

Abstract
Mitochondrial membrane fusion is a process essential for the maintenance of the structural integrity of the organelle. Since mitochondria are bounded by a double membrane, they face the challenge of fusing four membranes in a coordinated manner. We provide evidence that this is achieved by coupling of the mitochondrial outer and inner membranes by the mitochondrial fusion machinery. Fzo1, the first known mediator of mitochondrial fusion, spans the outer membrane twice, exposing a short loop to the intermembrane space. The presence of the intermembrane space segment is required for the localization of Fzo1 in sites of tight contact between the mitochondrial outer and inner membranes. Mutations in the intermembrane space domain of yeast Fzo1 relieve the association with the inner membrane. This results in a loss of function of the protein in vivo. We propose that the mitochondrial fusion machinery forms membrane contact sites that mediate mitochondrial fusion. A fusion machinery that is in contact with both mitochondrial membranes appears to be functionally important for coordinated fusion of four mitochondrial membranes.
Keywords: Fzo1, membrane contact sites, membrane fusion, mitochondria, organelle inheritance
Introduction
Intracellular membrane fusion is a process essential for the propagation and maintenance of the compartmental organization of eukaryotic cells (White 1992; Warren and Wickner 1996; Jahn and Südhof 1999). Fusion of mitochondrial membranes has been observed in a great variety of different cell types. These include budding yeast, filamentous fungi, mammalian cells in culture, and various plant cells (Bereiter-Hahn 1990; Bereiter-Hahn and Vöth 1994; Steinberg 1998). In yeast and some mammalian cells, the mitochondrial compartment consists of a branched tubular network, the continuity of which depends on a balanced frequency of fission and fusion events (Nunnari et al. 1997; Hermann and Shaw 1998; Yaffe 1999). Recently, the Fzo protein was identified in Drosophila as the first known mediator of mitochondrial fusion. At a certain developmental stage in spermatid formation, the Fzo protein is required for the fusion of mitochondria to the Nebenkern, a structure composed of two giant mitochondrial derivatives that are wrapped around each other (Hales and Fuller 1997). Although the precise role of Fzo in mitochondrial membrane fusion is not understood at the molecular level, it appears to be a key player in this process. The yeast homologue, Fzo1, is required for the maintenance of the branched mitochondrial network located below the cell cortex (Hermann et al. 1998; Rapaport et al. 1998). Mutants in the FZO1 gene harbor fragmented mitochondria and are respiratory deficient. Fzo1 is a component of a larger protein complex of ∼800 kD located in the mitochondrial outer membrane and possesses an NH2-terminal large cytosolic part containing a GTPase domain (Hermann et al. 1998; Rapaport et al. 1998).
Few protein machineries mediating membrane fusion have so far been characterized at the molecular level. Surprisingly, even unrelated fusion protein complexes that are clearly different in subunit composition share certain structural and mechanistic similarities (Skehel and Wiley 1998; Weber et al. 1998). The organelles of the secretory pathway are connected by a complex network of transport vesicles that bud from donor compartments and fuse with target membranes (heterotypic fusion). Some of these organelles, such as the vacuole or the endoplasmic reticulum, are able to fuse with themselves (homotypic fusion) (Pryer et al. 1992; Rothman 1994; Rothman and Wieland 1996; Pelham 1999; Wickner and Haas 2000). SNAREs are integral membrane proteins of intracellular transport vesicles and their respective target membranes. Cognate SNARE proteins on opposite membranes pair by forming a rod-shaped α-helical bundle. The membrane domains of the SNARE proteins on both membranes are located at the same end of this intermolecular parallel coiled coil (Hanson et al. 1997; Hohl et al. 1998; Sutton et al. 1998). Membrane fusion of enveloped animal viruses with cellular membranes constitutes the first step in the infectious process eventually leading to the transfer of viral genomes into host cells (Wiley and Skehel 1987; Hernandez et al. 1996). Viral fusion proteins, such as hemagglutinin, are integral proteins of viral membranes. Upon activation, a hydrophobic fusion peptide inserts into the target membrane. An extensive rearrangement of the molecule leads to the formation of an antiparallel coiled coil structure, again with both membrane-associated segments at the same end (Hughson 1995; Melikyan and Chernomordik 1997; Skehel and Wiley 2000). Thus, SNARE complexes and viral fusion proteins share two major features: they contain α-helical bundles composed of coiled coils and they have two membrane-associated regions located at the same end of the rod. It is thought that in both cases the formation of the rod structure draws the apposed membranes into close contact. At the same time, the fusion complexes might locally distort the membranes via their transmembrane segments and thereby initiate mixing of lipid bilayers (Skehel and Wiley 1998; Weber et al. 1998).
In contrast to these well characterized membrane fusion mediators, the mitochondrial fusion machinery has to merge four lipid bilayers. To obtain insight into the mechanism of fusion of a double membrane–bounded organelle, we asked whether Fzo1 might play a role in coordinating fusion of the outer and inner membranes. We show that Fzo1 has two transmembrane domains in the mitochondrial outer membrane and exposes a short loop to the intermembrane space. The presence of this loop is important for a location of Fzo1 in sites of intimate contact with the inner membrane. When the association of Fzo1 with the inner membrane is relieved by mutagenesis of the intermembrane space segment, mitochondrial fusion is defective. This suggests that an association of Fzo1 with the inner membrane is required for efficient fusion of the organelles. We propose that the fusion machinery in the outer membrane has to be in contact with components in the inner membrane in order to coordinate fusion of four mitochondrial membranes.
Materials and Methods
Recombinant DNA Techniques and Plasmid Constructions
Standard methods were used for the manipulation of DNA (Sambrook et al. 1989). The FZO1 gene including its own regulatory sequences was amplified from genomic DNA by PCR using Pfu polymerase (Stratagene) and oligonucleotides FZO1-5 (5′-CGC GGA TCC ACT ACC ATC CTT CTA GCC) and FZO1-3 (5′-CGC CTC GAG AAT GTT TAT GTA ATT TCG TGC). The PCR product was cloned into the pCRII-TOPO vector (Invitrogen) according to the manufacturer's instructions yielding plasmid pTOPO-FZO1. The insert was subcloned into the BamHI-XhoI sites of the yeast/Escherichia coli shuttle vectors pRS416 (Sikorski and Hieter 1989) and pRS425 (Christianson et al. 1992) yielding plasmids pRS416-FZO1 and pRS425-FZO1. Both yeast expression plasmids complemented the Δfzo1 deletion mutant.
For construction of plasmids for in vitro transcription/translation of Fzo1–dihydrofolate reductase (DHFR) fusion proteins and Fzo1 fragments, the region of the FZO1 gene encoding amino acids 601–810 was amplified from cloned DNA (pRS416-FZO1) by PCR using oligonucleotides FZO1 (5′-AGA GAA TTC AGA TCT ACC ATG ATT GGA AAA AAT GAA CTT GGT G) and FZO2 (5′-AGA AAG CTT CTA GGA TCC ATC CAT AAT TAT TTC ACA CGA C). The PCR product was cloned into the EcoRI and BamHI sites of plasmid pSu9(1–45)(1–45)-DHFR (Fölsch et al. 1998) to construct pGEM4-Fzo(600–810)-DHFR, and into the EcoRI and HindIII sites of vector pGEM4 (Promega) to construct pGEM4-Fzo(600–810).
For the construction of the fzo1-2 mutant allele, the COOH-terminal 120 codons and the terminator of the FZO1 gene were amplified by PCR using Pfu polymerase and oligonucleotides FZO1-735-N (5′-AAA GAA TTC AGT CGA CCA AGA AGT TAT CAG TTC CG) and FZO1-3 (see above). The PCR product was cloned into the EcoRI-XhoI sites of pBluescript II KS (Stratagene) yielding plasmid pBS-FZO1–C-term. The promoter and the NH2-terminal 733 codons of the FZO1 gene were amplified using oligonucleotides FZO1-5 (see above) and FZO1-733-C (5′-AAA GAA TTC CTG AGC TCC ACG ACG ATA A). The PCR product was cloned into the BamHI-EcoRI sites of pBS-FZO1–C-term yielding plasmid pBS-FZO1. A DNA fragment encoding two copies of the flexible linker segment KL(GGS)3 was amplified from plasmid pJM28-3 (McNew et al. 1999) using oligonucleotides sw-N (5′-AAA GAA TTC AAA GCA GGA AGA AGC TCG) and sw-C (5′-AAA AGT CGA CCC GAT CAT AAG CTT GGA AC). The PCR product was cloned into the EcoRI-SalI sites of pBS-FZO1 yielding plasmid pBS–fzo1-2. The insert was sequenced and subcloned into the BamHI-XhoI sites of the yeast/E. coli shuttle vectors pRS315 (Sikorski and Hieter 1989) and pRS425 yielding plasmids pRS315–fzo1-2 and pRS425–fzo1-2.
Yeast Strains and Construction of fzo1 Mutants
Standard genetic techniques were used for growth and manipulation of yeast strains (Sherman et al. 1986). Transformation of yeast was carried out as described (Gietz et al. 1992). To obtain parental strains for construction of fzo1 mutants, plasmid pRS416-FZO1 was transformed into the Δfzo1 deletion mutant which is rho 0, that is, it lacks mitochondrial DNA (Rapaport et al. 1998). This strain was crossed with its isogenic wild-type strain YPH500 (Sikorski and Hieter 1989). The resulting diploid strain was sporulated and tetrads were dissected. One rho + clone was isolated that carried a disrupted chromosomal allele of FZO1 and the wild-type gene on plasmid pRS416-FZO1. This strain, YBW18, was transformed with pYX232–mitochondria-targeted GFP (mtGFP) (Westermann and Neupert 2000), yielding strain YBW89 expressing mtGFP.
To obtain a strain expressing wild-type FZO1 from a multicopy plasmid (YBW114), YBW89 was transformed with pRS425-FZO1 and subsequently grown on medium containing 5-fluoro-orotic acid (5-FOA) (Boeke et al. 1984) to counterselect against pRS416-FZO1. To obtain mutant strains expressing the Fzo1-2 protein, YBW89 was transformed with pRS315–fzo1-2 and pRS425–fzo1-2, and the resulting strains were grown on 5-FOA, yielding strains YBW110 and YBW183. The Δfzo1 deletion mutant expressing mtGFP (YBW113) was obtained by transformation of YBW89 with the empty vector pRS315 and subsequent growth on 5-FOA. To obtain a strain overexpressing fzo1-2 from a multicopy plasmid in an FZO1 wild-type background (YBW210), YBW183 was transformed with pRS416-FZO1. Strains D273-10B (24657; American Type Culture Collection), YBW114, and YBW183 were used for the preparation of submitochondrial fractions. Mitochondrial morphology, and the fractionation behavior of Fzo1 proteins were identical in strains harboring single copy or multicopy plasmids.
Isolation and Subfractionation of Yeast Mitochondria
Mitochondria were prepared by differential centrifugation as described (Daum et al. 1982). For generation of mitoplasts by hypotonic swelling, mitochondria were resuspended at a concentration of 1 mg/ml in a buffer containing 3% (wt/vol) BSA, 0.5 M sorbitol, 80 mM KCl, 10 mM C4H6MgO4, 2 mM MnCl2, 50 mM Hepes/KOH, pH 7.2, diluted with 9 vol 20 mM Hepes/KOH, pH 7.2, and incubated for 30 min on ice. Protease treatment of mitochondria or mitoplasts was performed for 30 min on ice with the indicated concentrations of proteinase K or trypsin in a buffer containing 250 mM sucrose, 1 mM EDTA, 10 mM MOPS/KOH, pH 7.2, or in swelling buffer. Protease treatment was stopped either by adding 1 mM PMSF followed by 5 min incubation on ice and reisolation of the organelles by centrifugation at 12,000 g for 12 min at 2°C, or by precipitation of proteins with TCA. For carbonate extraction of imported proteins, organelles were resuspended in 0.1 M Na2CO3, 1 mM PMSF, incubated for 30 min on ice, and membranes were pelleted by centrifugation at 226,000 g for 1 h in a Beckman Coulter TLA45 rotor. For carbonate extraction of COOH-terminal Fzo1 fragments, protease-treated mitoplasts were washed two times in a buffer containing 250 mM sucrose, 1 mM EDTA, 1 mM PMSF, 10 mM MOPS/KOH, pH 7.2, resuspended in 0.1 M Na2CO3, and incubated for 30 min on ice. Then, 2.4 M sucrose was added to a final concentration of 1.5 M (final volume 266 μl). This was overlaid with 250 μl 1.4 M sucrose and 200 μl 250 mM sucrose in a buffer containing 1 mM EDTA, 1 mM PMSF, 20 mM Hepes, pH 7.4. The gradient was centrifuged for 4 h at 336,840 g in a Beckman Coulter SW60 rotor at 2°C. Then, 350 μl from the top, 150 μl from the middle, and 216 μl from the bottom of the gradient were harvested and proteins were precipitated with TCA. The pellet of the gradient was dissolved directly in sample buffer, and all fractions were analyzed by SDS-PAGE and Western blotting.
Fractionation of mitochondria with digitonin (Hartl et al. 1986) was performed as follows: 100 μg mitochondria was incubated for 3 min on ice in 20 μl SEMK buffer (250 mM sucrose, 1 mM EDTA, 80 mM KCl, 10 mM MOPS/KOH, pH 7.2) containing 0, 0.125, 0.15, 0.2, 0.3, 0.4, or 0.6% digitonin. Then, 800 μl SEMK buffer containing 500 μg/ml proteinase K was added, and the samples were incubated for 30 min on ice. Protease treatment was stopped by addition of 1 mM PMSF, mitochondria were harvested by centrifugation for 5 min at 17,500 g, and mitochondrial proteins were analyzed by SDS-PAGE and Western blotting.
For separation of submitochondrial membrane fragments, the mitochondrial matrix was first condensed by incubation of mitochondria (17.5 mg/ml) in a hyperosmotic buffer containing 1.2 M sorbitol, 80 mM KCl, 10 mM magnesium acetate, 2 mM MnCl2, 50 mM Hepes/KOH, pH 7.2 for 45 min on ice. Mitochondria were then subjected to hypoosmotic swelling by dilution with 18 vol of 20 mM Hepes/KOH, pH 7.2 and incubation for 30 min at 4°C under agitation. For protease treatment of samples, 100 μg/ml proteinase K was added during swelling. Protease treatment was stopped by the addition of 1 mM PMSF. Submitochondrial membrane vesicles were generated by extensive treatment with a tight fitting Dounce homogenizer for 20 min on ice. For urea treatment of samples, 2 M urea was added at this stage. Mitochondrial membranes were loaded on top of a 1.0–1.5 M sucrose gradient (volume 9 ml) in a buffer containing 2.5 mM EDTA, 100 mM KCl, 20 mM Hepes, pH 7.2. Centrifugation was performed for 16 h at 134,000 g in a Beckman Coulter SW41 rotor at 2°C. The gradient was harvested in 960-μl fractions. Proteins were precipitated with TCA and analyzed by Western blotting.
Miscellaneous Methods
Specific antibodies raised against the COOH-terminal 12 amino acid residues of Fzo1 (Rapaport et al. 1998) were affinity purified according to published procedures (Harlow and Lane 1988). Synthesis of radiolabeled precursor proteins and import into isolated mitochondria were carried out as described (Westermann et al. 1995). Gel filtration was performed as described (Rapaport et al. 1998). Detection of proteins after blotting onto nitrocellulose was performed using the ECL detection system (Amersham Pharmacia Biotech). Standard fluorescence and phase–contrast microscopy were performed as described (Westermann and Neupert 2000).
Results
The COOH Terminus of Fzo1 Faces the Cytosol
Fzo1 is an integral membrane protein of the mitochondrial compartment with its large NH2-terminal domain facing the cytosol (Hermann et al. 1998; Rapaport et al. 1998). To date, there has been no experimental evidence for the location of the COOH terminus. To determine which part(s) of the protein could be in contact with the inner membrane, we examined the topology of the protein.
Yeast Fzo1 as well as the Fzo protein of Drosophila and other related sequences found in the databases contain a predicted transmembrane domain close to the COOH terminus. Hydropathy analysis revealed that this putative transmembrane domain in fact consists of two distinct hydrophobic segments which are separated by a stretch of 6–10 amino acids containing 1–3 positive charges (Fig. 1 A; Hales and Fuller 1997). These predictions suggest three different possibilities. (a) Two neighboring transmembrane domains could span both mitochondrial membranes. In this case, the COOH terminus would be located in the matrix space, as was first suggested by Hales and Fuller 1997. (b) Alternatively, the hydrophobic region could span the mitochondrial outer membrane twice, and the COOH-terminal domain would thus face the cytosol. (c) If there is only one transmembrane domain, the COOH-terminal end of Fzo1 would be located in the intermembrane space.
Figure 1.
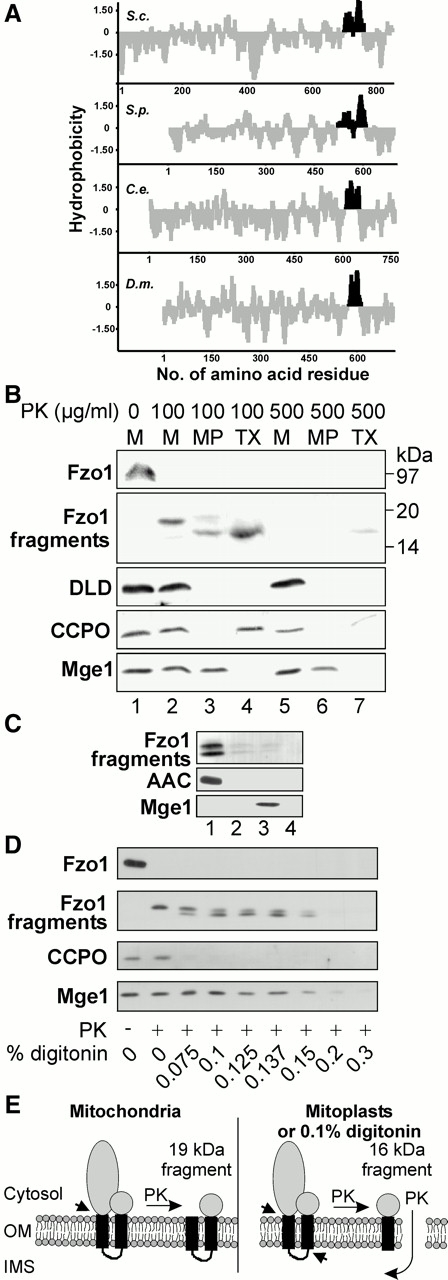
The COOH-terminal end of Fzo1 is exposed to the cytosol. (A) Members of the Fzo1 protein family contain two closely neighboring putative transmembrane segments. The hydrophobicity profile of the Fzo1 proteins of Saccharomyces cerevisiae (Sc) (available from GenBank/EMBL/DDBJ under accession no. Z36048), Schizosaccharomyces pombe (Sp) (CAA19004), Caenorhabditis elegans (Ce) (U29244, ORF14), and Drosophila melanogaster (Dm) (Hales and Fuller 1997) were plotted according to Kyte and Doolittle 1982. Hydrophobic regions are depicted in black. (B) Subfractionation and protease treatment of wild-type mitochondria. Mitochondria (M), mitoplasts (MP), or mitochondria solubilized with Triton X-100 (TX) were treated with the indicated amounts of proteinase K (PK). Then proteins were precipitated with TCA and analyzed by SDS-PAGE and immunoblotting. Fzo1 and COOH-terminal Fzo1 fragments were detected using an antiserum directed against the COOH-terminal 12 amino acid residues. Markers: D-lactate dehydrogenase (DLD), an integral protein of the inner membrane that exposes its major part to the intermembrane space; cytochrome c peroxidase (CCPO), a soluble protein of the intermembrane space; and Mge1, a soluble matrix protein. (C) Carbonate extraction of COOH-terminal Fzo1 fragments. Mitoplasts generated from strain YBW114 were treated with PK and extracted with 0.1 M Na2CO3. Then, membranes were floated in a sucrose gradient. Proteins were harvested from the gradient, precipitated with TCA, and analyzed by immunoblotting. Lane 1, floated membranes; lane 2, middle fraction; lane 3, bottom fraction containing soluble proteins; lane 4, pellet fraction. Markers: AAC, an integral protein of the inner membrane; and Mge1. (D) Digitonin fractionation of mitochondria. Isolated mitochondria of strain YBW114 were treated with the indicated concentrations of digitonin and then incubated in the absence or presence of 500 μg/ml PK. Mitochondria were reisolated by centrifugation and analyzed by immunoblotting as described for A. (E) Topology of Fzo1 and generation of COOH-terminal fragments. Left, topology of Fzo1 in the outer membrane (OM) and generation of the COOH-terminal 19-kD fragment by PK in intact mitochondria. Protease cleavage is indicated by an arrow. Right, generation of the 16-kD fragment when the intermembrane space (IMS) is accessible to protease. Two protease cleavage sites are indicated by arrows.
To discriminate between the different possible topologies, we examined the intramitochondrial location of the COOH terminus of Fzo1. Isolated yeast mitochondria were treated with proteinase K. Using a specific antiserum recognizing the COOH-terminal 12 amino acid residues of Fzo1, a protease-resistant fragment of ∼19 kD was detected by Western blotting (Fig. 1 B, lane 2). The calculated size of a COOH-terminal Fzo1 fragment including both predicted transmembrane domains (amino acid residues 704–855) is 17.4 kD. When mitochondria were converted to mitoplasts by selectively opening the outer membrane by hypotonic swelling, the size of the protected fragment was reduced to ∼16 kD (Fig. 1 B, lane 3). The calculated size of a COOH-terminal fragment including only the second predicted transmembrane domain (amino acid residues 737–855) is 13.8 kD. Thus, the observed reduction in size by ∼3 kD matches well the calculated values, indicating that the smaller fragment was generated because the part between the transmembrane segments became accessible to protease by opening of the outer membrane. When the mitochondrial membranes were solubilized with detergent, the 16-kD fragment was still observed (Fig. 1 B, lane 4). This indicates that the formation of this fragment was due to a protease-resistant conformation, rather than protection by a membrane. When intact mitochondria or mitoplasts were treated with very high amounts of protease, the Fzo1 fragments became protease sensitive (Fig. 1 B, lanes 5 and 6), indicating that the COOH terminus of Fzo1 is exposed to the outside of the organelle. Both fragments fractionated with mitochondrial membranes (see below) and were resistant to alkaline extraction (Fig. 1 C), indicating that they were integral parts of the membrane.
The same results were obtained when mitochondria were subjected to digitonin fractionation. The mitochondrial membranes were sequentially opened by incubation with increasing amounts of digitonin. The organelles were then treated with protease and reisolated by centrifugation. Again, the 19-kD fragment was formed in intact mitochondria, and the 16-kD fragment was generated as soon as the outer membrane was opened. The Fzo1 fragments disappeared from the mitochondrial pellet at digitonin concentrations that were high enough to solubilize also the inner membrane (Fig. 1 D).
We conclude that Fzo1 has an NH2out/COOHout topology in the mitochondrial outer membrane with two membrane-spanning segments that are connected by a short loop in the intermembrane space. Protease treatment of mitochondria removes the large GTPase-containing part of the protein and generates a 19-kD fragment consisting of both transmembrane segments and the COOH-terminal domain. When the intermembrane space is made accessible to protease, a protease-resistant 16-kD fragment is formed that consists of only the second transmembrane segment and the COOH-terminal domain. The topology of Fzo1 in the outer membrane and the generation of the fragments are schematically depicted in Fig. 1 E.
In Vitro Import of an Fzo1-DHFR Fusion Protein
We tested the in vitro import of an Fzo1-DHFR fusion protein as another approach to determine the topology of Fzo1. To construct Fzo(600–810)–DHFR, the NH2-terminal 600 amino acids of Fzo1 were replaced by a methionine, and mouse DHFR was fused to the COOH terminus of the Fzo1 fragment at amino acid residue 810. This chimeric protein includes both predicted transmembrane regions of Fzo1 with flanking regions of ∼100 amino acid residues at the NH2-terminal side and ∼60 residues at the COOH-terminal side (Fig. 2 A). Upon in vitro translation in the presence of [35S]methionine and SDS-PAGE, five distinct bands were observed (Fig. 2 B, lane 1). The apparent molecular mass of the largest band corresponds to the predicted size of the full length fusion protein (45.6 kD). The second band corresponds to an internal translation start at Met679 of Fzo1 (36.5 kD). This residue is located NH2-terminal of the transmembrane domains of Fzo1. The third band corresponds to a translation start at Met763 of Fzo1 (27.2 kD) which is located COOH-terminal of the transmembrane domains. The fourth band corresponds to a translation initiation at Met809 of Fzo1 (21.7 kD), and the fifth band corresponds to an initiation within the DHFR part (20.1 kD). The locations of the translation initiation sites within the fusion protein are schematically depicted in Fig. 2 A.
Figure 2.
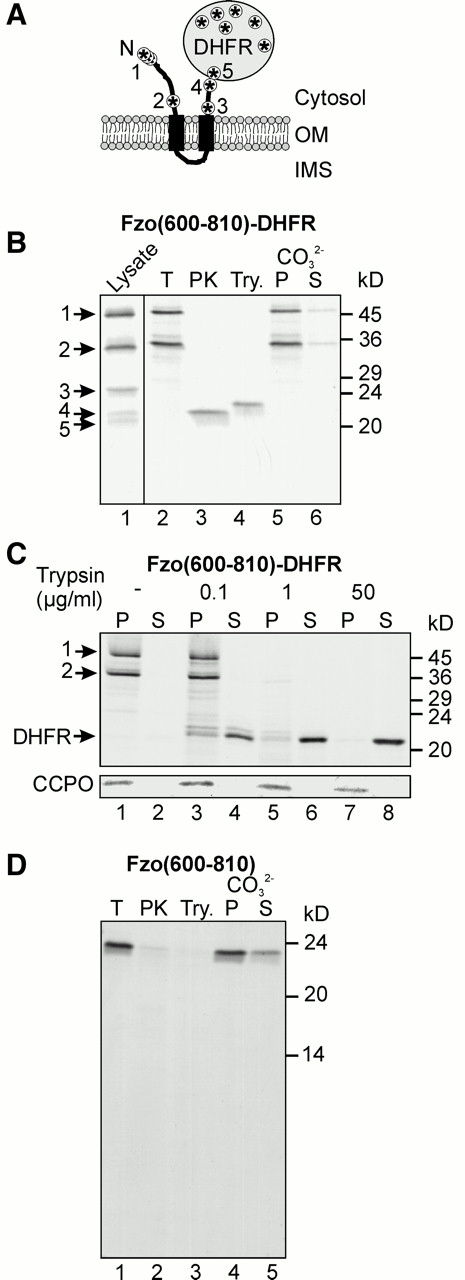
Import of Fzo(600–810)–DHFR and Fzo(600–810) in vitro. (A) Topology of Fzo(600–810)–DHFR in the outer membrane (OM). The distribution of methionine residues in the fusion protein is symbolized by asterisks. The methionines functioning as translation starts are numbered. N, NH2 terminus of the fusion protein. (B) In vitro translation and import of Fzo(600–810)–DHFR. Fzo(600–810)–DHFR was synthesized in reticulocyte lysate in the presence of [35S]methionine (lane 1, Lysate). For in vitro import, the protein was incubated with isolated mitochondria and the organelles were reisolated by centrifugation. Equal aliquots were either left untreated on ice (lane 2, total [T]), treated with proteinase K (lane 3, PK) or trypsin (lane 4, Try.), or were extracted with carbonate (CO3 2−) and separated into pellet (lane 5, P) and supernatant (lane 6, S) fractions. All samples were precipitated with TCA and analyzed by SDS-PAGE and autoradiography. The numbering of the different translation products corresponds to the numbers indicated in A. (C) Localization of the DHFR domain of imported Fzo(600–810)–DHFR. Import was performed as in B, and mitochondria were treated with the indicated concentrations of trypsin. Mitochondria were then sedimented by centrifugation, and pellet (P) and supernatant (S) fractions were precipitated with TCA and analyzed by SDS-PAGE and autoradiography. The size of the DHFR domain is indicated. The intactness of the outer membrane was controlled by immunoblotting using antiserum against cytochrome c peroxidase (CCPO). (D) Import of Fzo(600–810) was performed as described for B.
Upon incubation with isolated mitochondria, only the two largest translation products were bound to mitochondria, whereas the smaller proteins lacking the transmembrane segments did not associate with the organelles (Fig. 2 B, lane 2). Both species containing the transmembrane segments were resistant to extraction with carbonate, indicating insertion of the proteins into the membrane (Fig. 2 B, lanes 5 and 6). When mitochondria were treated with protease after the import reaction, fragments of ∼22 kD were generated (Fig. 2 B, lanes 3 and 4). These correspond to the folded DHFR domain which is known to be present in a protease-resistant conformation (Westermann et al. 1995). To test whether the DHFR moiety of the fusion protein was located on the outside of mitochondria, Fzo(600–810)–DHFR was imported and mitochondria were reisolated and treated with increasing amounts of trypsin. Then, the organelles were sedimented by centrifugation, and pellet and supernatant fractions were analyzed. It was observed that the folded DHFR fragment was released into the supernatant upon trypsin treatment, indicating that it was located on the outside of mitochondria (Fig. 2 C).
To control whether the folded DHFR domain might have prevented translocation of the COOH terminus of the fusion protein across the mitochondrial outer membrane, we imported the Fzo(600–810) fragment without an added DHFR moiety. This fragment was also inserted into the mitochondrial membranes as judged by carbonate extraction (Fig. 2 D, lanes 4 and 5). No protease-resistant fragments were observed after protease treatment, indicating that all parts harboring methionines were exposed to the outside (Fig. 2 D, lanes 2 and 3).
These observations are consistent with an NH2out/COOHout topology of Fzo1 in the mitochondrial outer membrane. The smallest chimeric protein that was efficiently imported was translation product 2; the largest translation product that was not associated with mitochondria was 3 (see Fig. 2 A). Thus, the mitochondrial targeting signal of Fzo1 is apparently contained within a relatively small region flanking the transmembrane domains, presumably between amino acid residues Met679 and Met763.
Fzo1 Is in Peripheral Contact with the Mitochondrial Inner Membrane
It was reported that Fzo1 fractionates in sucrose gradients with submitochondrial membrane fragments of an intermediate density overlapping with, but distinct from, the distribution of marker proteins of the inner and outer membranes (Hermann et al. 1998). The authors concluded that Fzo1 associates with both mitochondrial membranes. Furthermore, they pointed out that this fractionation pattern is characteristic of trapped translocation intermediates that span both mitochondrial membranes at translocation contact sites, as reported by Pon et al. 1989. However, such a double membrane–spanning topology would be in contradiction to an NH2out/COOHout topology of Fzo1. We asked what is the nature of the interaction of Fzo1 with the mitochondrial inner membrane.
Mitochondria were subfractionated by hypotonic swelling, subsequent treatment with a Dounce homogenizer, and sucrose density gradient centrifugation (Daum et al. 1982; Schwaiger et al. 1987; Pon et al. 1989). This procedure produces inner membrane fragments of high density and outer membrane vesicles of low density. The intermediate density fraction contains right side out, sealed inner membranes attached to inside out outer membranes that are mostly leaky, that is, accessible to protease from both sides (Pon et al. 1989). This fraction is thought to contain the contact site proteins that stably connect the mitochondrial inner and outer membranes (Schwaiger et al. 1987; Pon et al. 1989; Brdiczka 1991; Lithgow et al. 1991).
Upon subfractionation according to the standard procedure, the outer membrane protein porin was found in light fractions near the top of the gradient representing outer membrane fragments as well as in heavy fractions near the bottom of the gradient together with the inner membrane protein ADP/ATP carrier (AAC). The partial cofractionation of porin with the AAC can be explained by the fact that these two proteins form complexes at contact sites between the mitochondrial outer and inner membranes (Brdiczka et al. 1998; Crompton et al. 1998; Crompton 1999) or are present in submitochondrial fragments containing both membranes. Fzo1 was recovered exclusively from fractions that contained the inner membrane protein AAC. The outer membrane fragments near the top of the gradient were essentially free of Fzo1 (Fig. 3 A). This indicates that Fzo1 is located in outer membrane fragments that are in association with the inner membrane.
Figure 3.
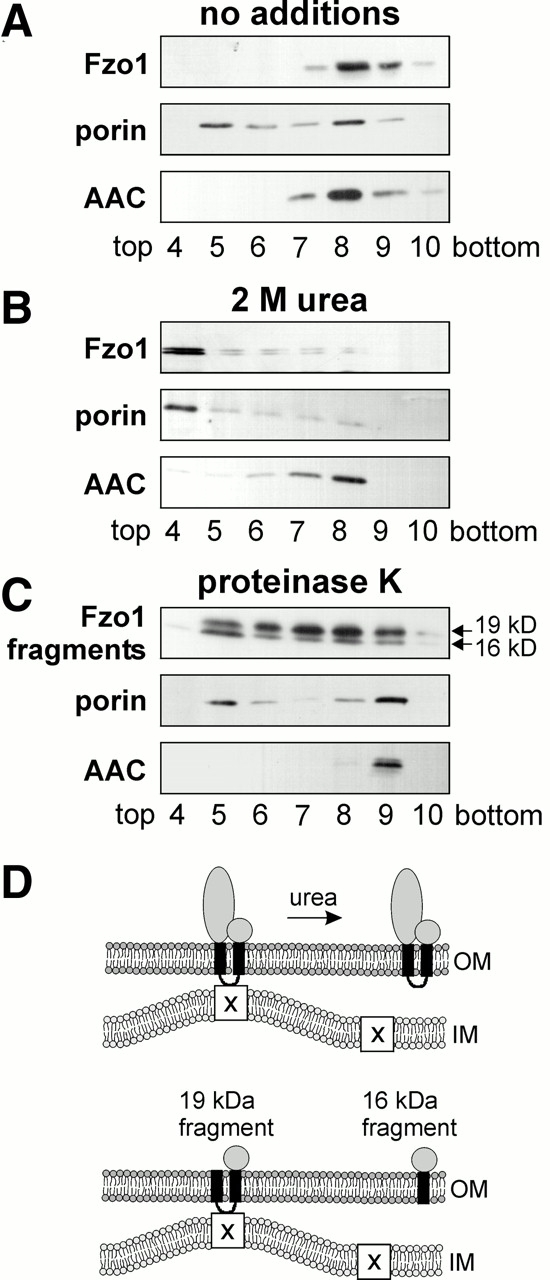
Localization of Fzo1 in contact sites. (A) Association of Fzo1 with the inner membrane. Mitochondrial membrane fragments were generated and separated on a sucrose gradient as described in Materials and Methods. Proteins from fractions 4–10 were precipitated with TCA and analyzed by Western blotting. Porin was used as a marker for the outer membrane, and AAC was used as a marker for the inner membrane. (B) Release of Fzo1 from the inner membrane by aqueous perturbant. Mitochondrial membrane fragments were generated in the presence of 2 M urea and analyzed as in A. (C) Localization of COOH-terminal Fzo1 fragments. Mitochondrial membrane fragments were prepared in the presence of 100 μg/ml proteinase K during the swelling step and analyzed as in A. (D) Location of Fzo1 and COOH-terminal fragments in membrane contact sites. OM, outer membrane; IM, inner membrane; and X, unknown component in the inner membrane.
We further asked whether the association of Fzo1 with the inner membrane is sensitive to protein denaturants. Aqueous perturbants such as urea are known to extract peripheral membrane proteins but not integral membrane proteins from the lipid bilayer (Steck and Yu 1973; Gilmore and Blobel 1985). When mitochondria were homogenized in the presence of 2 M urea, Fzo1 was exclusively recovered from the outer membrane fractions (Fig. 3 B). Under these conditions, the interaction of porin with the inner membrane was also relieved. This result indicates that Fzo1 is peripherally associated with the inner membrane.
To test which part of Fzo1 is responsible for the interaction with the inner membrane, we examined the fractionation of the protease-generated COOH-terminal fragments. The outer membrane of mitochondria was opened by hypotonic swelling, and mitoplasts were treated with protease to generate COOH-terminal Fzo1 fragments. Under the conditions used, both the 19-kD fragment and the 16-kD fragment were generated. After subfractionation of mitochondria, most of the 19-kD fragment was recovered from heavy fractions, similar to the full length protein. In contrast, most of the 16-kD fragment fractionated with the outer membrane (Fig. 3 C), indicating that it did not firmly interact with components of the inner membrane. This suggests that the presence of the intermembrane space segment of Fzo1 is critical for an association with the inner membrane.
We conclude that Fzo1 peripherally interacts with the inner membrane since this interaction can be relieved by urea treatment (Fig. 3 D, top). Furthermore, the association with the inner membrane appears to be mediated by the intermembrane space domain since the presence of this part of Fzo1 correlates with a fractionation with heavy membrane fragments (Fig. 3 D, bottom).
An Intact Intermembrane Space Segment Is Critical for a Tight Association of Fzo1 with the Inner Membrane
To investigate the function of the intermembrane space domain, we inserted a flexible linker segment, (KL[GGS]3)2, between the transmembrane domains of Fzo1. We reasoned that this will grossly alter the structure of the intermembrane space segment without affecting targeting, topology, and complex assembly of the mutant protein. However, the interaction with components in the inner membrane might be severely compromised in such a mutant (Fig. 4 A). The mutant protein, Fzo1-2, was expressed to the same level as the wild-type protein (not shown). Protease treatment of mitochondria generated a COOH-terminal fragment that was ∼3 kD larger than the corresponding fragment of the wild-type protein. Protease treatment of mitoplasts generated the same 16-kD fragment that was observed for the wild-type protein. Both fragments were sensitive to high concentrations of protease. This indicates that the mutant protein acquired the correct topology (Fig. 4 B). Similar to the wild-type protein (Rapaport et al. 1998), the mutant protein migrated at 800 kD in gel filtration, indicating that assembly into a high molecular weight complex was not affected (Fig. 4 C). To test an interaction of the mutant protein with the inner membrane, submitochondrial fragments were separated by density centrifugation. In contrast to the wild-type protein (see Fig. 3 A), the Fzo1-2 protein was not associated with the inner membrane (Fig. 4 D). We cannot formally exclude the possibility that the fzo1-2 mutation causes structural alterations that disrupt the protein's activity directly. However, we consider this possibility unlikely because such alterations would have to be effective in a part of the protein that is located on the other side of the membrane. We conclude that an intact intermembrane space segment is critical for an interaction of Fzo1 with the inner membrane.
Figure 4.
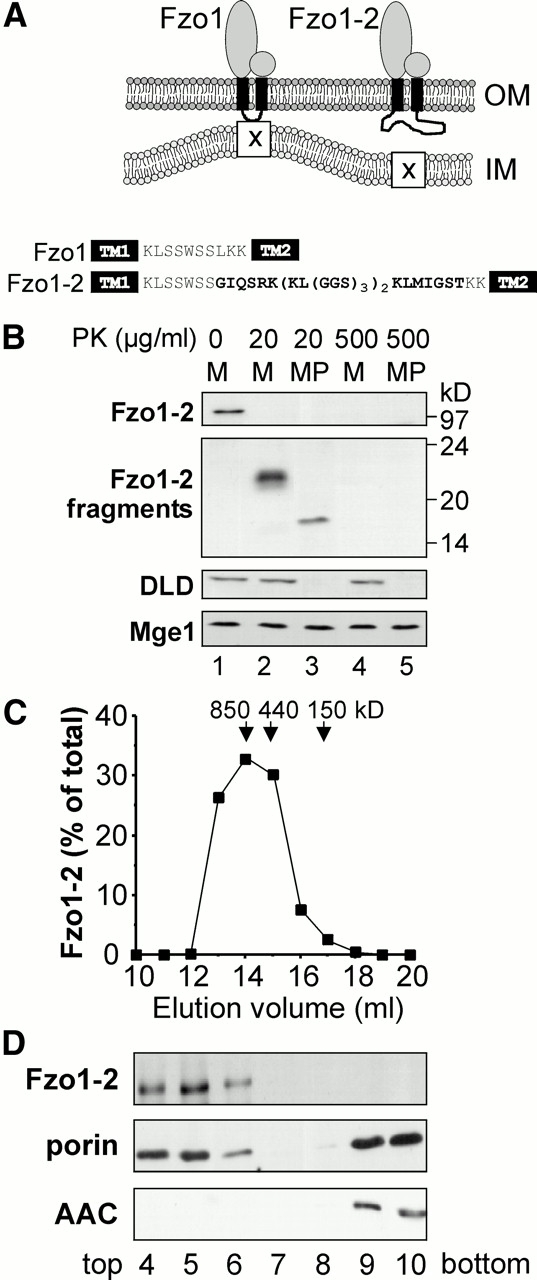
Insertion of a linker between the transmembrane segments of Fzo1 relieves its association with the inner membrane. (A) A mutant version of Fzo1, Fzo1-2, was constructed by insertion of a linker sequence between the transmembrane segments (TM). Depicted is the topology of the mutant protein and the amino acid sequence of the intermembrane space segment. (B) Topology of the Fzo1-2 protein. Mitochondria harboring the Fzo1-2 mutant protein were analyzed as in the legend to Fig. 1 B. (C) Complex assembly of the Fzo1-2 protein. A Triton X-100 extract of fzo1-2 mitochondria was loaded on a Superose-6 column. Fractions were collected and analyzed by immunoblotting and densitometry scanning. The elution peaks and molecular masses of marker proteins are indicated (Hsp60, 850 kD; apoferritin, 440 kD; ADH, 150 kD). (D) Submitochondrial localization of the Fzo1-2 protein. fzo1-2 mitochondria were subfractionated and analyzed as in the legend to Fig. 3 A.
Association of Fzo1 with the Inner Membrane Is Essential for Efficient Mitochondrial Fusion
The fzo1-2 mutant allowed us to investigate whether an interaction of Fzo1 with the inner membrane is important for the function of the protein in vivo. The Δfzo1 deletion mutant harbors fragmented mitochondria because organellar fusion is blocked, whereas mitochondrial fission is still active. Due to their abnormal mitochondrial morphology, Δfzo1 cells are defective in inheritance of mitochondrial DNA and therefore become respiratory deficient (Hermann et al. 1998; Rapaport et al. 1998; Bleazard et al. 1999; Sesaki and Jensen 1999). To test whether the fzo1-2 mutant has a similar phenotype, 10-fold serial dilutions of cell cultures were spotted onto plates containing either the fermentable carbon source glucose (yeast extract–peptone-dextrose [YPD]) or the nonfermentable carbon source glycerol (yeast extract–peptone-glycerol [YPG]). Growth of fzo1-2 cells was clearly affected on YPG medium (Fig. 5 A), indicating that the mutant protein is only partially functional.
Figure 5.
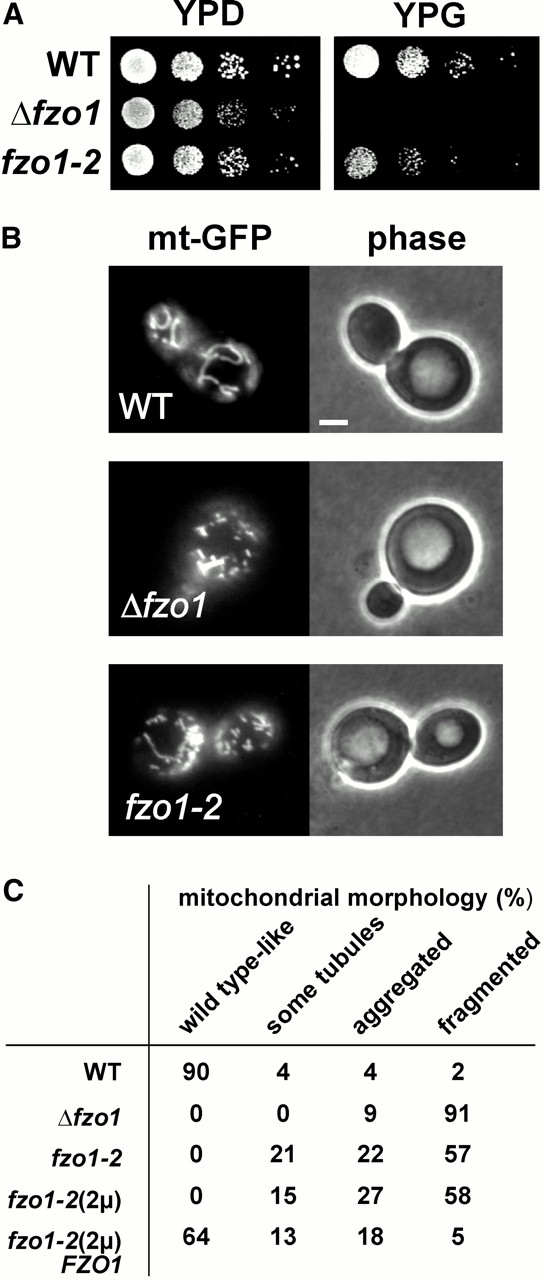
Mutation of the intermembrane space segment compromises the function of Fzo1. (A) Growth phenotype of the fzo1-2 mutant. The fzo1-2 mutant, the Δfzo1 deletion strain, and the isogenic wild type (WT) were grown overnight in liquid minimal medium selective for the maintenance of the plasmid encoding the mutant allele (SD; 2% glucose). Then, 10-fold serial dilutions were spotted onto YPD plates (2% glucose) and YPG plates (3% glycerol). YPD plates were incubated for 3 d at 30°C; YPG plates were incubated for 4 d at 30°C. (B) Mitochondrial morphology of the fzo1-2 mutant. The fzo1-2 mutant, the Δfzo1 strain, and the isogenic wild-type expressing mtGFP were grown overnight in galactose-containing liquid minimal medium (SGal; 2% galactose), selective for maintenance of the plasmid encoding the mutant allele. Living cells were subjected to fluorescence microscopy. Left, the mitochondrial morphology of representative cells is shown; and right, the corresponding phase–contrast images are shown. (C) Quantification of mitochondrial morphology in fzo1 mutants. The following strains were grown to mid-logarithmic growth phase in liquid minimal medium under selection for the plasmids: wild-type (WT; YBW89), Δfzo1 (YBW113), fzo1-2 (YBW117), an Fzo1-2–overexpressing strain (fzo1-2[2μ]; YBW183), and an Fzo1-2–overexpressing strain complemented with a single copy FZO1 wild-type gene (fzo1-2[2μ]FZO1; YBW210). More than 100 cells per culture were examined by fluorescence microscopy and grouped into the following phenotypic classes: wild-type like (mitochondrial reticulum below the cell cortex), some tubules (mostly fragmented mitochondria with a few tubular structures present), aggregated (clustered mitochondrial fragments), and fragmented (evenly distributed mitochondrial fragments). Bar, 2 μm.
Cells expressing mtGFP were grown to mid-logarithmic phase, and mitochondria were visualized in living cells by fluorescence microscopy. Wild-type cells displayed the characteristic tubular mitochondrial network, whereas the Δfzo1 deletion strain harbored highly fragmented mitochondria. The majority of fzo1-2 cells were indistinguishable from the deletion strain, and only few cells contained some tubular mitochondrial structures (Fig. 5B and Fig. C). Quantification of the mitochondrial morphology phenotypes (Fig. 5 C) revealed a significant proportion of fzo1-2 cells containing aggregated mitochondrial fragments, which could represent organellar fusion intermediates. Transformation of an Fzo1-2–overexpressing strain (fzo1-2[2μ]) with a single copy of the FZO1 wild-type gene (fzo1-2[2μ]FZO1) restored formation of the mitochondrial reticulum to a large extent (Fig. 5 C). However, the fact that a relatively large fraction of cells still contained aggregated mitochondria might indicate that the Fzo1-2 protein possibly has some negative effects on mitochondrial fusion even in the presence of the wild-type protein.
We conclude that alteration of the intermembrane space segment of Fzo1 by insertion of a flexible linker sequence severely affects the function of the protein. Since this segment appears to be critical for an interaction with the inner membrane, the mutant phenotype can be ascribed to a lack of such an interaction. We propose that coupling of the outer and inner membranes by the mitochondrial fusion machinery is critical for efficient mitochondrial fusion.
Discussion
Membrane fusion is a process of fundamental importance for life of eukaryotic cells. During evolution, Nature has developed several nonrelated proteinaceous machineries to overcome the energy barriers for fusion of lipid bilayers. These fusion machineries must dock the apposing membranes, draw them into close proximity, and finally mediate mixing of lipid bilayers via transmembrane segments or fusion peptides which are inserted in the membranes. As double membrane–bounded organelles, mitochondria are faced with the unique challenge of fusing four membranes. How is this accomplished? The simplest solution to this problem is a machinery that fuses the mitochondrial outer and inner membranes at the same time in a coordinated manner. Alternatively, inner membrane fusion might be uncoupled from outer membrane fusion. In this case, an independent fusion machinery in the inner membrane would initiate docking and fusion after merging of the outer membranes is complete. The results reported here provide evidence to discriminate between these possibilities.
We established by three independent lines of evidence that Fzo1 is a bona fide outer membrane protein. The biochemical subfractionation shows that the protein has an NH2out/COOHout topology in the outer membrane. This was confirmed by analysis of import of Fzo-DHFR fusion proteins. Moreover, endogenous Fzo1 fractionates exclusively with outer membrane vesicles upon urea treatment. These results exclude the possibility that Fzo spans both mitochondrial membranes as originally suggested by Hales and Fuller 1997. Thus, only the intermembrane space segment is available for an interaction with inner membrane components. Even though we cannot exclude the possibility that additional factors are involved, we consider it likely that it interacts directly with the inner membrane. Its presence is required for a cofractionation of COOH-terminal Fzo1 fragments with the inner membrane, and this interaction is relieved in a mutant, fzo1-2, containing an alteration in the loop exposed to the intermembrane space. Most importantly, using this mutant we show that a connection of the Fzo1 complex to the inner membrane not only exists, but is required for function. This strongly suggests that a connection of the outer and inner membranes is mechanistically important for mitochondrial fusion.
A hypothetical model for the mechanism of mitochondrial fusion is presented in Fig. 6. The first steps of membrane fusion might be not very much different from the mechanisms proposed for SNARE- or hemagglutinin-mediated membrane fusion (Skehel and Wiley 1998; Weber et al. 1998). First, parts of the fusion machinery exposed to the cytosol could dock the apposing outer membranes of two mitochondria approaching each other. Next, assembly of a fusion complex, possibly together with inter- or intramolecular rearrangements, would lead to outer membrane lipid mixing. These initial steps would correspond to the formation of the membrane-bound α-helical rod-like structures of viral fusion proteins or SNARE complexes (Skehel and Wiley 1998). They might well be reversible, as is for example the fusion pore opening in exocytosis (Zimmerberg et al. 1993). The following reactions will be unique to double membrane–bounded organelles. After fusion of the outer membranes, a coupling of the mitochondrial outer and inner membranes by a fusion machinery located in contact sites might be required to initiate fusion of the inner membranes. This would obviate a new round of docking and drawing membranes into apposition by an inner membrane fusion machinery because these components would be already ideally positioned for fusion. It is conceivable that an uncoupling of the membranes would make inner membrane lipid mixing significantly less efficient, and would therefore favor the backwards reaction. This prediction is in good accordance with the observed fragmentation of mitochondria in the fzo1-2 mutant. Finally, separation of the fused inner membranes and mixing of matrix contents would complete the organellar fusion process.
Figure 6.
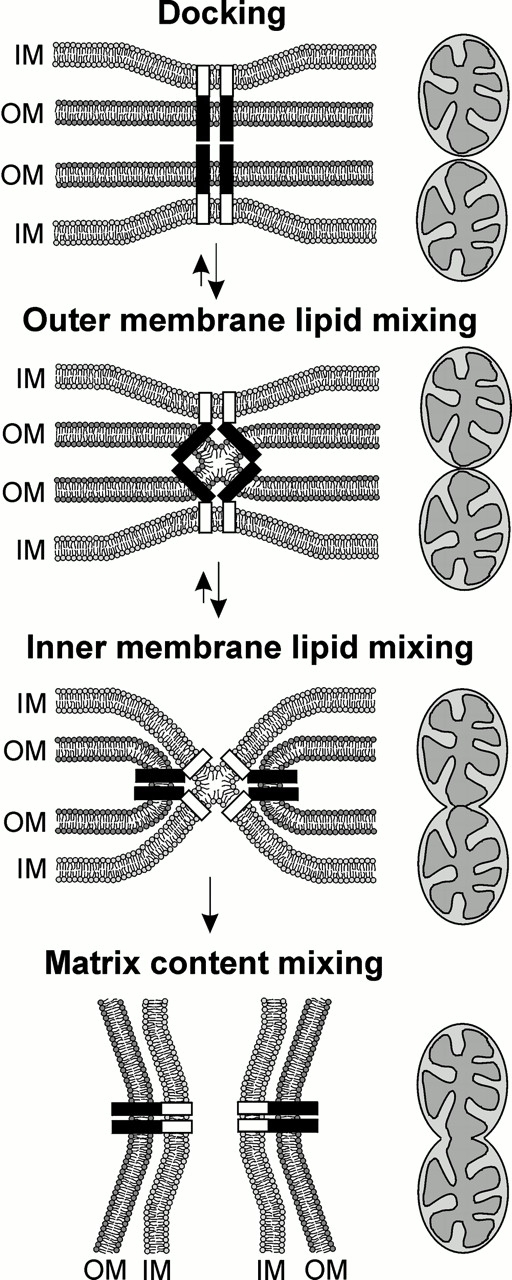
Hypothetical model of mitochondrial fusion mediated by a fusion machinery located in contact sites. Coordinated fusion of mitochondria might be achieved by a mechanism which requires tight contact between the outer membranes (OM) and inner membranes (IM) (see text for details). The fusion complex in the outer membrane is depicted in black; and putative interaction partners in the inner membrane are indicated by white boxes.
From electron microscopic studies, it has been known for a long time that mitochondria possess relatively stable zones of close apposition of the outer and inner membranes with the membranes being still separated from each other (Hackenbrock 1968; van der Klei et al. 1994; Perkins et al. 1997; Nicastro et al. 2000). Several different functions were ascribed to these mitochondrial membrane contact sites. These include protein translocation across the mitochondrial membranes (Schleyer and Neupert 1985; Schwaiger et al. 1987; Pon et al. 1989; Rassow et al. 1989; Donzeau et al. 2000), intramitochondrial translocation of phospholipids (Simbeni et al. 1991), and energy metabolism (Brdiczka et al. 1990, Brdiczka et al. 1998; Rojo et al. 1991). However, it should be pointed out that the contact sites involved in these different processes presumably comprise different protein components (Brdiczka 1991; Pfanner et al. 1992). Consistent with this notion, a fraction of the outer membrane protein porin remained in contact with the inner membrane in the fzo1-2 mutant (Fig. 4 D). Interestingly, observations with the electron microscope provided evidence that mitochondrial fusion is initiated at sites of close apposition of the outer and inner membranes (Bereiter-Hahn and Vöth 1994). The localization of Fzo1 in contact sites reported here provides a biochemical basis for these morphological observations. We propose that the mitochondrial fusion machinery forms contact sites mediating mitochondrial membrane fusion.
At present, the precise role of Fzo1 in the complex process of mitochondrial fusion is unclear. One possibility is that Fzo1 is a regulatory factor that recruits fusion proteins in the outer and inner membranes to the sites of membrane fusion in contact sites. Alternatively, it might play a direct role in the fusion of the outer membranes. Fzo1 possesses all structural elements that are predicted to be required for membrane fusion. It contains several putative coiled coil regions that could be involved in protein–protein interactions, the GTPase domain might provide energy to overcome the activation energy barrier of membrane fusion, and the transmembrane domains might mechanistically couple this energy to lipid bilayer mixing. In this case, the intermembrane space segment might well be important for coordinating outer membrane fusion with putative factors fusing the inner membranes. It will be a major challenge for the future to identify the interaction partners of Fzo1 and to analyze the function of Fzo1 in a reconstituted system.
Acknowledgments
The excellent technical assistance of Gabi Ludwig and Petra Heckmeyer is gratefully acknowledged. We thank Drs. Johannes Herrmann and Michael Brunner for helpful discussions, and Hanno Wolters for his contributions to some experiments. We thank Dr. James A. McNew for plasmid pJM28-3.
Research was supported by the Deutsche Forschungsgemeinschaft (grant WE 2174/2-1) and the Fonds der Chemischen Industrie.
Footnotes
Abbreviations used in this paper: AAC, ADP/ATP carrier; DHFR, dihydrofolate reductase; 5-FOA, 5-fluoro-orotic acid; GFP, green fluorescent protein; mtGFP, mitochondria-targeted GFP; YPD, yeast extract–peptone-dextrose; YPG, yeast extract–peptone-glycerol.
References
- Bereiter-Hahn J. Behavior of mitochondria in the living cell. Int. Rev. Cytol. 1990;122:1–63. doi: 10.1016/s0074-7696(08)61205-x. [DOI] [PubMed] [Google Scholar]
- Bereiter-Hahn J., Vöth M. Dynamics of mitochondria in living cellsshape changes, dislocations, fusion, and fission of mitochondria. Microsc. Res. Tech. 1994;27:198–219. doi: 10.1002/jemt.1070270303. [DOI] [PubMed] [Google Scholar]
- Bleazard W., McCaffery J.M., King E.J., Bale S., Mozdy A., Tieu Q., Nunnari J., Shaw J. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1999;1:298–304. doi: 10.1038/13014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeke J.D., LaCroute F., Fink G.R. A positive selection for mutants lacking orotidine-5-phosphate decarboxylase activity in yeast5-fluoro-orotic acid resistance. Mol. Gen. Genet. 1984;197:345–346. doi: 10.1007/BF00330984. [DOI] [PubMed] [Google Scholar]
- Brdiczka D. Contact sites between mitochondrial envelope membranesstructure and function in energy- and protein-transfer. Biochim. Biophys. Acta. 1991;1071:291–312. doi: 10.1016/0304-4157(91)90018-r. [DOI] [PubMed] [Google Scholar]
- Brdiczka D., Bücheler K., Kottke M., Adams V., Nalam V.K. Characterization and metabolic function of mitochondrial contact sites. Biochim. Biophys. Acta. 1990;1018:234–238. doi: 10.1016/0005-2728(90)90256-4. [DOI] [PubMed] [Google Scholar]
- Brdiczka D., Beutner G., Ruck A., Dolder M., Wallimann T. The molecular structure of mitochondrial contact sites. Their role in regulation of energy metabolism and permeability transition. Biofactors. 1998;8:235–242. doi: 10.1002/biof.5520080311. [DOI] [PubMed] [Google Scholar]
- Christianson T.W., Sikorski R.S., Dante M., Shero J.H., Hieter P. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992;110:119–122. doi: 10.1016/0378-1119(92)90454-w. [DOI] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Crompton M., Virji S., Ward J.M. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore. Eur. J. Biochem. 1998;258:729–735. doi: 10.1046/j.1432-1327.1998.2580729.x. [DOI] [PubMed] [Google Scholar]
- Daum G., Böhni P.C., Schatz G. Import of proteins into mitochondriacytochrome b2 and cytochrome c peroxidase are located in the intermembrane space of yeast mitochondria. J. Biol. Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- Donzeau M., Kaldi K., Adam A., Paschen S., Guiard B., Bauer M.F., Neupert W., Brunner M. Tim23 links inner and outer mitochondrial membranes. Cell. 2000;101:401–412. doi: 10.1016/s0092-8674(00)80850-8. [DOI] [PubMed] [Google Scholar]
- Fölsch H., Gaume B., Brunner M., Neupert W., Stuart R.A. C- to N-terminal translocation of preproteins into mitochondria. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6508–6515. doi: 10.1093/emboj/17.22.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz D., Jean A.S., Woods R.A., Schiestl R.H. Improved method for high efficiency transformation of intact yeast cells. Nucleic Acids Res. 1992;20:1425. doi: 10.1093/nar/20.6.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore R., Blobel G. Translocation of secretory proteins across the microsomal membrane occurs through an environment accessible to aqueous perturbants. Cell. 1985;42:497–505. doi: 10.1016/0092-8674(85)90107-2. [DOI] [PubMed] [Google Scholar]
- Hackenbrock C.R. Chemical and physical fixation of isolated mitochondria in low-energy and high-energy states. Proc. Natl. Acad. Sci. USA. 1968;61:598–605. doi: 10.1073/pnas.61.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales K.G., Fuller M.T. Developmentally regulated mitochondrial fusion mediated by a conserved, novel, predicted GTPase. Cell. 1997;90:121–129. doi: 10.1016/s0092-8674(00)80319-0. [DOI] [PubMed] [Google Scholar]
- Hanson P.I., Roth R., Morisaki H., Jahn R., Heuser J.E. Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell. 1997;90:523–535. doi: 10.1016/s0092-8674(00)80512-7. [DOI] [PubMed] [Google Scholar]
- Harlow E., Lane D. AntibodiesA Laboratory Manual 1988. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: pp. 725 pp [Google Scholar]
- Hartl F.-U., Schmidt B., Wachter E., Weiss E., Neupert W. Transport into mitochondria and intramitochondrial sorting of the Fe/S protein of ubiquinol-cytochrome c reductase. Cell. 1986;47:939–951. doi: 10.1016/0092-8674(86)90809-3. [DOI] [PubMed] [Google Scholar]
- Hermann G.J., Shaw J.M. Mitochondrial dynamics in yeast. Annu. Rev. Cell Dev. Biol. 1998;14:265–303. doi: 10.1146/annurev.cellbio.14.1.265. [DOI] [PubMed] [Google Scholar]
- Hermann G.J., Thatcher J.W., Mills J.P., Hales K.G., Fuller M.T., Nunnari J., Shaw J.M. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J. Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez L.D., Hoffman L.R., Wolfsberg T.G., White J.M. Virus-cell and cell-cell fusion. Annu. Rev. Cell Dev. Biol. 1996;12:627–661. doi: 10.1146/annurev.cellbio.12.1.627. [DOI] [PubMed] [Google Scholar]
- Hohl T.M., Parlati F., Wimmer C., Rothman J.E., Söllner T., Engelhardt H. Arrangement of subunits in 20 S particles consisting of NSF, SNAPs, and SNARE complexes. Mol. Cell. 1998;2:539–548. doi: 10.1016/s1097-2765(00)80153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughson F.M. Structural characterization of viral fusion proteins. Curr. Biol. 1995;5:265–274. doi: 10.1016/s0960-9822(95)00057-1. [DOI] [PubMed] [Google Scholar]
- Jahn R., Südhof T.C. Membrane fusion and exocytosis. Annu. Rev. Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Lithgow T., Timms M., Hoi P.B., Hoogengraad N.J. Identification of a GTP-binding protein in the contact sites between inner and outer mitochondrial membranes. Biochem. Biophys. Res. Commun. 1991;180:1453–1459. doi: 10.1016/s0006-291x(05)81359-2. [DOI] [PubMed] [Google Scholar]
- McNew J.A., Weber T., Engelman D.M., Söllner T.H., Rothman J.E. The length of the flexible SNAREpin juxtamembrane region is a critical determinant of SNARE-dependent fusion. Mol. Cell. 1999;4:415–421. doi: 10.1016/s1097-2765(00)80343-3. [DOI] [PubMed] [Google Scholar]
- Melikyan G.B., Chernomordik L.V. Membrane rearrangements in fusion mediated by viral proteins. Trends Microbiol. 1997;5:349–355. doi: 10.1016/S0966-842X(97)01107-4. [DOI] [PubMed] [Google Scholar]
- Nicastro D., Frangakis A.S., Typke D., Baumeister W. Cryo-electron tomography of Neurospora mitochondria. J. Struct. Biol. 2000;129:48–56. doi: 10.1006/jsbi.1999.4204. [DOI] [PubMed] [Google Scholar]
- Nunnari J., Marshall W.F., Straight A., Murray A., Sedat J.W., Walter P. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell. 1997;8:1233–1242. doi: 10.1091/mbc.8.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelham H.R.B. SNAREs and the secretory pathway—lessons from yeast. Exp. Cell Res. 1999;247:1–8. doi: 10.1006/excr.1998.4356. [DOI] [PubMed] [Google Scholar]
- Perkins G., Renken C., Martone M.E., Young S.J., Ellisman M., Frey T. Electron tomography of neuronal mitochondriathree-dimensional structure and organization of christae and membrane contacts. J. Struct. Biol. 1997;119:260–272. doi: 10.1006/jsbi.1997.3885. [DOI] [PubMed] [Google Scholar]
- Pfanner N., Rassow J., van der Klei I., Neupert W. A dynamic model of the mitochondrial protein import machinery. Cell. 1992;68:999–1002. doi: 10.1016/0092-8674(92)90069-o. [DOI] [PubMed] [Google Scholar]
- Pon L., Moll T., Vestweber D., Marshallsay B., Schatz G. Protein import into mitochondriaATP-dependent protein translocation activity in a submitochondrial fraction enriched in membrane contact sites and specific proteins. J. Cell Biol. 1989;109:2603–2616. doi: 10.1083/jcb.109.6.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryer N.K., Wuestehube L.J., Schekman R. Vesicle-mediated protein sorting. Annu. Rev. Biochem. 1992;61:471–516. doi: 10.1146/annurev.bi.61.070192.002351. [DOI] [PubMed] [Google Scholar]
- Rapaport D., Brunner M., Neupert W., Westermann B. Fzo1p is a mitochondrial outer membrane protein essential for the biogenesis of functional mitochondria in Saccharomyces cerevisiae . J. Biol. Chem. 1998;273:20150–20155. doi: 10.1074/jbc.273.32.20150. [DOI] [PubMed] [Google Scholar]
- Rassow J., Guiard B., Wienhues U., Herzog V., Hartl F.-U., Neupert W. Translocation arrest by reversible folding of a precursor protein imported into mitochondria. A means to quantitate translocation contact sites. J. Cell Biol. 1989;109:1421–1428. doi: 10.1083/jcb.109.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojo M., Hovius R., Demel R.A., Nicolay K., Wallimann T. Mitochondrial creatine kinase mediates contact formation between mitochondrial membranes. J. Biol. Chem. 1991;266:20290–29295. [PubMed] [Google Scholar]
- Rothman J.E. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Rothman J.E., Wieland F.T. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Molecular CloningA Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Schleyer M., Neupert W. Transport of proteins into mitochondriatranslocational intermediates spanning contact sites between outer and inner membranes. Cell. 1985;43:339–350. doi: 10.1016/0092-8674(85)90039-x. [DOI] [PubMed] [Google Scholar]
- Schwaiger M., Herzog V., Neupert W. Characterization of translocation contact sites involved in the import of mitochondrial proteins. J. Cell Biol. 1987;105:235–246. doi: 10.1083/jcb.105.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H., Jensen R.E. Division versus fusionDnm1p and Fzo1p antagonistically regulate mitochondrial shape. J. Cell Biol. 1999;147:699–706. doi: 10.1083/jcb.147.4.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F., Fink G.R., Hicks J. Methods in Yeast GeneticsA Laboratory Course Cold Spring Harbor Laboratory Press ColdSpring Harbor, NY. 198 pp1986. [Google Scholar]
- Sikorski R.S., Hieter P. A system of shuttle vectors and host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae . Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simbeni R., Pon L., Zinser E., Paltauf F., Daum G. Mitochondrial membrane contact sites of yeast. J. Biol. Chem. 1991;266:10047–10049. [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Coiled coils in both intracellular vesicle and viral membrane fusion. Cell. 1998;95:871–874. doi: 10.1016/s0092-8674(00)81710-9. [DOI] [PubMed] [Google Scholar]
- Skehel J.J., Wiley D.C. Receptor binding and membrane fusion in virus entrythe influenza hemagglutinin. Annu. Rev. Biochem. 2000;69:531–569. doi: 10.1146/annurev.biochem.69.1.531. [DOI] [PubMed] [Google Scholar]
- Steck T.L., Yu J. Selective solubilization of proteins from red blood cell membranes by protein perturbants. J. Supramol. Struct. 1973;1:220–248. doi: 10.1002/jss.400010307. [DOI] [PubMed] [Google Scholar]
- Steinberg G. Organelle transport and molecular motors in fungi. Fungal Genet. Biol. 1998;24:161–177. doi: 10.1006/fgbi.1998.1058. [DOI] [PubMed] [Google Scholar]
- Sutton R.B., Fasshauer D., Jahn R., Brunger A.T. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- van der Klei I.J., Veenhuis M., Neupert W. A morphological view on mitochondrial protein targeting. Microsc. Res. Tech. 1994;27:284–293. doi: 10.1002/jemt.1070270404. [DOI] [PubMed] [Google Scholar]
- Warren G., Wickner W. Organelle inheritance. Cell. 1996;84:395–400. doi: 10.1016/s0092-8674(00)81284-2. [DOI] [PubMed] [Google Scholar]
- Weber T., Zemelman B.V., McNew J.A., Westermann B., Gmachl M., Parlati F., Söllner T.H., Rothman J.E. SNAREpinsminimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Westermann B., Neupert W. Mitochondria-targeted green fluorescent proteinsconvenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae . Yeast. 2000;16:1421–1427. doi: 10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Westermann B., Prip-Buus C., Neupert W., Schwarz E. The role of the GrpE homologue, Mge1p, in mediating protein import and folding in mitochondria. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:3452–3460. doi: 10.1002/j.1460-2075.1995.tb07351.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.M. Membrane fusion. Science. 1992;258:917–924. doi: 10.1126/science.1439803. [DOI] [PubMed] [Google Scholar]
- Wickner W., Haas A. Yeast homotypic vacuole fusiona window on organelle trafficking mechanisms. Annu. Rev. Biochem. 2000;69:247–275. doi: 10.1146/annurev.biochem.69.1.247. [DOI] [PubMed] [Google Scholar]
- Wiley D.C., Skehel J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987;56:365–394. doi: 10.1146/annurev.bi.56.070187.002053. [DOI] [PubMed] [Google Scholar]
- Yaffe M.P. The machinery of mitochondrial inheritance and behavior. Science. 1999;283:1493–1497. doi: 10.1126/science.283.5407.1493. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J., Vogel S.S., Chernomordik L.V. Mechanisms of membrane fusion. Annu. Rev. Biophys. Biomol. Struct. 1993;22:433–466. doi: 10.1146/annurev.bb.22.060193.002245. [DOI] [PubMed] [Google Scholar]