anemia 4. The lack of pathogenic activities by these two IgM autoantibodies is likely to be related to their specificities, since they are unable to induce agglutination of mouse RBCs in vitro. Thus, the surface density of corresponding self-antigen epitopes may be too low to promote high-avidity binding and agglutination of mouse RBCs in vivo by these IgM anti-RBC autoantibodies.
Our demonstration here of the high pathogenic potency of low-affinity autoantibodies suggests that the affinity maturation of autoantibodies may not be a critical process for the generation of autoantibodies with immunopathologic consequences. This is consistent with the demonstration that low-affinity anti-IgG2a rheumatoid factors of the IgG3 isotype are able to induce remarkable pathology—immune complex–mediated skin vasculitis and lupus-like glomerulonephritis—owing to their cryoglobulin activity, a unique property of the IgG3 isotype 11 12. These results provide new insight into the cellular basis for the generation of pathogenic autoantibodies of IgM and IgG isotypes. It has already been shown that a fraction of B cells expressing the low-affinity 4C8 autoantibody can escape clonal deletion in the bone marrow and can be activated to produce pathogenic autoantibodies in the periphery, as a result of nonspecific activation of B cells 27 28. These autoantibodies may even be switched to IgG classes under certain conditions, possibly through the action of the cytokines, independently of the presence of autoantigen-specific T helper cells, as is the case of T cell–independent type II immune responses 29. Genetic abnormalities present in certain autoimmune-prone mice may favor the switching of IgM to IgG isotypes, as shown by a spontaneous class switch from IgM to IgG2a autoantibody by B cells derived from lupus-prone (NZB × NZW)F1 mice in the absence of functional CD4+ T helper cells 30 31. In addition, we have recently observed that the constitutive expression of the bcl-2 transgene in germinal center B cells is able to induce the spontaneous production of IgG autoantibodies (our unpublished results). Therefore, it is possible that autoreactive B cells can be generated as a result of somatic hypermutations in the germinal centers during immune responses against environmental antigens, and such B cells may persist, if they are defective in the process of apoptosis, which is likely to be one of the genetic defects present in autoimmune-prone mice 32 33. Thus, although the presence of somatic mutations in Ig variable regions, including CDR, of autoantibodies is likely to be CD4+ T cell dependent, several autoantibody-producing B cells may not have been driven by autoreactive T helper cells and self-antigens. This idea is consistent with a limited affinity maturation of clonally related rheumatoid factors, despite extensive somatic mutations, derived from a Fas-deficient MRL-lpr/lpr mouse 10. Clearly, in view of the high pathogenic potential of low-affinity autoantibodies and of the critical role of CD4+ T cells in the development of autoimmune diseases 34, better understanding of the nature of help provided by CD4+ T cells would help elucidate the cellular basis central to the development of autoantibody-mediated autoimmune diseases.
Acknowledgments
We thank Ms. G. Leyvraz, Ms. G. Lange, and Mr. G. Brighouse for their excellent technical assistance.
This work was supported by a grant from the Swiss National Foundation for Scientific Research, a grant from the Association pour la Recherche sur le Cancer, France, and grants for the Center of Excellence Program from the Ministry of Education, Science, Sports and Culture, Japan.
Footnotes
Abbreviations used in this paper: FcγR, Fc receptor for IgG; FcRγ, FcR γ chain; HE, hematoxylin and eosin; Ht, hematocrit(s); NHS-biotin, N-hydroxysuccinimide biotin.
References
- Cooke L.A., Staines N.A., Morgan A., Moorhouse C., Harris G. Haemolytic disease in mice induced by transplantation of hybridoma cells secreting monoclonal anti-erythrocyte autoantibodies. Immunology. 1982;47:569–572. [PMC free article] [PubMed] [Google Scholar]
- Ozaki S., Nagasawa R., Sato H., Shirai T. Hybridoma autoantibodies to erythrocytes from NZB mice and the induction of hemolytic anemia. Immunol. Lett. 1984;8:115–119. doi: 10.1016/0165-2478(84)90062-2. [DOI] [PubMed] [Google Scholar]
- Caulfield M.J., Stanko D., Calkins C. Characterization of the spontaneous autoimmune (anti-erythrocyte) response in NZB mice using a pathogenic monoclonal autoantibody and its anti-idiotype. Immunology. 1989;66:233–237. [PMC free article] [PubMed] [Google Scholar]
- Shibata T., Berney T., Reininger L., Chicheportiche Y., Ozaki S., Shirai T., Izui S. Monoclonal anti-erythrocyte autoantibodies derived from NZB mice cause autoimmune hemolytic anemia by two distinct pathogenic mechanisms. Int. Immunol. 1990;2:1133–1141. doi: 10.1093/intimm/2.12.1133. [DOI] [PubMed] [Google Scholar]
- Scott B.B., Sadigh S., Stow M., Mageed R.A.K., Andrew E.M., Maini R.N. Anti–mouse red blood cell monoclonal antibodies use functionally rearranged genes from the VH J558 family and are derived from the CD5− B-lymphocyte subpopulation. Immunology. 1993;79:568–573. [PMC free article] [PubMed] [Google Scholar]
- Shlomchik M.J., Marshak-Rothstein A., Wolfowicz C.B., Rothstein T.L., Weigert M.G. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- Shlomchik M., Mascelli M., Shan H., Radic M.Z., Pisetsky D.S., Marshak-Rothstein A., Weigert M. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J. Exp. Med. 1990;171:265–292. doi: 10.1084/jem.171.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillman D.M., Jou N.-T., Hill R.J., Marion T.N. Both IgM and IgG anti-DNA antibodies are the products of clonally selective B cell stimulation in (NZB × NZW)F1 mice. J. Exp. Med. 1992;176:761–779. doi: 10.1084/jem.176.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose S., Wakiya M., Kawano-Nishi Y., Yi J., Sanokawa R., Taki S., Shimamura T., Kishimoto T., Tsurui H., Nishimura H., Shirai T. Somatic diversification and affinity maturation of IgM and IgG anti-DNA antibodies in murine lupus. Eur. J. Immunol. 1993;23:2813–2820. doi: 10.1002/eji.1830231114. [DOI] [PubMed] [Google Scholar]
- Jacobson B.A., Sharon J., Shan H., Shlomchik M., Weigert M.G., Marshak-Rothstein A. An isotype switched and somatically mutated rheumatoid factor clone isolated from a MRL-lpr/lpr mouse exhibits limited intraclonal affinity maturation. J. Immunol. 1994;152:4489–4499. [PubMed] [Google Scholar]
- Berney T., Fulpius T., Shibata T., Reininger L., Van Snick J., Shan H., Weigert M., Marshak-Rothstein A., Izui S. Selective pathogenicity of murine rheumatoid factors of the cryoprecipitable IgG3 subclass. Int. Immunol. 1992;4:93–99. doi: 10.1093/intimm/4.1.93. [DOI] [PubMed] [Google Scholar]
- Fulpius T., Spertini F., Reininger L., Izui S. Immunoglobulin heavy chain constant region determines the pathogenicity and the antigen-binding activity of rheumatoid factor. Proc. Natl. Acad. Sci. USA. 1993;90:2345–2349. doi: 10.1073/pnas.90.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clynes R., Ravetch J.V. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Meyer D., Schiller C., Westermann J., Izui S., Hazenbos W.L.W., Verbeek J.S., Schmidt R.E., Gessner J.E. FcγRIII (CD16)-deficient mice show IgG isotype-dependent protection to experimental autoimmune hemolytic anemia. Blood. 1998;92:3997–4002. [PubMed] [Google Scholar]
- Takai T., Li M., Sylvestre D., Clynes R., Ravetch J.V. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Okamoto M., Honjo T. Nucleotide sequences of the gene/cDNA coding for anti–murine erythrocyte autoantibody produced by a hybridoma from NZB mouse. Nucleic Acids Res. 1990;18:1895. doi: 10.1093/nar/18.7.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuberger M.S. Expression and regulation of immunoglobulin heavy gene transfected into lymphoid cells. EMBO (Eur. Mol. Biol. Organ.) J. 1983;2:1373–1378. doi: 10.1002/j.1460-2075.1983.tb01594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon T., Rajewsky K. ‘Enhancer-constitutive’ vectors for the expression of recombinant antibodies. Nucleic Acids Res. 1988;16:354. doi: 10.1093/nar/16.1.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu A., Takahashi N., Yamawaki-Kataoka Y., Nishida Y., Kataoka T., Honjo T. Ordering of mouse immunoglobulin heavy chain genes by molecular cloning. Nature. 1981;289:149–153. doi: 10.1038/289149a0. [DOI] [PubMed] [Google Scholar]
- Labit C., Pierres M. Rat monoclonal antibodies to mouse IgG1, IgG2a, IgG2b, and IgG3 subclasses, and kappa chain isotypic determinants. Hybridoma. 1984;3:163–169. doi: 10.1089/hyb.1984.3.163. [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalez M., Bettinger S., Ott S., Olivier P., Kadouche J., Pouletty P. Purification of murine IgG3 and IgM monoclonal antibodies by euglobulin precipitation. J. Immunol. Methods. 1988;111:17–23. doi: 10.1016/0022-1759(88)90054-3. [DOI] [PubMed] [Google Scholar]
- Sanger F.S., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale G.L., Daniels R.B. Quantitation of immunoglobulin associated with senescent erythrocytes from the rabbit. Blood. 1991;77:1096–1099. [PubMed] [Google Scholar]
- Oliveira G.G.S., Izui S., Ravirajan C.T., Mageed R.A.K., Lydyard P.M., Elson C.J., Barker R.N. Diverse antigen specificity of erythrocyte-reactive monoclonal autoantibodies from NZB mice. Clin. Exp. Immunol. 1996;105:313–320. doi: 10.1046/j.1365-2249.1996.d01-772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshank R.L., Luster A.D., Ravetch J.V. Function and regulation of a murine macrophage-specific IgG Fc receptor, FcγR-α. J. Exp. Med. 1988;167:1909–1925. doi: 10.1084/jem.167.6.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears D.W., Osman N., Tate B., McKenzie I.F.C., Hogarth P.M. Molecular cloning and expression of the mouse high affinity Fc receptor for IgG. J. Immunol. 1990;144:371–378. [PubMed] [Google Scholar]
- Okamoto M., Murakami M., Shimizu A., Ozaki S., Tsubata T., Kumagai S.-I., Honjo T. A transgenic model of autoimmune hemolytic anemia. J. Exp. Med. 1992;175:71–79. doi: 10.1084/jem.175.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M., Tsubata T., Shinkura R., Nisitani S., Okamoto M., Yoshioka H., Usui T., Miyawaki S., Honjo T. Oral administration of lipopolysaccharides activates B-1 cells in the peritoneal cavity and lamina propria of the gut and induces autoimmune symptoms in an autoantibody transgenic mouse. J. Exp. Med. 1994;180:111–121. doi: 10.1084/jem.180.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snapper C.M., Mond J.J. Towards a comprehensive view of immunoglobulin class switching. Immunol. Today. 1993;14:15–17. doi: 10.1016/0167-5699(93)90318-F. [DOI] [PubMed] [Google Scholar]
- Reininger L., Radaszkiewicz T., Kosco M., Melchers F., Rolink A.G. Development of autoimmune disease in SCID mice populated with long-term in vitro proliferating (NZB × NZW)F1 pre-B cells. J. Exp. Med. 1992;176:1343–1353. doi: 10.1084/jem.176.5.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reininger L., Winkler T.H., Kalberer C.P., Jourdan M., Melchers F., Rolink A.G. Intrinsic B cell defects in NZB and NZW mice contribute to systemic lupus erythematosus in (NZB × NZW)F1 mice. J. Exp. Med. 1996;184:853–862. doi: 10.1084/jem.184.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P.L., Eisenberg R.A. Lpr and gldsingle gene models of systemic autoimmunity and lymphoproliferative disease. Annu. Rev. Immunol. 1991;9:243–269. doi: 10.1146/annurev.iy.09.040191.001331. [DOI] [PubMed] [Google Scholar]
- Tsubata T., Murakami M., Honjo T. Antigen-receptor cross-linking induces peritoneal B-cell apoptosis in normal but not autoimmunity-prone mice. Curr. Biol. 1994;4:8–17. doi: 10.1016/s0960-9822(00)00003-8. [DOI] [PubMed] [Google Scholar]
- Wofsy D., Seaman W.E. Successful treatment of autoimmunity in NZB/NZW F1 mice with monoclonal antibody to L3T4. J. Exp. Med. 1985;161:378–391. doi: 10.1084/jem.161.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]