

Abstract
Plasminogen activator inhibitor 1 (PAI-1) is a major inhibitor of urokinase-type plasminogen activator (uPA). In this study, we explored the role of PAI-1 in cell signaling. In MCF-7 cells, PAI-1 did not directly activate the mitogen-activated protein (MAP) kinases, extracellular signal–regulated kinase (ERK) 1 and ERK2, but instead altered the response to uPA so that ERK phosphorylation was sustained. This effect required the cooperative function of uPAR and the very low density lipoprotein receptor (VLDLr). When MCF-7 cells were treated with uPA–PAI-1 complex in the presence of the VLDLr antagonist, receptor-associated protein, or with uPA–PAI-1R76E complex, which binds to the VLDLr with greatly decreased affinity, transient ERK phosphorylation (<5 min) was observed, mimicking the uPA response. ERK phosphorylation was not induced by tissue-type plasminogen activator–PAI-1 complex or by uPA–PAI-1 complex in the presence of antibodies that block uPA binding to uPAR. uPA–PAI-1 complex induced tyrosine phosphorylation of focal adhesion kinase and Shc and sustained association of Sos with Shc, whereas uPA caused transient association of Sos with Shc.
By sustaining ERK phosphorylation, PAI-1 converted uPA into an MCF-7 cell mitogen. This activity was blocked by receptor-associated protein and not observed with uPA–PAI-1R76E complex, demonstrating the importance of the VLDLr. uPA promoted the growth of other cells in which ERK phosphorylation was sustained, including β3 integrin overexpressing MCF-7 cells and HT 1080 cells. The MEK inhibitor, PD098059, blocked the growth-promoting activity of uPA and uPA–PAI-1 complex in these cells. Our results demonstrate that PAI-1 may regulate uPA-initiated cell signaling by a mechanism that requires VLDLr recruitment. The kinetics of ERK phosphorylation in response to uPAR ligation determine the function of uPA and uPA–PAI-1 complex as growth promoters.
Keywords: urokinase-type plasminogen activator, plasminogen activator inhibitor 1, urokinase receptor, VLDL receptor, extracellular signal–regulated kinase
Introduction
Urokinase-type plasminogen activator (uPA) is a serine proteinase that binds with high affinity to the glycosylphosphatidylinositol-anchored receptor, uPAR, and activates a cascade of extracellular proteinases that includes plasmin and matrix metalloproteinases (Mignatti and Rifkin 2000). These activated proteinases degrade extracellular matrix proteins in basement membranes and other tissue boundaries, facilitating cellular migration in vivo. Polarization of uPAR to the leading edge of the migrating cell may optimally localize proteinase activity (Estreicher et al. 1990; Gyetko et al. 1994). Not surprisingly, uPA has been implicated in diverse processes that require cellular migration, including inflammation, neointima formation, healing of myocardial infarcts, cancer invasion, and metastasis (Carmeliet et al. 1994; Gyetko et al. 1996; Shapiro et al. 1996, Shapiro et al. 1997; Kim et al. 1998; Lijnen et al. 1998; Heymans et al. 1999). However, not all of the activities demonstrated by uPA in vivo may be attributed to the function of uPA as a proteinase (Waltz et al. 2000).
Plasminogen activator inhibitor 1 (PAI-1) is a member of the Serpin gene family and a major inhibitor of uPA (Mignatti and Rifkin 2000). PAI-1 reacts rapidly with free and uPAR-associated uPA (Ellis et al. 1990). Thus, PAI-1 might be expected to inhibit cellular migration through tissues and prevent cancer invasion (Mignatti and Rifkin 2000). However, there is substantial evidence that PAI-1 may promote cancer progression. Coexpression of PAI-1 with uPA is necessary for optimal invasion of lung carcinoma cells through Matrigel in vitro (Liu et al. 1995). In at least one study, PAI-1 deficiency in mice inhibited cancer cell invasion and decreased cancer angiogenesis (Bajou et al. 1998). Furthermore, high PAI-1 levels have been correlated with cancer progression in patients (Jänicke et al. 1991; Kuhn et al. 1994; Nekarda et al. 1994; Costantini et al. 1996; Schmitt et al. 1997; Knoop et al. 1998). Taken together, these results suggest that PAI-1 may express activities in vivo that are more complex than simple proteinase inhibition.
In its active conformation, PAI-1 binds directly to vitronectin and blocks the binding sites for αVβ3 and uPAR, inhibiting cell adhesion to vitronectin and altering cell migration (Deng et al. 1996; Stefansson and Lawrence 1996; Waltz et al. 1997). PAI-1 also binds sequentially to uPAR-associated uPA and then to receptors in the low density lipoprotein (LDL) receptor family, including low density lipoprotein receptor-related protein and the very low density lipoprotein receptor (VLDLr) (Conese et al. 1995; Webb et al. 1999). In the resulting quaternary complex, uPA–PAI-1 functions as a structural bridge so that uPAR undergoes endocytosis with the constitutively recycling LDL receptor homologues (Conese et al. 1995; Nykjær et al. 1997; Webb et al. 1999). Thus, the function of PAI-1 in vivo reflects diverse integrated activities, including regulation of cell surface proteolysis, cell adhesion, and uPAR catabolism.
A novel mechanism whereby PAI-1 may regulate cell physiology regards its potential to impact on cell signaling. Although there is no evidence that PAI-1 directly initiates cell signaling or modulates the response to other agonists, receptors for the uPA–PAI-1 complex, including uPAR and LDL receptor homologues, have been implicated in cell signaling (Trommsdorf et al., 1998; Blasi 1999; Hiesberger et al. 1999). Binding of uPA to uPAR activates p56/p59hck (Resnati et al. 1996; Konakova et al. 1998), the JAK–STAT pathway (Koshelnick et al. 1997; Dumler et al. 1998), focal adhesion kinase (FAK) (Tang et al. 1998; Yebra et al. 1999, Nguyen et al. 2000), protein kinase Cε (Busso et al. 1994), casein kinase 2 (Dumler et al. 1999), and the mitogen-activated protein (MAP) kinases, extracellular signal–regulated kinase (ERK) 1 and ERK2 (Kanse et al. 1997; Konakova et al. 1998; Nguyen et al. 1998; Tang et al. 1998; Ghiso et al. 1999). Since uPAR is glycosylphosphatidylinositol-anchored, uPA-initiated cell signaling probably requires one or more adaptor proteins with transmembrane domains (Chapman et al. 1999).
When MCF-7 breast cancer cells are treated with uPA, ERK is activated, and this signaling event is necessary for promoting cell migration (Nguyen et al. 1998, Nguyen et al. 1999). Interestingly, ERK activation in MCF-7 cells by uPA does not promote cell growth. This result may reflect the fact that ERK phosphorylation is highly transient in uPA-treated MCF-7 cells with levels returning to baseline in <5 min. In the present investigation, we explored the effects of PAI-1 on the Ras–ERK signaling pathway in MCF-7 cells. Although PAI-1 did not signal directly, it modulated the response to uPA so that ERK activation was sustained. The mechanism involved bridging of two essential receptors, uPAR and the VLDLr, by the uPA–PAI-1 complex. As a consequence of sustained ERK activation, PAI-1 converted uPA into an MCF-7 cell mitogen. In the absence of PAI-1, uPA still functioned as a mitogen towards cell lines that demonstrate sustained ERK phosphorylation in response to uPA, including HT 1080 fibrosarcoma cells and β3 integrin overexpressing MCF-7 cells. The ability of PAI-1 to function as a uPA response modifier at the level of cell signaling represents a novel mechanism whereby PAI-1 may affect cell physiology.
Materials and Methods
Reagents and Proteins
Two-chain uPA was provided by Drs. Jack Henkin and Andrew Mazar (Abbott Laboratories, Abbott Park, IL) and inactivated with diisopropyl fluorophosphate to form diisopropyl phospho (DIP)–uPA as described previously (Nguyen et al.. 1998). PAI-1 was provided by Dr. Duane Day (Molecular Innovations, Southfield, MI). PAI-1R76E was provided by Dr. Daniel Lawrence (American Red Cross, Rockville, MD). This mutant form of PAI-1 binds to receptors in the LDL receptor family with greatly decreased affinity (Stefansson et al. 1998). uPA-specific monoclonal antibody, which is directed against the amino-terminal fragment of uPA (No. 3471), and uPAR-specific antibody 399R were from American Diagnostica. Both of these antibodies, at a concentration of 25 μg/ml, inhibited specific binding of 125I-DIP–uPA to MCF-7 cells by >95% as demonstrated previously (Nguyen et al. 1999). The MEK inhibitor, PD098059, and the myosin light chain kinase (MLCK) inhibitor, ML-7, were from Calbiochem-Novabiochem. Phosphorylated ERK-specific antibody was from Promega or Calbiochem-Novabiochem. The polyclonal antibody that recognizes total ERK was from Zymed Laboratories. Phosphotyrosine-specific monoclonal antibody 4G10 was from Upstate Biotechnology. Sos1-specific polyclonal antibody (C-23) was from Santa Cruz Biotechnology, Inc. FLAG-specific monoclonal antibody was from Stratagene. Monoclonal anti-HA antibody 12CA5 was from Babco. Shc-specific polyclonal antibody was from Transduction Laboratories. Monoclonal antibody LM609, which is specific for αVβ3, was from Chemicon International. The expression construct that encodes glutathione-S-transferase (GST)–receptor-associated protein (RAP) was obtained from Dr. Joachim Herz (University of Texas Southwestern Medical Center, Dallas, TX). GST-RAP was expressed and purified as described previously (Webb et al.. 1999).
The uPA–PAI-1 complex was prepared by reacting 0.25 μM two-chain uPA with an equimolar concentration of PAI-1 for 10 min at 37°C. The activity of uPA in the uPA–PAI-1 complex was decreased to undetectable levels as determined by the rate of hydrolysis of L-pyroglutamyl-glycyl-arginine-p-nitroanilide. Two-chain tissue-type plasminogen activator (tPA) was purchased from American Diagnostica and reacted with PAI-1 using the strategy described for uPA.
Cell Culture and Transfection
Low passage MCF-7 cells were provided by Dr. Richard Santen (University of Virginia, Richmond, VA) and cultured in RPMI supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. HT 1080 fibrosarcoma cells were obtained from the American Type Culture Collection and cultured in MEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were passaged at subconfluence using enzyme-free cell dissociation buffer (Life Technologies) and maintained in culture for at least 48 h before performing experiments.
The full-length cDNA encoding the β3 integrin was provided by Dr. David Cheresh (Scripps Research Institute, La Jolla, CA) and subcloned into pBK–CMV. This construct was linearized and transfected into MCF-7 cells using Superfect (QIAGEN). After selection in G418 (1 mg/ml) for 3 wk, cells were single-cell cloned by serial dilution. β3 integrin expression was confirmed by FACS® analysis using antibody LM609 (Nguyen et al. 1999). Two clones were selected for further study.
Analysis of ERK Activation
MCF-7 and HT 1080 cells were cultured in 60-mm dishes. When the cultures were 80% confluent, the cells were incubated in serum-free medium for 12 h and then treated with 5 nM uPA–PAI-1 complex, 5 nM DIP–uPA, 0.4 μM GST-RAP, or 10 ng/ml EGF for the indicated times. HT 1080 cells were incubated with 10 nM DIP–uPA. To terminate an incubation, the medium was aspirated and replaced with ice cold 20 mM sodium phosphate, 150 mM NaCl, pH 7.4 (PBS) containing 1 mg/ml sodium orthovanadate. The cultures were then extracted in 1.0% Nonidet P-40, 50 mM Hepes, 100 mM NaCl, 2 mM EDTA, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 0.4 mg/ml sodium orthovanadate, 0.4 mg/ml sodium fluoride, and 5 mg/ml dithiothreitol, pH 7.4. Extracts were subjected to SDS-PAGE on 12% slabs. Proteins were transferred to nitrocellulose membranes and probed with antibodies that detect phosphorylated and total ERK.
Tyrosine Phosphorylation of FAK and Shc
Constructs encoding FLAG-tagged FAK and HA-tagged Shc were obtained from Drs. J. Thomas Parsons and Kodi Ravichandran (University of Virginia, Charlottesville, VA), respectively. The constructs were transfected into MCF-7 cells (10 μg of DNA/2 × 106 cells) using Superfect. Transfected cultures were maintained in serum-containing medium for 36 h, serum starved for 4 h, and then stimulated with uPA–PAI-1 complex (5 nM) for the indicated times. The medium was aspirated and replaced with ice cold PBS containing 1 mg/ml sodium orthovanadate. The cells were then extracted and the protein content in each extract determined. FLAG-tagged FAK and HA-tagged Shc were isolated from equal amounts of cellular protein by immunoprecipitation. The immunoprecipitated proteins were subjected to SDS-PAGE and electrotransferred to nitrocellulose membranes. Tyrosine-phosphorylated FAK and Shc were detected by immunoblot analysis. Total levels of FAK and Shc were also determined.
Coimmunoprecipitation of Sos1 with Shc
MCF-7 cells that were 80% confluent were cultured in serum-free RPMI for 12 h and then treated with 5 nM uPA–PAI-1 complex or vehicle at 37°C for the indicated times. Some cultures were pre-incubated with 0.4 μM RAP for 15 min to neutralize the activity of the VLDLr and then treated with uPA–PAI-1 complex. The cells were washed with ice cold PBS containing 1 mg/ml sodium orthovanadate and extracted in N-octyl glucoside as described previously (Nguyen et al. 2000). Shc was isolated by immunoprecipitation from equal amounts of cellular protein. The immunoprecipitates were subjected to SDS-PAGE in the presence of dithiothreitol and electrotransferred to nitrocellulose. Sos1 was detected by immunoblot analysis using antibody C-23.
Cell Migration Assays
Cell migration was studied using tissue culture–treated 6.5-mm Transwell chambers with 8.0 μm pore membranes (Costar). The bottom surface of each membrane was coated with 20% FBS as described previously (Nguyen et al. 1998). MCF-7 cells were dissociated from monolayer culture, washed with serum-free medium, and transferred to the top chamber of each Transwell at a density of 106 cells/ml (100 μl). uPA–PAI-1 complex (1 nM), DIP–uPA (1 nM), or PAI-1 (1–500 nM) were preincubated with the cells in suspension for 15 min and then added to both chambers. The bottom chamber contained RPMI + 10% FBS. Migration was allowed to proceed for 6 h at 37°C. Nonmigrating cells were removed from the top surface of each membrane using a cotton swab. The membranes were then fixed and stained with Diff-Quik (Dade Diagnostics). Cells that penetrated to the lower surface of each membrane were counted. We demonstrated previously that MCF-7 cells migrate equivalently on serum- or vitronectin-coated surfaces (Nguyen et al.. 1999) which is anticipated since vitronectin serves as the major cell attachment and spreading factor in serum (Hayman et al.. 1985). Single-chain uPA and DIP–uPA are equally effective at promoting MCF-7 cell migration and the response is not affected by proteinase inhibitors that are present in the FBS in the lower chamber (Nguyen et al. 1998, Nguyen et al. 1999).
Cell Growth Experiments
DNA synthesis was assessed in MCF-7 cells by measuring [3H]thymidine incorporation. MCF-7 cells were plated at a density of 2.5 × 104 cells/well in 24-well plates and cultured in serum-supplemented medium for 48 h. The cells were then washed two times with serum-free RPMI (no phenol red) containing 300 μg/ml glutamine, 5 μg/ml transferrin, and 38 nM selenium and cultured in the same medium supplemented with DIP–uPA (5 nM), uPA–PAI-1 complex (5 nM), PAI-1 (5 nM), or vehicle. After 30 h, the cells were pulse-exposed to [3H]thymidine (1 μCi/ml) for 1 h at 37°C and washed two times with Earle's balanced salt solution, 25 mM Hepes, pH 7.4. The cultures were then incubated with 10% trichloroacetic acid for 10 min at 4°C followed by a second incubation for 10 min at 22°C. Cell-associated radioactivity was recovered by incubation in 1.0 M NaOH for 12 h. The pH was neutralized with 1.0 M HCl, and the cell extracts were combined with Ready-Safe scintillation fluid (Beckman Coulter) for counting in a Beckman Coulter scintillation counter.
The effects of uPA–PAI-1 complex on total viable cell number was assessed in cultures of MCF-7, HT 1080, and β3 integrin overexpressing MCF-7 cells by 3-[4,5 dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay using the Cell Proliferation Kit I (Boehringer). MTT measures the activity of the mitochondrial enzyme, succinyl dehydrogenase, which is expressed only in living cells and is not subject to regulation (Mosmann 1983). MCF-7 cells and β3 integrin overexpressing MCF-7 cells (5 × 103) were cultured in 96-well plates in serum-supplemented medium for 48 h, washed two times, and treated with DIP–uPA (5 nM), uPA–PAI-1 complex (5 nM), or PAI-1 (5 nM) in serum-free RPMI (no phenol red) containing 300 μg/ml glutamine, 5 μg/ml transferrin, and 38 nM selenium. Some cultures were simultaneously treated with PD098059 (50 μM), or ML-7 (3 μM). After culturing for an additional 36 h, MTT assays were performed. Experiments with HT 1080 cells were performed identically except that incubations with DIP–uPA and uPA–PAI-1 complex were performed in serum-free MEM.
Results
uPA–PAI-1 Complex Activates ERK in a uPAR-dependent Manner
We demonstrated previously that ERK1 and ERK2 are rapidly phosphorylated and activated in uPA-treated MCF-7 cells (Nguyen et al. 1998, Nguyen et al. 1999). The response requires uPA binding to uPAR and is highly transient since levels of phosphorylated ERK return to baseline in <5 min (Nguyen et al. 1998). In the present study, we confirmed our previous results, demonstrating rapid but transient ERK phosphorylation in MCF-7 cells treated with 5 nM DIP–uPA (Fig. 1 A). ERK phosphorylation was not observed in MCF-7 cells that were treated with free PAI-1 (5 nM) in its active conformation (results not shown).
Figure 1.



Activation of ERK1 and ERK2 in uPA–PAI-1 complex-treated MCF-7 cells. MCF-7 cells were serum starved for 12 h and then treated with 5 nM DIP–uPA (A), 5 nM uPA–PAI-1 complex (B), or vehicle (Control; A and B) for the indicated times. (C) Cultures were incubated with uPA- or uPAR-specific antibodies (+) or vehicle (−) for 15 min at 37°C and then exposed to uPA–PAI-1 complex (5 nM) or vehicle for 5 min. Phosphorylated ERK1 and ERK2 were detected by immunoblot analysis. The nitrocellulose membranes were then stripped and reprobed with a separate antibody that detects total ERK.
When two-chain uPA was incubated with an equimolar concentration of PAI-1, stoichiometric uPA inactivation was demonstrated by chromogenic substrate hydrolysis. Preformed uPA–PAI-1 complex was immediately added to MCF-7 cell cultures, and ERK phosphorylation was monitored as a function of time. As was the case with free uPA, uPA–PAI-1 complex (5 nM) induced rapid ERK phosphorylation. However, the response to uPA–PAI-1 complex was sustained throughout the course of the 30-min incubation (Fig. 1 B). Equivalent results were obtained when the concentration of uPA–PAI-1 complex was decreased to 1.0 or 0.2 nM (results not shown). These experiments demonstrate that PAI-1 does not directly activate ERK in MCF-7 cells but instead alters the kinetics of ERK phosphorylation/dephosphorylation in response to uPA.
uPA–PAI-1 complex binds with high affinity to two separate receptors in MCF-7 cells, uPAR and the VLDLr (Cubellis et al. 1989; Jensen et al. 1990; Argraves et al. 1995; Heegaard et al. 1995). MCF-7 cells express ∼4,000 copies of uPAR/cell (Nguyen et al. 1998) and ≥60,000 copies of the VLDLr/cell (Webb et al. 1999). To determine whether ERK activation by uPA–PAI-1 complex requires uPAR, MCF-7 cells were treated for 15 min with uPA- or uPAR-specific antibodies (25 μg/ml) that block uPA binding to uPAR and then with uPA–PAI-1 complex. As shown in Fig. 1 C, both antibodies blocked ERK phosphorylation in response to uPA–PAI-1 complex, whereas nonimmune IgG (25 μg/ml) had no effect (results not shown). These results demonstrate that binding of uPA–PAI-1 complex to uPAR is necessary for ERK activation.
tPA binding to PAI-1 exposes the VLDLr recognition site in PAI-1 analogously to uPA. However, tPA–PAI-1 complex does not associate with uPAR (Kasza et al. 1997). To further test the role of uPAR in the activation of ERK by uPA–PAI-1 complex, we compared the activity of purified tPA–PAI-1 complex. In two separate experiments, 5 nM tPA–PAI-1 complex failed to induce ERK phosphorylation in MCF-7 cells (results not shown). These results support our conclusion that uPAR ligation is necessary for ERK activation by uPA–PAI-1 complex.
In MCF-7 cells, FAK, Shc, and Ras serve as necessary upstream components in the pathway by which uPA binding to uPAR leads to ERK phosphorylation (Nguyen et al. 1999, Nguyen et al. 2000). FAK phosphorylation also results from uPA treatment of uPAR-expressing LNCap cells and endothelial cells (Tang et al. 1998; Yebra et al. 1999). To determine whether the pathways that link uPA and uPA–PAI-1 complex to ERK involve the same upstream factors, we examined the phosphorylation of FAK and Shc in uPA–PAI-1 complex-treated MCF-7 cells. As shown in Fig. 2, FAK and Shc were tyrosine phosphorylated when MCF-7 cells were treated with 5 nM uPA–PAI-1 complex. Both responses were sustained for at least 30 min.
Figure 2.

Tyrosine phosphorylation of FAK and Shc in uPA–PAI-1 complex-treated MCF-7 cells. MCF-7 cells were transfected to express FLAG-tagged FAK or HA-tagged Shc. The cultures were then serum starved for 4 h and stimulated with uPA–PAI-1 complex (5 nM) for the indicated times at 37°C. Control cells were not treated with uPA–PAI-1 complex. FLAG-tagged FAK was immunoprecipitated with FLAG-specific monoclonal antibody, and Shc was immunoprecipitated with HA-specific antibody 12CA5. Phosphorylated FAK and Shc were detected by immunoblot analysis using phosphotyrosine-specific antibody (P-tyr). Total levels of FAK and Shc were detected by immunoblot analysis using FLAG-specific antibody and Shc-specific antibody, respectively.
Sustained ERK Activation by uPA–PAI-1 Complex Requires the VLDLr
Free and uPAR-associated uPA–PAI-1 complex both bind to the VLDLr (Argraves et al. 1995; Conese et al. 1995; Heegaard et al. 1995; Webb et al. 1999). To determine whether the VLDLr is required for ERK activation by uPA–PAI-1 complex, MCF-7 cells were treated with 5 nM uPA–PAI-1 complex in the presence of RAP. RAP binds to LDL receptor homologues, including the VLDLr and inhibits the binding of all other known ligands, including uPA–PAI-1 complex (Battey et al. 1994; Argraves et al. 1995; Heegaard et al. 1995, Webb et al. 1999). As shown in Fig. 3 A, uPA–PAI-1 complex induced ERK phosphorylation in RAP-treated MCF-7 cells, suggesting that the VLDLr is not required for this response. However, in the presence of RAP, ERK phosphorylation was transient. Within 5 min, the level of phosphorylated ERK returned to baseline, mimicking the kinetics observed with DIP–uPA. RAP did not promote ERK phosphorylation in the absence of uPA–PAI-1 complex or inhibit ERK phosphorylation in response to EGF (Fig. 3 B). These results are consistent with a model in which uPAR is necessary for ERK phosphorylation by uPA–PAI-1 complex and the VLDLr is necessary for sustaining the response.
Figure 3.


RAP blocks the sustained phosphorylation of ERK1 and ERK2 in uPA–PAI-1 complex-treated cells. (A) Serum-starved MCF-7 cells were incubated with GST-RAP (0.4 μM) for 15 min at 37°C. Cultures were then treated with 5 nM uPA–PAI-1 complex for the indicated times. Control cells were not treated with RAP or uPA–PAI-1 complex. (B) MCF-7 cells were serum starved and then treated with GST-RAP (+) for the indicated times at 37°C. Cultures were also incubated with GST-RAP (+) or vehicle (−) for 15 min at 37°C and then treated with EGF for 5 min at 37°C. Control cells were not treated with RAP or EGF. Phosphorylated and total ERK1 and ERK2 were detected by immunoblot analysis.
To further test the role of the VLDLr in uPA–PAI-1 complex-promoted ERK phosphorylation, uPA–PAI-1 complex was prepared using a mutant form of PAI-1 (PAI-1R76E) that binds to LDL receptor homologues with greatly decreased affinity (Stefansson et al. 1998). uPA–PAI-1R76E complex (5 nM) induced ERK phosphorylation in MCF-7 cells. However, the response was transient (Fig. 4). These results support the conclusion that cooperation between uPAR and the VLDLr is necessary for sustained ERK phosphorylation in MCF-7 cells.
Figure 4.

uPA–PAI-1 complex formed with a mutant form of PAI-1 that binds with greatly decreased affinity to the VLDLr induces transient ERK phosphorylation. MCF-7 cells were cultured in serum-free medium for 12 h and then exposed to 5 nM uPA–PAI-1R76E complex for the indicated times. Control cells were not incubated with uPA–PAI-1R76E complex. Phosphorylated and total ERK1 and ERK2 were detected by immunoblot analysis.
Sos Association with Shc Is Sustained in uPA–PAI-1 Complex-treated Cells
The Ras–ERK signaling pathway includes negative feedback loops that may be responsible for transient ERK activation. An example is the MEK-dependent phosphorylation of Sos, which promotes dissociation of Sos from Grb2 (Langlois et al. 1995). When MCF-7 cells are stimulated with uPA, Sos initially coimmunoprecipitates with Shc, probably reflecting the formation of Shc–Grb2–Sos complex. However, by 10 min, the complex is no longer observed even though Shc remains tyrosine phosphorylated (Nguyen et al. 2000). Sos dissociation from Grb2 and Shc may functionally dissociate Ras from upstream activators.
Sos1 coimmunoprecipitated with Shc in MCF-7 cells that were treated with uPA–PAI-1 complex (Fig. 5) as was previously observed with uPA (Nguyen et al. 2000). However, in the uPA–PAI-1 complex-treated cells, Sos1 remained associated with Shc for at least 30 min (the last time point in the experiment, n = 3). When MCF-7 cells were treated with RAP to block the function of the VLDLr and then with uPA–PAI-1 complex, Sos1 still associated with Shc. However, by 10 min, the complex was no longer observed. Thus, sustained ERK phosphorylation in uPA–PAI-1 complex-treated MCF-7 cells correlates with the continued presence of Shc–Grb2–Sos complex and requires the VLDLr.
Figure 5.

Association of Sos with Shc is sustained in uPA–PAI-1 complex-treated cells. MCF-7 cells were incubated in serum-free medium for 12 h. The cells were then treated with 5 nM uPA–PAI-1 complex for the indicated times (top panel). Some cultures were treated with GST-RAP for 15 min and then with vehicle for 1 min or with uPA–PAI-1 complex for the indicated times (bottom panel). Shc was isolated by immunoprecipitation. The immunoprecipitates were then subjected to immunoblot analysis to detect Sos1. Control cells were not treated with uPA–PAI-1 complex or RAP.
Sustained ERK Phosphorylation Requires the Continuous Presence of uPA–PAI-1 Complex
The VLDLr plays a critical role in cell signaling initiated by the extracellular matrix protein, reelin (D'Arcangelo et al. 1999; Hiesberger et al. 1999). This activity may be explained by the ability of reelin to bind simultaneously to the VLDLr and to a separate coreceptor with tyrosine kinase activity so that an intracellular kinase is brought into close juxtaposition with a VLDLr-associated substrate such as disabled 1 (Hiesberger et al. 1999). Similarly, uPA–PAI-1 complex may induce sustained ERK phosphorylation by a “receptor-bridging” mechanism.
A second model to explain the sustained activation of ERK in uPA–PAI-1 complex-treated MCF-7 cells involves the ability of the VLDLr to mediate uPAR endocytosis and recycling when uPA–PAI-1 complex binds to uPAR (Conese et al. 1995; Nykjær et al. 1997; Webb et al. 1999). Because uPA-initiated ERK activation in MCF-7 cells is transient, due to processes such as Shc–Grb2–Sos dissociation, uPAR endocytosis and recycling may provide a constant pool of unliganded uPAR to initiate new signaling events. If this model is correct, then a continuous source of free uPA–PAI complex should be necessary to sustain ERK phosphorylation. To test this hypothesis, MCF-7 cells were pulse-exposed to 5 nM uPA–PAI-1 complex for 1 min, washed, and then incubated in fresh medium without uPA–PAI-1 complex. As shown in Fig. 6, ERK was phosphorylated in the early time points. However, by 15 min, the level of phosphorylated ERK returned to baseline. Thus, a continuous source of free uPA–PAI-1 complex is necessary to sustain ERK phosphorylation in MCF-7 cells.
Figure 6.

Sustained ERK phosphorylation requires the continuous presence of uPA–PAI-1 complex. MCF-7 cells were serum starved for 12 h and then pulse-exposed to 5 nM uPA–PAI-1 complex for 1 min at 37°C. Cultures were then processed for ERK analysis (1 min) or washed and incubated in fresh medium without uPA–PAI-1 complex for the indicated times. Control cells were not treated with uPA–PAI-1 complex. Phosphorylated and total ERK1 and ERK2 were detected by immunoblot analysis.
uPA–PAI-1 Complex Promotes MCF-7 Cell Migration
uPA promotes cell migration by a mechanism that requires ERK activation in parental MCF-7 cells, HT 1080 cells, and in MCF-7 cells that are transfected to overexpress uPAR (Nguyen et al. 1998, Nguyen et al. 1999). In the present study, Transwell membranes were coated on the underside surface with FBS to form a haptotactic vitronectin gradient (Hayman et al. 1985; Nguyen et al. 1998). DIP–uPA, PAI-1, or uPA–PAI-1 complex (each at 1 nM) was added to both Transwell chambers, and migration was allowed to proceed for 6 h. As shown in Fig. 7 A, DIP–uPA and uPA–PAI-1 complex promoted MCF-7 cell migration equivalently, whereas free PAI-1 had no effect. In control experiments, we demonstrated that neither DIP–uPA or uPA–PAI-1 complex affect MCF-7 cell growth (MTT assay) in the time frame of the migration experiments (6 h). In other studies that are not shown, we demonstrated that uPA–PAI-1 complex and DIP–uPA promote MCF-7 cell migration equivalently when both Transwell membrane surfaces are coated with FBS and migration is allowed to proceed for 24 h. However, due to the longer incubation time, we cannot rule out the possibility that cell proliferation affected these results.
Figure 7.
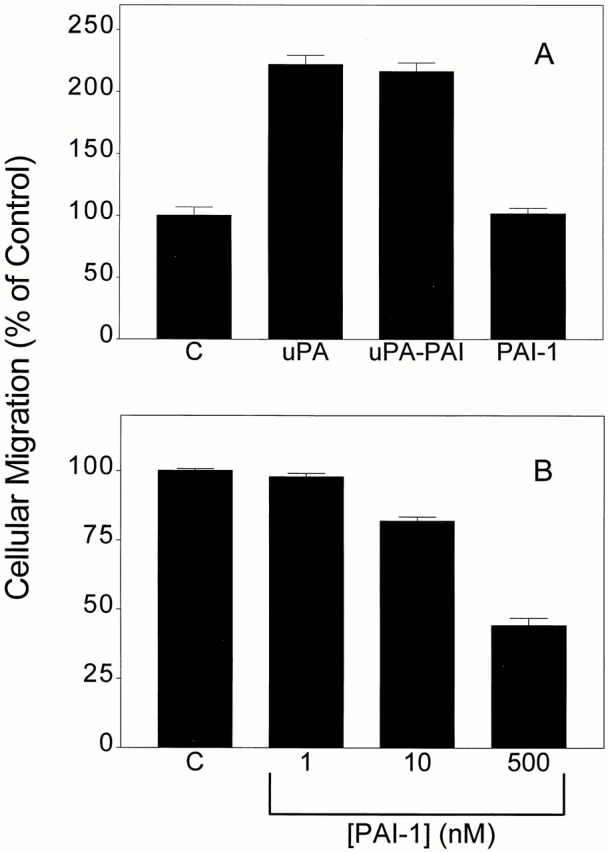
uPA–PAI-1 complex promotes MCF-7 cell migration. (A) MCF-7 cells were treated with DIP–uPA, uPA–PAI-1 complex, PAI-1, or vehicle (“C”) for 15 min and then added to Transwells in the presence of the same proteins or complexes. The cells were allowed to migrate for 6 h. (B) Transwell membranes were coated with FBS and then preincubated with the indicated concentrations of PAI-1 for 15 min before adding cells to the upper chamber.
Previous studies have demonstrated that PAI-1 regulates cell migration on vitronectin; however, high concentrations of PAI-1 may be necessary (Stefansson and Lawrence 1996; Kjøller et al. 1997; Waltz et al. 1997). Furthermore, the function of PAI-1 in cell migration may depend on whether αV integrins or uPAR function as the primary adhesion receptor (Deng et al. 1996; Stefansson and Lawrence 1996; Kjøller et al. 1997; Waltz et al. 1997). To understand more fully the role of free PAI-1 in the regulation of MCF-7 cell migration, we studied increasing concentrations of PAI-1. As shown in Fig. 7 B, MCF-7 cell migration was significantly inhibited when the Transwell membranes were preincubated for 15 min with 10–500 nM PAI-1 (p < 0.01). However, when the PAI-1 was added to the Transwell chambers simultaneously with the cells, migration was not affected (results not shown). We interpret these results to indicate that PAI-1 inhibits MCF-7 cell migration by binding to the immobilized vitronectin and blocking cell adhesion (Stefansson and Lawrence 1996).
uPA–PAI-1 Complex Promotes MCF-7 Cell Growth
The length of time that ERK phosphorylation is sustained may determine whether active ERK translocates to the nucleus and promotes cell growth (Lenormand et al. 1993). We demonstrated previously that uPA does not stimulate MCF-7 cell proliferation (Nguyen et al. 1998). To determine whether uPA–PAI-1 complex functions as a mitogen, [3H]thymidine incorporation experiments were conducted. Fig. 8 A shows that [3H]thymidine incorporation was increased 3.5 ± 0.2-fold (n = 4) in MCF-7 cells that were exposed to 5 nM uPA–PAI-1 complex for 30 h. Under equivalent conditions, DIP–uPA and free PAI-1 had no effect on [3H]thymidine incorporation.
Figure 8.



uPA–PAI-1 complex promotes MCF-7 cell growth. (A) MCF-7 cells were cultured in serum-supplemented medium for 48 h. The cells were then washed and cultured in serum-free medium supplemented with DIP–uPA, uPA–PAI-1 complex, PAI-1, or vehicle (“C”). After 30 h, the cells were pulse-exposed to [3H]thymidine for 1 h at 37°C. The cells were then washed and incubated with 10% trichloroacetic acid. Cell-associated radioactivity was recovered in 1 M NaOH. (B) MCF-7 cells were cultured in serum-supplemented medium for 48 h, washed, and treated with uPA, uPA–PAI-1 complex, PAI-1, uPA–PAI-1R76E complex, or vehicle (“C”) in serum free–defined medium. After culturing for an additional 36 h, MTT assays were performed. The graph shows the increase in cell number relative to the increase observed in control cultures. (C) MCF-7 cells were pulse-exposed to uPA–PAI-1 complex for 4 h, in the presence or absence of RAP. After culturing for an additional 32 h, cell growth was determined by MTT assay.
We also assessed the effects of uPA–PAI-1 complex on MCF-7 cell growth using the MTT assay, which measures total viable cell number (Fig. 8 B). MCF-7 cells that were incubated for 36 h in serum free–defined medium (control cells) demonstrated a 48 ± 7% increase in cell number, which was arbitrarily designated 100% growth. DIP–uPA (5 nM) did not cause a statistically significant increase in cell growth compared with the control. Similarly, free PAI-1 (5 nM) had no effect on cell growth. However, when uPA–PAI-1 complex was added, cell growth increased 3.0 ± 0.1-fold compared with the control, corresponding to ∼1.25 doublings in 36 h. The increase in growth measured by the MTT assay may reflect an increased rate of cell proliferation and/or a decreased rate of apoptosis.
To determine whether activated ERK is required for uPA–PAI-1 complex-induced MCF-7 cell growth, the MEK antagonist, PD098059 (50 μM), was added. In four separate experiments, PD098059 did not significantly affect MCF-7 cell growth in serum free–defined medium or in medium supplemented with DIP–uPA. However, PD098059 completely blocked the response to uPA–PAI-1 complex (results not shown). Interestingly, the MLCK inhibitor, ML-7 (3 μM), had no effect on the response to uPA–PAI-1 complex. In the presence of ML-7, uPA–PAI-1 complex increased cell growth 2.9 ± 0.2-fold compared with the control, which was not significantly different than that observed with uPA–PAI-1 complex alone. The same concentration of ML-7 completely blocked uPA-promoted MCF-7 cell migration (Nguyen et al. 1999).
The VLDLr Is Required for uPA–PAI-1 Complex-induced MCF-7 Cell Growth
We hypothesized that uPA–PAI-1 complex induces cell growth based on its ability to sustain ERK phosphorylation, which is dependent on cooperation between uPAR and the VLDLr. To test this hypothesis, we studied the effects of uPA–PAI-1R76E complex on cell growth. uPA–PAI-1R76E complex does not bind to the VLDLr (Stefansson et al. 1998) and elicits highly transient ERK phosphorylation. As shown in Fig. 8 B, uPA–PAI-1R76E complex failed to promote MCF-7 cell growth compared with the control, supporting our model.
To further test our hypothesis regarding the function of the VLDLr in uPA–PAI-1 complex-promoted MCF-7 cell growth, we examined the effects of RAP. When cells are cultured in the presence of RAP for long periods of time (1–5 d), uPAR endocytosis is inhibited and cell-surface uPAR levels slowly increase (Conese et al. 1995; Weaver et al. 1997; Webb et al. 1999). To avoid this complication, we executed a pulse-exposure protocol. MCF-7 cells were pretreated with 0.4 μM RAP or vehicle for 15 min and then with uPA–PAI-1 complex in the presence or absence of RAP for 4 h. The cells were then washed and incubated in serum free–defined medium in the absence of RAP and uPA–PAI-1 complex for an additional 32 h. Total cell number was determined by MTT assay. As shown in Fig. 8 C, pulse exposure of MCF-7 cells to RAP for 4.25 h did not affect cell growth. By contrast, a 3.2 ± 0.5-fold increase in cell growth was observed when MCF-7 cells were pulse-exposed to uPA–PAI-1 complex. This result suggests that the growth-promoting activity of uPA–PAI-1 complex is sustained even after the complex is removed. RAP blocked the growth-promoting activity of uPA–PAI-1 complex, again supporting our model in which the VLDLr and sustained ERK phosphorylation are required for cell growth in response to uPAR ligation.
uPAR-promoted Cell Growth Correlates with the Duration of ERK Phosphorylation
HT 1080 cells demonstrate increased migration when treated with DIP–uPA, and the response is completely blocked by PD098059 (Nguyen et al. 1999). Although HT 1080 cells express significant amounts of uPA (Tsuboi and Rifkin 1990; Laug et al. 1992), we demonstrated previously that under our culturing conditions, the amount of endogenously produced uPA is sufficient to saturate <5% of the available uPAR, accounting for the ability of these cells to respond to exogenously added uPA (Nguyen et al. 1999; Webb et al. 2000).
Fig. 9 A shows that ERK was phosphorylated in HT 1080 cells that were treated with 10 nM DIP–uPA. ERK phosphorylation was observed within 1 min. However, the response was sustained for at least 30 min. DIP–uPA also induced HT 1080 cell growth (Fig. 9 B). The magnitude of the response was comparable with that observed using an equal concentration of uPA–PAI-1 complex. PD098059 inhibited the growth-promoting activity of DIP–uPA and uPA–PAI-1 complex without affecting basal HT 1080 cell growth, indicating an essential role for activated ERK.
Figure 9.


uPA stimulates sustained ERK phosphorylation and growth in HT 1080 cells. (A) HT 1080 cells were serum starved for 12 h and then treated with 10 nM DIP–uPA for the indicated times. Cell extracts were isolated and subjected to immunoblot analysis to detect phosphorylated and total ERK. Control cells were not treated with DIP–uPA. (B) HT 1080 cells were treated with DIP–uPA or uPA–PAI-1 complex in serum-free medium for 36 h. Some cultures were treated with PD098059. Cell growth was determined by MTT assay. The increase in cell number is standardized against that observed in control cultures.
β3 integrin expression sustains ERK phosphorylation in response to a number of agonists in cells that express αVβ5 (Elicieri et al. 1998; Nguyen et al. 1999). The reason for this remains unclear. In MCF-7 cells that are transfected to overexpress β3 and treated with DIP–uPA (10 nM), ERK phosphorylation is observed within 1 min and remains elevated for at least 40 min (Nguyen et al. 1999). As shown in Fig. 10 A, MCF-7 cells that were transfected to overexpress β3 and single-cell cloned, expressed cell-surface αVβ3 as determined by FACS analysis with antibody LM609. β3 overexpressing MCF-7 cells also demonstrated increased growth when treated with DIP–uPA or uPA–PAI-1 complex (Fig. 10 B). Identical results were obtained with a second distinct β3 overexpressing clone (results not shown). When β3 overexpressing MCF-7 cells were treated with uPA or uPA–PAI-1 complex in the presence of PD098059, the increase in cell growth was blocked.
Figure 10.


uPA stimulates growth in β3 integrin overexpressing MCF-7 cells. (A) αVβ3 expression was demonstrated in β3 integrin overexpressing MCF-7 cells by FACS® analysis. (B) These cells were treated with DIP–uPA or uPA–PAI-1 complex for 36 h in the presence and absence of PD098059. Cell growth was determined by MTT assay. The increase in cell number is plotted relative to the increase observed in control cultures.
Discussion
The Ras–ERK signaling pathway regulates diverse cellular processes, including proliferation, differentiation, apoptosis, and migration. Whether activated ERK stimulates cell growth may depend on a number of factors, including the duration of the response (Meloche et al. 1992; Lenormand et al. 1993; Sergeant et al. 2000). We demonstrated previously that uPA activates ERK in MCF-7 cells without promoting cell growth, probably reflecting the highly transient nature of the signaling response (Nguyen et al. 1998, Nguyen et al. 1999). We now demonstrate that free PAI-1 does not independently activate ERK but instead modifies the response obtained when uPA ligates uPAR so that ERK phosphorylation is sustained. In MCF-7 cells, sustained ERK phosphorylation is necessary to convert uPA into a mitogen.
The ability of uPA–PAI-1 complex to induce sustained ERK phosphorylation in MCF-7 cells requires the recruitment of uPAR and the VLDLr, which function cooperatively. All of our evidence suggests that cell signaling is initiated when uPA–PAI-1 complex binds to uPAR and follows a pathway that involves FAK and Shc as was described previously for uPA (Nguyen et al. 2000). The function of the VLDLr in uPA–PAI-1 complex-promoted ERK activation is to sustain the response, probably by engaging uPA–PAI-1 complex after it has bound uPAR. The MCF-7 cell provided an excellent model system to demonstrate cooperativity between uPAR and the VLDLr given the highly transient nature of uPA-induced ERK phosphorylation in this cell line. In HT 1080 cells and MCF-7 cells that overexpress β3 integrin, uPA induced more sustained ERK phosphorylation and, as a result, functioned comparably to uPA–PAI-1 complex in cell growth assays.
The possibility that PAI-1, the VLDLr, and uPAR cooperate to convert uPA into a cancer cell mitogen in vivo represents an appealing model to explain why PAI-1 expression adversely affects breast cancer progression. All of the components of uPA–uPAR system have been identified in breast cancer. The malignant epithelial cells in breast cancers express variable amounts of uPAR. However, uPAR is almost always detectable, at least at the level of mRNA (Jankun et al. 1993; Hildenbrand et al. 1998). Furthermore, malignant breast epithelia tend to be strongly immunoreactive for the VLDLr (Martensen et al. 1997). uPA is expressed at high levels by benign cells that are intermixed with malignant epithelia in breast cancers, establishing the context for a paracrine interaction (Nielsen et al. 1996). We propose that uPA–PAI-1 complex may promote growth of malignant cells in some breast cancers, whereas uPA does not as was observed in MCF-7 cells. To explore the feasibility of this argument, it will be important to gain a better understanding of the factors that control the kinetics of ERK phosphorylation and dephosphorylation in breast cancer cells and in other cells in general. The results reported here suggest that stability of Shc–Grb2–Sos complex is a key determinant of whether ERK phosphorylation is sustained in MCF-7 cells. Other pathways, including those that lead to the activation of dual specificity phosphatases, may also be involved (Keyse 1995).
The mitogenic activity of uPA–PAI-1 complex towards MCF-7 cells, β3 integrin overexpressing MCF-7 cells, and HT 1080 cells and the mitogenic activity of DIP–uPA towards the latter two cell lines was completely blocked by the MEK antagonist, PD098059, supporting an essential role for the Ras–ERK pathway in uPA promoted cell proliferation. In other cell types, blocking the activity of casein kinase 2 completely neutralizes the mitogenic activity of uPA (Dumler et al. 1999). Although it is possible that separate signaling pathways are operational in different cell types, we think it is much more likely that uPAR ligation triggers signaling through distinct but cooperative pathways that must be simultaneously activated in order to observe cell growth. According to this model, inhibiting either casein kinase 2 or ERK would be expected to independently inhibit mitogenesis.
Once activated, ERK translocates to multiple subcellular compartments, including the nucleus, the cytoskeleton, the plasma membrane, and newly formed focal adhesions (Chen et al. 1992; Gonzalez et al. 1993; Lenormand et al. 1993; Reszka et al. 1995; Fincham et al. 2000). Failure of ERK to translocate to the nucleus may partially explain why transient ERK phosphorylation in MCF-7 cells does not stimulate cell growth (Meloche et al. 1992; Lenormand et al. 1993; Sergeant et al. 2000). By contrast, MLCK is activated by ERK or an ERK-dependent kinase rapidly and effectively in DIP–uPA-treated MCF-7 cells, and this reaction appears to be responsible for the stimulation of cell migration (Nguyen et al. 1999). MLCK remains phosphorylated, and levels of serine-phosphorylated myosin II regulatory light chain remain elevated long after the level of phosphorylated ERK returns to baseline (Nguyen et al. 1999). Rapid and sustained activation of MLCK explains why DIP–uPA and uPA–PAI-1 complex are comparable promoters of MCF-7 cell migration.
An NPXY motif in the cytoplasmic tail of the VLDLr binds adaptor proteins, such as disabled 1, that function in cell signaling (Trommsdorff et al. 1998; Hiesberger et al. 1999). By bridging the VLDLr and uPAR, uPA–PAI-1 complex brings the intracytoplasmic tail of the VLDLr into close proximity with the uPAR-associated signaling machinery and may thereby alter the character of the signaling response induced by uPAR ligation. An alternative model to explain the function of PAI-1 as a uPA response modifier involves the ability of the VLDLr to mediate uPAR internalization in association with uPA–PAI-1 complex (Webb et al. 1999). In this model, each cell-signaling event is viewed as highly transient, perhaps due to the instability of Shc–Grb2–Sos complex in uPA-treated MCF-7 cells. Clearance of uPA–PAI-1–uPAR complex from the cell surface may allow for the formation of new multicomponent signaling receptors in which unliganded uPAR associates with adaptor proteins that are available in short supply. Binding of uPA–PAI-1 complex to each newly formed receptor complex induces transient ERK activation. However, due to uPAR endocytosis and the continuous reformation of signaling–receptor complexes, the integrated response to uPA–PAI-1 complex is sufficient to maintain a stable pool of Shc–Grb2–Sos complex and ERK at an increased level of phosphorylation. In support of this model, a continuous source of free uPA–PAI-1 complex was necessary for sustained ERK phosphorylation in MCF-7 cells. The uPAR that enters into newly formed signaling–receptor complexes may be derived from distinct plasma membrane microdomains, intracytoplasmic vesicles, and/or recycling pools.
Based on this and previous studies, PAI-1 emerges as a complex regulator of the uPA–uPAR system. By binding directly to vitronectin, PAI-1 may alter cell adhesion and migration (Deng et al. 1996; Stefansson and Lawrence 1996; Kjøller et al. 1997; Waltz et al. 1997). By binding uPAR-associated two-chain uPA, PAI-1 inhibits activation of the cell surface proteinase cascade and may thereby inhibit cellular penetration of tissue boundaries. As a result of the same reaction, PAI-1 promotes uPAR endocytosis by a mechanism that depends on uPAR bridging to LDL receptor homologues (Conese et al. 1995; Nykjær et al. 1997; Webb et al. 1999). Since the recycling efficiency of uPAR is <100%, continuous uPA–PAI-1 complex-promoted uPAR endocytosis results in the re-equilibration of cell surface uPAR levels below that observed in the absence of LDL receptor homologues (Weaver et al. 1997; Webb et al. 1999, Webb et al. 2000). However, this reequilibration occurs very slowly. In a more rapid time frame, uPA–PAI-1 complex may alter the character of uPAR initiated cell signaling compared with free uPA. By sustaining the activity of the Ras–ERK pathway, PAI-1 may promote cell growth and potentially alter other physiologic processes such as apoptosis. The ability of PAI-1 to modulate uPAR-initiated cell signaling represents a novel mechanism whereby PAI-1 may influence cancer progression in vivo.
Acknowledgments
This work was supported by grant HL60551 from the National Institutes of Health and the Susan G. Komen Breast Cancer Research Foundation.
Footnotes
Abbreviations used in this paper: DIP, diisopropyl phospho; ERK, extracellular signal–regulated kinase; FAK, focal adhesion kinase; GST, glutathione-S-transferase; LDL, low density lipoprotein; MAP, mitogen-activated protein; MLCK, myosin light chain kinase; PAI, plasminogen activator inhibitor; RAP, receptor-associated protein; tPA, tissue-type plasminogen activator; uPA, urokinase-type plasminogen activator; uPAR, uPA receptor; VLDL, very low density lipoprotein; VLDLr, VLDL receptor.
References
- Argraves K.M., Battey F.D., MacCalman D.D., McCrae K.R., Gåfvels M., Kozarsky K.F., Chappell D.A., Strauss J.F., III, Strickland D.K. The very low density lipoprotein receptor mediates the cellular catabolism of lipoprotein lipase and urokinase-plasminogen activator inhibitor type I complexes. J. Biol. Chem. 1995;270:26550–26557. doi: 10.1074/jbc.270.44.26550. [DOI] [PubMed] [Google Scholar]
- Bajou K., Noël A., Gerard R.D., Masson V., Brunner N., Holst-Hansen C., Skobe M., Fusenig N.E., Carmeliet P., Collen D., Foidart J.M. Absence of host plasminogen activator inhibitor 1 prevents cancer invasion and vascularization. Nature Med. 1998;4:924–928. doi: 10.1038/nm0898-923. [DOI] [PubMed] [Google Scholar]
- Battey F.D., Gåfvels M.E., Fitzgerald D.J., Argraves W.S., Chappell D.A., Strauss J.F., Strickland D.K. The 39-kDa receptor associated protein regulates ligand binding by the very low density lipoprotein receptor. J. Biol. Chem. 1994;269:23268–23273. [PubMed] [Google Scholar]
- Blasi F. Proteolysis, cell adhesion, chemotaxis, and invasiveness are regulated by the uPA-u-PAR-PAI-1 system. Thromb. Haemost. 1999;82:298–304. [PubMed] [Google Scholar]
- Busso N., Masur S.K., Lazega D., Waxman S., Ossowski L. Induction of cell migration by pro-urokinase binding to its receptorpossible mechanism for signal transduction in human epithelial cells. J. Cell Biol. 1994;126:259–270. doi: 10.1083/jcb.126.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P., Schoonjans L., Kieckens L., Ream B., Degen J., Bronson R., De Vos R., van den Oord J.J., Collen D., Mulligan R.C. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- Chapman H.A., Wei Y., Simon D.I., Waltz D.A. Role of urokinase receptor and caveolin in regulation of integrin signaling. Thromb. Haemost. 1999;82:291–297. [PubMed] [Google Scholar]
- Chen R.H., Sarnecki C., Blenis J. Nuclear localization and regulation of ERK- and RSK-encoded protein kinases. Mol. Cell. Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conese M., Nykjaer A., Petersen C.M., Cremona O., Pardi R., Andreasen P.A., Gleimann J., Christensen E.I., Blasi F. alpha-2 Macroglobulin receptor/LDL receptor-related protein (LRP)-dependent internalization of the urokinase receptor. J. Cell Biol. 1995;131:1609–1622. doi: 10.1083/jcb.131.6.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini V., Sidoni A., Deveglia R., Cazzato O.A., Bellezza G., Ferri I., Bucciarelli E., Nenci G.G. Combined overexpression of urokinase, urokinase receptor, and plasminogen activator inhibitor-1 is associated with breast cancer progression. Cancer. 1996;77:1079–1088. doi: 10.1002/(sici)1097-0142(19960315)77:6<1079::aid-cncr12>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Cubellis M.V., Andreasen P., Ragno P., Mayer M., Dano K., Blasi F. Accessibility of receptor-bound urokinase to type-1 plasminogen activator inhibitor. Proc. Natl. Acad. Sci. USA. 1989;86:4828–4832. doi: 10.1073/pnas.86.13.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G.D., Homayouni R., Keshvara L., Rice D.S., Sheldon M., Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;26:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- Deng G., Curriden S.A., Wang S., Rosenberg S., Loskutoff D.J. Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J. Cell Biol. 1996;134:1563–1571. doi: 10.1083/jcb.134.6.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumler I., Weis A., Mayboroda O.A., Maasch C., Jerke U., Haller H., Gulba D.C. The Jak/Stat Pathway and urokinase receptor signaling in human aortic vascular smooth muscle cells. J. Biol. Chem. 1998;273:315–321. doi: 10.1074/jbc.273.1.315. [DOI] [PubMed] [Google Scholar]
- Dumler I., Stepanova V., Jerke U., Mayboroda O.A., Vogel F., Bouvet P., Tkachuk V., Haller H., Gulba D.C. Urokinase-induced mitogenesis is mediated by casein kinase 2 and nucleolin. Current Biol. 1999;9:1468–1476. doi: 10.1016/s0960-9822(00)80116-5. [DOI] [PubMed] [Google Scholar]
- Elicieri B.P., Klemke R., Stromblad S., Cheresh D.A. Integrin alphavbeta3 requirement for sustained mitogen-activated protein kinase activity during angiogenesis. J. Cell Biol. 1998;140:1255–1263. doi: 10.1083/jcb.140.5.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis V., Wun T.C., Behrendt N., Ronne E., Dano K. Inhibition of receptor-bound urokinase by plasminogen-activator inhibitors. J. Biol. Chem. 1990;265:9904–9908. [PubMed] [Google Scholar]
- Estreicher A., Muhlhauser J., Carpentier J.L., Orici L., Vassalli J.D. The receptor for urokinase type plasminogen activator polarizes expression of the protease to the leading edge of migrating monocytes and promotes degradation of enzyme inhibitor complexes. J. Cell Biol. 1990;111:783–792. doi: 10.1083/jcb.111.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincham V.J., James M., Frame M.C., Winder S.J. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO (Eur. Mol. Biol. Organ.) J. 2000;19:2911–2923. doi: 10.1093/emboj/19.12.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiso J.A.A., Kovalski K., Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J. Cell Biol. 1999;147:89–103. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez F.A., Seth A., Raden D.L., Bowman D.S., Fay F.S., Davis R.J. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J. Cell Biol. 1993;122:1089–1101. doi: 10.1083/jcb.122.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyetko M.R., Todd R.F., III, Wilkison C.C., Citrin R.G. The urokinase receptor is required for human monocyte chemotaxis in vitro . J. Clin. Invest. 1994;93:1380–1387. doi: 10.1172/JCI117114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyetko M.R., Chen G.H., McDonald R.A., Goodman R., Huffnagle G.B., Wilkinson C.C., Fuller J.A., Toews G.B. Urokinase is required for the pulmonary inflammatory response to Cryptococcus neoformans . J. Clin. Invest. 1996;97:1818–1826. doi: 10.1172/JCI118611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayman E.G., Pierschbacker M.D., Suzuki S., Ruoslahti E. Vitronectin—a major cell attachment-promoting protein in fetal bovine serum. Exp. Cell Res. 1985;60:245–258. doi: 10.1016/0014-4827(85)90173-9. [DOI] [PubMed] [Google Scholar]
- Heegaard C.W., Simonsen A.C.W., Oka K., Kjøller L., Christensen A., Madsen B., Ellgaard L., Chan L., Andreasen P.A. Very low density lipoprotein receptor binds and mediates endocytosis of urokinase-type plasminogen activator-type-1 plasminogen activator inhibitor complex. J. Biol. Chem. 1995;270:20855–20861. doi: 10.1074/jbc.270.35.20855. [DOI] [PubMed] [Google Scholar]
- Heymans S., Luttun A., Nuyens D., Theilmeier G., Creemers E., Moons L., Dyspersin G.D., Cleutjens J.P.M., Shipley M., Angellilo A. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat. Med. 1999;5:1135–1142. doi: 10.1038/13459. [DOI] [PubMed] [Google Scholar]
- Hiesberger T., Trommsdorff M., Howell B.W., Goffinet A., Mumby M.C., Cooper J.A., Herz J. Direct binding of reelin to VLDL receptor and apoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hildenbrand R., Glienk W., Magdolen V., Graeff H., Stutte H.J., Schmitt M. Urokinase receptor localization in breast cancer and benign lesions assessed by in situ hybridization and immunohistochemistry. Histochem. Cell Biol. 1998;110:27–32. doi: 10.1007/s004180050261. [DOI] [PubMed] [Google Scholar]
- Jänicke F., Schmitt M., Graeff H. Clinical relevance of the urokinase-type and tissue-type plasminogen activators and of their type 1 inhibitor in breast cancer. Sem. Thromb. Hemostas. 1991;17:303–312. doi: 10.1055/s-2007-1002624. [DOI] [PubMed] [Google Scholar]
- Jankun J., Merrick H.W., Goldblatt P.J. Expression and localization of elements of the plasminogen activation system in benign breast disease and breast cancers. J. Cell. Biochem. 1993;53:135–144. doi: 10.1002/jcb.240530206. [DOI] [PubMed] [Google Scholar]
- Jensen P.H., Christensen E.I., Ebbesen P., Gliemann J., Anreasen P.A. Lysosomal degradation of receptor-bound urokinase-type plasminogen activator is enhanced by its inhibitors in human trophoblastic choriocarcinoma cells. Cell Regul. 1990;1:1043–1056. doi: 10.1091/mbc.1.13.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanse S.M., Benzakour O., Kanthou C., Kost C., Lijnen H.R., Preissner K.T. Induction of vascular SMC proliferation by urokinase indicates a novel mechanism of action in vasoproliferative disorders. Arterioscler. Thromb. Vasc. Biol. 1997;17:2848–2854. doi: 10.1161/01.atv.17.11.2848. [DOI] [PubMed] [Google Scholar]
- Kasza A., Petersen H.H., Heegaard C.W., Oka K., Christensen A., Dubin A., Chan L., Andreasen P.A. Specificity of serine proteinase/serpin complex binding to very-low-density lipoprotein receptor and alpha2-macroglobulin receptor/low-density-lipoprotein-receptor-related protein. Eur. J. Biochem. 1997;248:270–281. doi: 10.1111/j.1432-1033.1997.00270.x. [DOI] [PubMed] [Google Scholar]
- Keyse S.M. An emerging family of dual specificity MAP kinase phosphatases. Biochim. Biophys. Acta. 1995;1265:152–160. doi: 10.1016/0167-4889(94)00211-v. [DOI] [PubMed] [Google Scholar]
- Kim J., Yu W., Kovalski K., Ossowski L. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell. 1998;94:353–362. doi: 10.1016/s0092-8674(00)81478-6. [DOI] [PubMed] [Google Scholar]
- Kjøller L., Kanse S.M., Kirkegaard T., Rodenburg K.W., Rønne E., Goodman S.L., Preissner K.T., Ossowski L., Andreasen P.A. Plasminogen activator inhibitor-1 represses integrin- and vitronectin-mediated cell migration independently of its function as an inhibitor of plasminogen activator. Exp. Cell Res. 1997;232:420–429. doi: 10.1006/excr.1997.3540. [DOI] [PubMed] [Google Scholar]
- Knoop A., Andreasen P.A., Andersen J.A., Hansen S., Lænkholm A.V., Simonsen A.C.W., Andersen J., Overgaard J., Rose C. Prognostic significance of urokinase-type plasminogen activator and plasminogen activator inhibitor-1 in primary breast cancer. Brit. J. Cancer. 1998;77:932–940. doi: 10.1038/bjc.1998.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konakova M., Hucho F., Schleuning W.D. Downstream targets of urokinase-type plasminogen-activator-mediated signal transduction. Eur. J. Biochem. 1998;253:421–429. doi: 10.1046/j.1432-1327.1998.2530421.x. [DOI] [PubMed] [Google Scholar]
- Koshelnick Y., Ehart M., Hufnagl P., Heinrich P.C., Binder B.R. Urokinase receptor is associated with the components of the JAK1/STAT1 signaling pathway and leads to activation of this pathway upon receptor clustering in the human kidney epithelial tumor cell line TCL-598. J. Biol. Chem. 1997;272:28563–28567. doi: 10.1074/jbc.272.45.28563. [DOI] [PubMed] [Google Scholar]
- Kuhn W., Pache L., Schmalfeldt B., Dettmar P., Schmitt M., Jänicke F., Graeff H. Urokinase (uPA) and PAI-1 predict survival in advanced ovarian cancer patients (FIGO III) after radical surgery and platinum-based chemotherapy. Gynecol. Oncol. 1994;55:401–409. doi: 10.1006/gyno.1994.1313. [DOI] [PubMed] [Google Scholar]
- Langlois W.J., Sasaoka T., Saltiel A.R., Olefsky J.M. Negative feedback regulation and desensitization of insulin- and epidermal growth factor-stimulated p21ras activation. J. Biol. Chem. 1995;270:25320–25323. doi: 10.1074/jbc.270.43.25320. [DOI] [PubMed] [Google Scholar]
- Laug W.E., Wang K., Mundi R., Rideout W., Kruithof E.K.O., Bogenmann E. Clonal variation of expression of the genes coding for plasminogen activators, their inhibitors and the urokinase receptorin HT 1080 sarcoma cells. Int. J. Cancer. 1992;52:298–304. doi: 10.1002/ijc.2910520224. [DOI] [PubMed] [Google Scholar]
- Lenormand P., Sardet C., Pages G., Allemain G.L., Brunet A., Pouyssegur J. Growth factors induce nuclear translocation of MAP kinases (p42mapk and p44mapk) but not their activator MAP kinase kinase (p45mapkk) in fibroblasts. J. Cell Biol. 1993;122:1079–1088. doi: 10.1083/jcb.122.5.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lijnen H.R., Van Hoef B., Lupu F., Moons L., Carmeliet P., Collen C. Function of the plasminogen/plasmin and matrix metalloproteinase systems after vascular injury in mice with targeted inactivation of fibrinolytic system genes. Arterioscler. Thromb. Vasc. Biol. 1998;18:1035–1045. doi: 10.1161/01.atv.18.7.1035. [DOI] [PubMed] [Google Scholar]
- Liu G., Shuman M.A., Cohen R.L. Co-expression of urokinase, urokinase receptor and PAI-1 is necessary for optimum invasiveness of cultured lung cancer cells. Int. J. Cancer. 1995;60:501–506. doi: 10.1002/ijc.2910600413. [DOI] [PubMed] [Google Scholar]
- Martensen P.M., Oka K., Christensen L., Rettenberger P.M., Petersen H.H., Christensen A., Chan L., Heegard C.W., Andreasen P.A. Breast carcinoma epithelial cells express a very low-density lipoprotein receptor variant lacking the O-linked glycosylation domain encoded by exon 16, but with full binding activity for serine proteinase/serpin complexes and Mr-40,000 receptor-associated protein. Eur. J. Biochem. 1997;248:583–591. doi: 10.1111/j.1432-1033.1997.00583.x. [DOI] [PubMed] [Google Scholar]
- Meloche S., Seuwen K., Pages G., Pouysségur J. Biphasic and synergistic activation of p44mapk (ERK1) by growth factorscorrelation between late phase activation and mitogenicity. Mol. Endocrinol. 1992;6:845–854. doi: 10.1210/mend.6.5.1603090. [DOI] [PubMed] [Google Scholar]
- Mignatti P., Rifkin D.B. Non-enzymatic interactions between proteinases and the cell surfacenovel roles in normal and malignant cell physiology. Adv. Cancer Res. 2000;65:103–157. doi: 10.1016/s0065-230x(08)61024-6. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survivalapplication to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nekarda H., Schmitt M., Ulm K., Wenninger A., Vogelsang H., Becker K., Roder J.D., Fink U., Siewert J.R. Prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in completely resected gastric cancer. Cancer Res. 1994;54:2900–2907. [PubMed] [Google Scholar]
- Nielsen B.S., Sehested M., Timshel S., Pyke C., Dano K. Messenger RNA for urokinase plasminogen activator is expressed in myofibroblasts adjacent to cancer cells in human breast cancer. Lab. Invest. 1996;74:168–177. [PubMed] [Google Scholar]
- Nguyen D.H.D., Hussaini I.M., Gonias S.L. Binding of urokinase-type plasminogen activator to its receptor in MCF-7 cells activates extracellular signal-regulated kinase 1 and 2 which is required for increased cellular motility. J. Biol. Chem. 1998;273:8502–8507. doi: 10.1074/jbc.273.14.8502. [DOI] [PubMed] [Google Scholar]
- Nguyen D.H.D., Catling A.D., Webb D.J., Sankovic M., Waler L.A., Somlyo A.V., Weber M.J., Gonias S.L. Myosin light chain kinase functions downstream of Ras/ERK to promote migration of urokinase-type plasminogen activator-stimulated cells in an integrin-selective manner. J. Cell Biol. 1999;146:149–164. doi: 10.1083/jcb.146.1.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.H.D., Catling A.D., Webb D.J., Dhakephalkar A., Weber M.J., Ravichandran K.S., Gonias S.L. Urokinase-type plasminogen activator stimulates the Ras/ERK signaling pathway and MCF-7 cell migration by a mechanism that requires FAK, Src, and Shcrapid dissociation of Grb2/Sos-Shc complex is associated with the transient phosphorylation of ERK in urokinase-treated cells. J. Biol. Chem. 2000;275:19382–19388. doi: 10.1074/jbc.M909575199. [DOI] [PubMed] [Google Scholar]
- Nykjær A., Conese M., Christensen E.I., Olson D., Cremona O., Gliemann J., Blasi F. Recycling of the urokinase receptor upon internalization of the uPAserpin complexes. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:2610–2620. doi: 10.1093/emboj/16.10.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnati M., Guttinger M., Valcamonica S., Sidenius N., Blasi F., Fazioli F. Proteolytic cleavage of the urokinase receptor substitutes for the antagonist-induced chemotactic effect. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:1572–1582. [PMC free article] [PubMed] [Google Scholar]
- Reszka A.A., Seger R., Diltz C.D., Krebs E.G., Fischer E.H. Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc. Natl. Acad. Sci. USA. 1995;92:8881–8885. doi: 10.1073/pnas.92.19.8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt M., Harbeck N., Thomssen C., Wilhelm O., Magdolen V., Reuning U., Ulm K., Höfler H., Jänicke F., Graeff H. Clinical impact of the plasminogen activation system in tumor invasion and metastasisprognostic relevance and target for therapy. Thromb. Haemost. 1997;78:285–296. [PubMed] [Google Scholar]
- Sergeant N., Lyon M., Rudland P.S., Fernig D.G., Delehedde M. Stimulation of DNA synthesis and cell proliferation of human mammary myoepithelial-like cells by hepatocyte growth factor/scatter factor depends on heparan sulfate proteoglycans and sustained phosphorylation of mitogen-activated protein kinases p42/44. J. Biol. Chem. 2000;275:17094–17099. doi: 10.1074/jbc.M000237200. [DOI] [PubMed] [Google Scholar]
- Shapiro R.L., Duquette J.G., Roses D.F., Nunes I., Harris M.N., Kamino H., Wilson E.L., Rifkin D.B. Induction of primary cutaneous melanocytic neoplasms in urokinase-type plasminogen activator (uPA)-deficient and wild-type micecellular blue nevi invade but do not progress to malignant melanoma in uPA-deficient animals. Cancer Res. 1996;56:3597–3604. [PubMed] [Google Scholar]
- Shapiro R.L., Duquette J.G., Nunes I., Roses D.F., Harris M.N., Wilson E.L., Rifkin D.B. Urokinase-type plasminogen activator-deficient mice are predisposed to staphylococcal botryomycosis, pleuritis, and effacement of lymphoid follicles. Am. J. Pathol. 1997;150:359–369. [PMC free article] [PubMed] [Google Scholar]
- Stefansson S., Lawrence D.A. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature. 1996;383:441–443. doi: 10.1038/383441a0. [DOI] [PubMed] [Google Scholar]
- Stefansson S., Muhammad S., Cheng X.F., Battey F.D., Strickland D.K., Lawrence D.A. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J. Biol. Chem. 1998;273:6358–6366. doi: 10.1074/jbc.273.11.6358. [DOI] [PubMed] [Google Scholar]
- Tang H., Kerins D.M., Hao Q., Inagami T., Vaughan D.E. The urokinase-type plasminogen activator receptor mediates tyrosine phosphorylation of focal adhesion proteins and activation of mitogen-activated protein kinase in cultured endothelial cells. J. Biol. Chem. 1998;273:18268–18272. doi: 10.1074/jbc.273.29.18268. [DOI] [PubMed] [Google Scholar]
- Trommsdorff M., Borg J.P., Margolis B., Herz J. Interaction of the cytosolic adapter proteins with neuronal apolipoprotein E receptors and the amyloid precursor protein. J. Biol. Chem. 1998;273:33556–33560. doi: 10.1074/jbc.273.50.33556. [DOI] [PubMed] [Google Scholar]
- Tsuboi R., Rifkin D.B. Bimodal relationship between invasion of amniotic membrane and plasminogen activator activity. Int. J. Cancer. 1990;46:56–60. doi: 10.1002/ijc.2910460112. [DOI] [PubMed] [Google Scholar]
- Waltz D.A., Fujita R.M., Wei Y., Chapman H.A. Plasmin and plasminogen activator inhibitor type 1 promote cellular motility by regulating the interaction between the urokinase receptor and vitronectin. J. Clin. Invest. 1997;100:58–67. doi: 10.1172/JCI119521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltz D.A., Fujita R.M., Yang X., Natkin L., Zhuo S., Gerard C.J., Rosenberg S., Chapman H.A. Non-proteolytic role for the urokinase receptor in cellular migration in vivo . Am. J. Respir. Cell Mol. Biol. 2000;22:316–322. doi: 10.1165/ajrcmb.22.3.3713. [DOI] [PubMed] [Google Scholar]
- Weaver A.M., Hussaini I.M., Mazar A., Henkin J., Gonias S.L. Embryonic fibroblasts that are genetically deficient in low density lipoprotein receptor-related protein demonstrate increased activity of the urokinase receptor system and accelerated migration on vitronectin. J. Biol. Chem. 1997;272:14372–14379. doi: 10.1074/jbc.272.22.14372. [DOI] [PubMed] [Google Scholar]
- Webb D.J., Nguyen D.H.D., Sankovic M., Gonias S.L. The very low density lipoprotein receptor regulates urokinase receptor catabolism and breast cancer cell motility in vitro . J. Biol. Chem. 1999;274:7412–7420. doi: 10.1074/jbc.274.11.7412. [DOI] [PubMed] [Google Scholar]
- Webb D.J., Nguyen D.H.D., Gonias S.L. Extracellular signal-regulated kinase functions in the urokinase receptor-dependent pathway by which neutralization of low density lipoprotein receptor-related protein promotes fibrosarcoma cell migration and matrigel invasion. J. Cell Sci. 2000;113:123–134. doi: 10.1242/jcs.113.1.123. [DOI] [PubMed] [Google Scholar]
- Yebra M., Goretzki L., Pfeifer M., Mueller B.M. Urokinase-type plasminogen activator binding to its receptor stimulates tumor cell migration by enhancing integrin-mediated signal transduction. Exp. Cell Res. 1999;250:231–240. doi: 10.1006/excr.1999.4510. [DOI] [PubMed] [Google Scholar]