

Abstract
In light chain (LC) amyloidosis an immunoglobulin LC assembles into fibrils that are deposited in various tissues. Little is known about how these fibrils form in vivo. We previously showed that a known amyloidogenic LC, SMA, can give rise to amyloid fibrils in vitro when a segment of one of its β sheets undergoes a conformational change, exposing an Hsp70 binding site. To examine SMA aggregation in vivo, we expressed it and its wild-type counterpart, LEN, in COS cells. While LEN is rapidly oxidized and subsequently secreted, newly synthesized SMA remains in the reduced state. Most SMA molecules are dislocated out of the ER into the cytosol, where they are ubiquitinylated and degraded by proteasomes. A parallel pathway for molecules that are not degraded is condensation into perinuclear aggresomes that are surrounded by vimentin-containing intermediate filaments and are dependent upon intact microtubules. Inhibition of proteasome activity shifts the balance toward aggresome formation. Intracellular aggregation is decreased and targeting to proteasomes improved by overexpression of the cytosolic chaperone Hsp70. Importantly, transduction into the cell of an Hsp70 target peptide, derived from the LC sequence, also reduces aggresome formation and increases SMA degradation. These results demonstrate that an amyloidogenic LC can aggregate intracellularly despite the common presentation of extracellular aggregates, and that a similar molecular surface mediates both in vitro fibril formation and in vivo aggregation. Furthermore, rationally designed peptides can be used to suppress this aggregation and may provide a feasible therapeutic approach.
Keywords: amyloidosis, BiP, fibril assembly, immunoglobulin, proteasome
Introduction
Amyloidosis is a family of diseases characterized by the assembly of a particular protein into fibrils that are deposited in various organs and tissues, often resulting in organ failure and death. Each type of amyloidosis is defined by the constituent protein, and >20 different proteins are known to be able to polymerize into fibrils (Westermark et al. 1999). This group of proteins does not share a common native structure. Rather, they have in common the ability to undergo a conformational change into a partially folded intermediate that is capable of ordered assembly. The molecular mechanisms governing this transition have been studied primarily in vitro and only few studies have related the in vitro findings to cell physiology and pathological manifestations.
Of the many proteins known to aggregate in fibrillar form, immunoglobulin light chains (LCs), the causative agents of light chain amyloidosis, are unique: they are designed to be highly polymorphic as a way of maximizing the diversity of the immune repertoire. By contrast, many other amyloidogenic proteins contain specific, often heritable mutations that explain their increased tendency to shift from the native state to a fibrillogenic form (Perutz 1994; Zoghbi and Orr 1995; Kelly et al. 1997). Immunoglobulin LCs are encoded by multiple gene segments that rearrange in combinatorial fashion, and the variable domain (VL) gene segments also undergo somatic hypermutation. As an inevitable consequence of this sequence diversification, LCs are produced that are inherently unstable and prone to form fibrils. In fact, as little as one or two amino acid substitutions have been found to suffice for the in vitro conversion of a soluble LC to an amyloidogenic variant (Raffen et al. 1999; Davis et al. 2000).
Typically, LC amyloid deposits are extracellular, and only rarely do patients present with intracellular LC aggregates (Kjeldsberg et al. 1977; Ishihara et al. 1991). This observation is puzzling from a biological perspective because proteins with altered conformations or unassembled subunits of oligomeric proteins usually do not transit from the ER to the Golgi. Rather, they are retained in the ER (Bole et al. 1986; Gething et al. 1986), dislocated to the cytosol, and targeted for degradation by proteasomes (Werner et al. 1996). LC mutants that fit this pattern have been described (Ma et al. 1990; Gardner et al. 1993). A second coping mechanism that was recently reported involves the extraction of misfolded integral membrane proteins from the ER membrane, followed by their aggregation into a perinuclear inclusion body (Johnston et al. 1998). With either mechanism, the question remains how an unstable mutant LC might escape the cellular quality control system, reach the extracellular space, and form fibrils.
Certain LC families, such as κ4 and λ6, have been shown to be overrepresented in LC deposits (Solomon et al. 1982; Denoroy et al. 1994; Stevens and Argon 1999). Earlier work from our laboratories has focused on the in vitro fiber-forming capability of two members of the κ4 family, LEN and SMA. LEN differs from the germline sequence by only one somatic mutation, and was isolated as a Bence Jones protein from a patient with no evidence of amyloid deposits (Wilkins Stevens et al. 1995). SMA has eight amino acid substitutions relative to LEN and was isolated from a patient with LC amyloidosis (Wilkins Stevens et al. 1995). In vitro, LEN is a thermodynamically stable protein. SMA, however, is less thermodynamically stable than LEN (Raffen et al. 1999). Furthermore, we have shown that the purified, recombinant VL of SMA forms fibers upon incubation with shaking at 37°C (Raffen et al. 1999; Davis et al. 2000). These results support the hypothesis that a conformational change in the protein is a prerequisite for fiber formation.
Based on our in vitro studies, we have proposed a molecular model for LC fiber assembly (Davis et al. 2000). The identification of a VL-derived peptide that prevents fiber formation suggested that an intermolecular loop-swapping event is important for fiber growth. The peptide is hypothesized to serve as a competitive inhibitor of fiber formation by binding to a crevice in the VL vacated by a mobile loop containing the peptide. The physiological relevance of these in vitro experiments is currently unknown.
To begin to address these questions in vivo, we have expressed the complete LC of SMA or LEN in COS cells and examined their fates. Our data show that an amyloidogenic protein can aggregate intracellularly. We find that, unlike LEN, SMA is slow to fold and is readily recognized as an unstable protein. It is dislocated into the cytosol, where it either aggregates into inclusion bodies or is degraded by proteasomes. The chaperone Hsp70 interacts with SMA in the cytosol and plays an important role in its fate. Overexpression of Hsp70 improves the solubility of SMA and decreases its aggregation. Furthermore, we find that the same molecular surface required for fibril formation in vitro is important for aggregation in vivo. The same Hsp70-binding peptide that we showed to inhibit fibril assembly in vitro prevents aggregation of SMA in cells. This ability to modulate the degree of intracellular LC aggregation with chaperones and peptides suggests that rationally designed peptides may provide effective treatment modalities for a variety of amyloidoses.
Materials and Methods
Plasmids
Constructs directing expression of the complete SMA and LEN LC, wild-type BiP and T19G BiP have been described elsewhere (Davis et al. 2000). An Hsp70 expression plasmid was constructed by PCR amplification of the inducible gene from the pETWThsp70 vector (Abravaya et al. 1992), a gift of Dr. R. Morimoto (Northwestern University, Evanston, IL). Amplified material was inserted into the vector pcDNA3.1 (Invitrogen). An epitope-tagged derivative of this plasmid was generated by an in-frame insertion of the hemagglutinin (HA) peptide sequence (YPYDYPDYA) at the NH2 terminus of Hsp70. An expression plasmid encoding HA-tagged serum and glucocorticoid-inducible kinase (SGK) was a gift of Dr. Suzanne Conzen (The University of Chicago).
Transfections
COS-1 cells were transiently transfected with FuGENE 6 transfection reagent (Roche Molecular Biochemicals) according to manufacturer's instructions, and analyzed 2 d later. Overnight incubations with various concentrations of N-Acetyl-Leu-Leu-Norleu-Al (ALLN; Sigma-Aldrich), 10 μM lactacystin (Calbiochem), 10 μg/ml nocodazole (Sigma-Aldrich), or various concentrations of peptides (University of Chicago Peptide Synthesis Facility) were begun the day after transfection.
Metabolic Labeling and Immunoprecipitation
Culture, metabolic labeling, lysis, and immunoprecipitation of COS-1 cells were performed as previously described (Dul et al. 1992). Eluates from equal cell equivalents were resolved on 11% reducing SDS-PAGE gels. The following antibodies were used in immunoprecipitations: rabbit anti–human kappa light chain (Bethyl Labs), goat anti–human kappa light chain (Caltag), and mouse anti–ubiquitin (StressGen).
Immunoblotting
Cells were transfected in six-well dishes and lysed in the wells with 200 μl each of the following buffer (mM): 50 Tris-HCl, pH 8.0, 150 NaCl, 5 KCl, 5 MgCl2, 0.5% Triton X-100, 0.5% deoxycholate, and 20 NEM, supplemented with protease inhibitors (1 μg/ml each of leupeptin and pepstatin A; 10 μg/ml each of TLCK, TPCK, and soybean trypsin inhibitor). The lysates were passed through a 25-gauge needle 10× on ice. Insoluble material was recovered by centrifugation at 16,000 g for 15 min. Pellets were solubilized in 50 μl 60 mM Tris-HCl, pH 6.8, 5% SDS, 10% glycerol with 1 min of sonication in a cuphold sonicator, followed by 10 min of boiling. Unless otherwise noted, equal cell equivalents from all samples were separated on 11% reducing SDS-PAGE gels and transferred to nitrocellulose. Membranes were incubated with goat anti–human kappa light chain (BioSource) and HRP-conjugated donkey anti–goat IgG (Santa Cruz Biotechnology), rabbit anti–VSV G (J. Burkhardt, University of Chicago) and HRP-conjugated goat anti–rabbit IgG (Jackson ImmunoResearch Laboratories), mouse anti–raf (Transduction Labs) or mouse anti–HA (Roche Molecular Biochemicals) and HRP-conjugated goat anti–mouse IgG (Jackson ImmunoResearch Laboratories). Bound antibodies were detected with SuperSignal West Pico Chemiluminescent Substrate (Pierce Chemical Co.). Blots were quantitated using scanning densitometry and NIH Image 1.6 software.
Fluorescence Microscopy
Cells grown on coverslips were transfected as described above, fixed in 3.7% formaldehyde/PBS, permeabilized, and stained as previously described (Burkhardt et al. 1993). The following antibodies were used: FITC-conjugated goat anti–human kappa light chain (BioSource), mouse anti–vimentin (Clone V9; Sigma-Aldrich), and Texas red–conjugated rabbit anti–mouse IgG (Jackson ImmunoResearch Laboratories).
Electron Microscopy
Cells were fixed with 1.6% paraformaldehyde/2.5% glutaraldehyde, dehydrated, and embedded according to standard procedure. After specimen preparation, ultrathin sections (60–80 nm) were cut using a Reichert FC-D ultramicrotome, and picked up on uncoated, 300 mesh, Cu-Rh grids. Sections were stained for 8 min with 1.5% uranyl acetate and counter stained for 10 min with lead citrate. Grids were viewed with a JEOL 100CX-II electron microscope, operated at 80 kV.
Synthesis and Treatment of Cells with TAT Fusion Peptides
Peptides were synthesized at the University of Chicago peptide synthesis facility. An 11-mer sequence (YGRKKRRQRRR) from the HIV TAT protein has been shown to transduce peptides and proteins into cells in a receptor-independent fashion (Schwarze et al. 1999). A peptide was synthesized that fused this TAT sequence with the VL-derived sequence we have shown to inhibit fiber formation in vitro (Davis et al. 2000). To ensure adequate flexibility and solubility, additional VL sequence was added on both sides of the active 7-mer, generating the following sequence for the TAT-TISS peptide: NH2-YGRKKRRQRRRSGSGTDFTLTISSLQAED-OH. A control peptide was synthesized (TAT-PASS) in which the critical hydrophobic residues were substituted with alanines and the extended structure of the peptide destroyed by the addition of a proline (NH2-YGRKKRRQRRRSGSGTDATAPASSLQAED-OH). Cells were treated with either peptide 24 h after transfection and incubated overnight in the presence of the indicated concentrations of ALLN. Cells were then harvested for Western blotting or immunofluorescence as described above.
Results
SMA Is a Nonsecreted Light Chain that Is Degraded by Proteasomes
Unlike most subunits of oligomeric proteins, LCs are secretion-competent even when expressed alone (Shapiro et al. 1966; Mosmann and Williamson 1980). As with all proteins, however, they must be correctly folded to exit the ER and transit the secretory pathway (reviewed by Gething and Sambrook 1992). Thus, secretability is a direct measure of a LCs ability to fold into the native state. Since LEN was isolated as a Bence Jones protein from the urine of a patient, it follows that it should be secretion competent, but it is not known whether the amyloidogenic SMA was secreted, and then aggregated in the extracellular space. For the purpose of expressing SMA and LEN in vivo, expression plasmids were constructed that direct the synthesis of complete kappa chains, containing either of these two variable region sequences followed by the constant region of human Cκ (Hieter et al. 1980). Pulse-chase analysis of COS cells transiently expressing these κ chains revealed that LEN was secreted as a wild-type LC, but that SMA was not secreted (Fig. 1). Furthermore, SMA was immunoprecipitated from the cell lysates only immediately after the pulse, and disappeared rapidly during the chase. This suggested that SMA was recognized as a mutant LC by the cellular quality control system, and was either rapidly degraded and/or aggregated into a detergent insoluble fraction.
Figure 1.

SMA is not secretion competent. COS cells were transfected with expression plasmids encoding either LEN or SMA human kappa chains. 2 d later, cells were pulse-labeled with 35S-methionine for 30 min (P) and chased for 1 or 2 h (1 and 2). Intracellular LCs (INT) were immunoprecipitated from the detergent soluble lysates; secreted LCs (EXT) from the cell culture media. The eluates were separated on reducing SDS-PAGE. LEN is secreted. SMA is recovered only from the pulse lysate sample and is not secreted.
Degradation of ER proteins is primarily controlled by the ERAD system (reviewed by Brodsky and McCracken 1999). Such proteins are exported to the cytosol, where they are tagged with polyubiquitin and directed to proteasomes for degradation. To determine whether SMA undergoes ERAD-mediated degradation, we treated transfected cells with the proteasome inhibitors ALLN (Fig. 2) or lactacystin (data not shown) and analyzed the detergent soluble and insoluble fractions by immunoblotting. In untreated cells, very little SMA was detected in either fraction (Fig. 2 A), again consistent with rapid degradation. Overnight incubation with proteasome inhibitors dramatically increased the yield of SMA, the majority of which was recovered in the detergent-insoluble pellet. LEN and VSV G protein (an unrelated protein that also transits through the secretory pathway) were found primarily in the soluble fractions, with only a small quantity becoming insoluble in the presence of ALLN. Long exposures of the blots showed that a small proportion of the insoluble pool consisted of a ladder of higher molecular weight forms of SMA, indicative of a post-translational modification (Fig. 2 B). The spacing of these bands was consistent with progressive addition of ubiquitin to SMA, which is known to target proteins to the ERAD pathway. To test this possibility, soluble and insoluble fractions were immunoprecipitated with anti–ubiquitin antibody, and then blotted with anti–kappa (Fig. 2 C) or, conversely, immunoprecipitated with anti–kappa and blotted with anti–ubiquitin (D). In the anti–ubiquitin immunoprecipitates, insoluble SMA was detected in a high molecular weight smear, as well as in a ladder whose spacing was equivalent to that seen in the total insoluble fraction of the lysate (Fig. 2 C, compare lane 2 with 5). When anti–kappa immunoprecipitates were blotted with anti–ubiquitin, a similar high molecular weight smear was observed, while the smaller molecular weight ladder was no longer evident. Thus, the detergent-insoluble fraction contains three pools of SMA molecules: the majority are unmodified monomers, a second pool is polyubiquitinylated, high molecular weight SMA, and the third pool appears as a regularly spaced ladder that is either ubiquitinylated or modified by an alternative mechanism. Taken together, these experiments demonstrate that SMA molecules are ubiquitinylated and targeted to proteasomal degradation. However, when the degradative machinery cannot keep pace with its synthesis, SMA forms detergent-insoluble aggregates.
Figure 2.
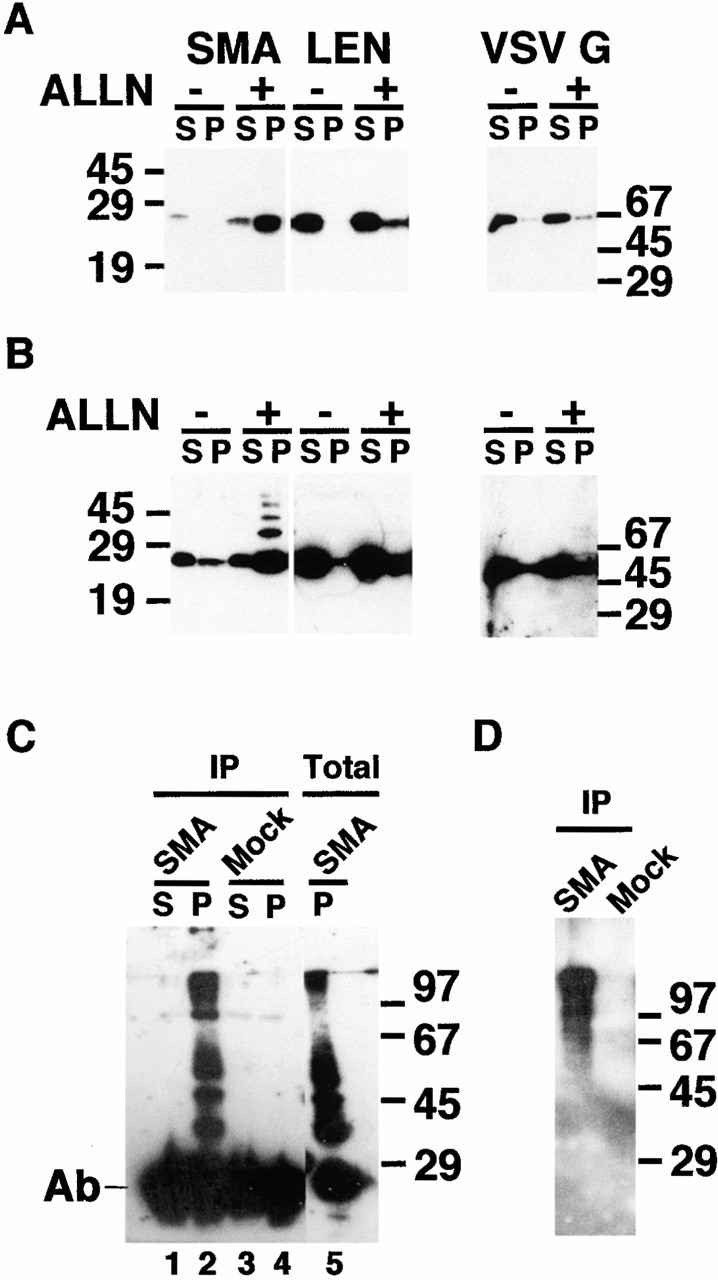
Proteasome inhibitors increase steady state levels of SMA, most of which is found in insoluble aggregates. (A) Transfected cells expressing SMA, LEN, or VSV G proteins were incubated overnight in the absence (−) or presence (+) of 10 μg/ml ALLN. Detergent soluble (S) and pelleted, insoluble (P) fractions were prepared and analyzed by Western blotting with anti–kappa antibody. The SMA fractions were loaded equally; two times more insoluble material was loaded for LEN and VSV G. Insoluble SMA, but not LEN or VSV G, accumulates in the presence of ALLN. (B) Upon long exposure, a ladder of bands is visible in the insoluble fraction, reflecting post-translational modification of SMA. (C) After overnight incubation with 10 μg/ml ALLN, soluble (S) and insoluble (P) fractions from SMA (lanes 1 and 2) or mock-transfected (3 and 4) cells were immunoprecipitated with anti–ubiquitin antibody. Precipitated material was analyzed by Western blotting with anti–kappa antibody. For comparison, total insoluble lysate (lane 5) was also analyzed by Western blotting with anti–kappa antibody. The major, 29-kD band in 1–4 is the light chain from the immunoprecipitating antibody; the 29-kD band in 5 is the monomeric, unmodified form of SMA. (D) After overnight incubation with 10 μg/ml ALLN, insoluble fractions from SMA- or mock-transfected cells were immunoprecipitated with anti–kappa antibody. Precipitated material was analyzed by Western blotting with anti–ubiquitin antibody. Ubiquitinated forms of kappa are detected only in the insoluble fraction from SMA-expressing cells.
SMA and Insoluble LEN Remain Permanently Reduced
The SMA and LEN proteins differ by only eight amino acids (Wilkins Stevens et al. 1995), but are processed in vivo along vastly different pathways. Therefore, we asked whether a difference in folding between SMA and LEN could be detected. In reducing gels, LCs migrate as one band lacking both disulfide bonds (F0). Under nonreducing conditions, two additional forms of LC are distinguishable: a folding intermediate that contains one disulfide (F1) and the completely oxidized form (Fox) (Hendershot et al. 1996). With other wild-type LCs, the F0 and F1 forms are only visible after short pulse labeling and lysis in the presence of alkylating agents (Melnick et al. 1994; Aviel, S., J. Melnick, T. Gidalevitz, F.J. Stevens, J.L. Dul, and Y. Argon, submitted for publication), indicating that these intermediates are usually short lived. Fox is the only LC form that is secreted from cells, and hence represents the fully folded, native state.
To examine the folding of SMA and LEN, soluble and insoluble fractions from ALLN-treated cells were resolved by SDS-PAGE and analyzed by Western blotting (Fig. 3). All SMA molecules, whether in the detergent soluble or insoluble fractions, were found to be in the F0 form. There was no detectable F1 (which migrates between F0 and Fox) or Fox since the migration of SMA was similar under both non-reducing and reducing conditions. Identical patterns were obtained with cells that were not treated with proteasome inhibitors (data not shown). This indicates that SMA has an early and profound block in folding, failing to oxidize either of its two domains. In contrast to SMA, the migration of LEN was consistent with the majority of the molecules having progressed to the completely oxidized, more compact form (Fig. 3). Only a small fraction of soluble LEN remained in the unfolded, F0 form. However, all LEN molecules found in the detergent-insoluble fraction were reduced, indicating that unfolded molecules had been segregated from those that were correctly folded. Thus, SMA is an early folding mutant that fails to oxidize even its constant domain, despite the fact that all its somatic mutations are in the variable domain. This phenotype is unusual, as the effects of previously described mutations in the variable domain seem to be limited to the folding of only this domain (Hendershot et al. 1996; Skowronek et al. 1998; Aviel, S., J. Melnick, T. Gidalevitz, F.J. Stevens, J.L. Dul, and Y. Argon, submitted for publication).
Figure 3.

SMA and insoluble LEN do not progress in folding. Transfected cells were treated with 10 μg/ml ALLN overnight. Soluble (S) and pelleted, insoluble (P) fractions were separated by SDS-PAGE under reducing (R) and nonreducing (NR) conditions and analyzed by Western blotting. Twofold more insoluble than soluble material was loaded. F0, completely reduced LC; Fox, LC containing two disulfide bonds. Only soluble LEN folds into the Fox state. All of SMA, insoluble LEN, and a small percentage of soluble LEN is found in the F0 state.
SMA Accumulates in Aggresomes
Commonly, mutations in secretory proteins prevent exit from the ER such that the proteins accumulate in that organelle (Hammond and Helenius 1995). We previously determined that the intracellular localization of SMA did not fit this pattern (Davis et al. 2000). Rather, as shown again in Fig. 4, it displayed features that were unique. First, there was some ER staining, but it was frequently obscured by diffuse cytoplasmic staining. Second, in most cells, anti–kappa staining was prevalent throughout the nucleus, but the nucleoli were negative. Third, roughly one third to one half of SMA-expressing cells exhibited concentrated perinuclear staining that at first appeared similar to the Golgi staining observed in LEN-expressing cells (Fig. 4A and Fig. B). However, we have determined, through a series of experiments described in detail below, that these structures were instead perinuclear inclusions. Only one such SMA-induced perinuclear inclusion body was observed in each cell, and the frequency of cells with these inclusions was increased substantially when proteasomal activity was inhibited (see Fig. 6 and Fig. 8).
Figure 4.
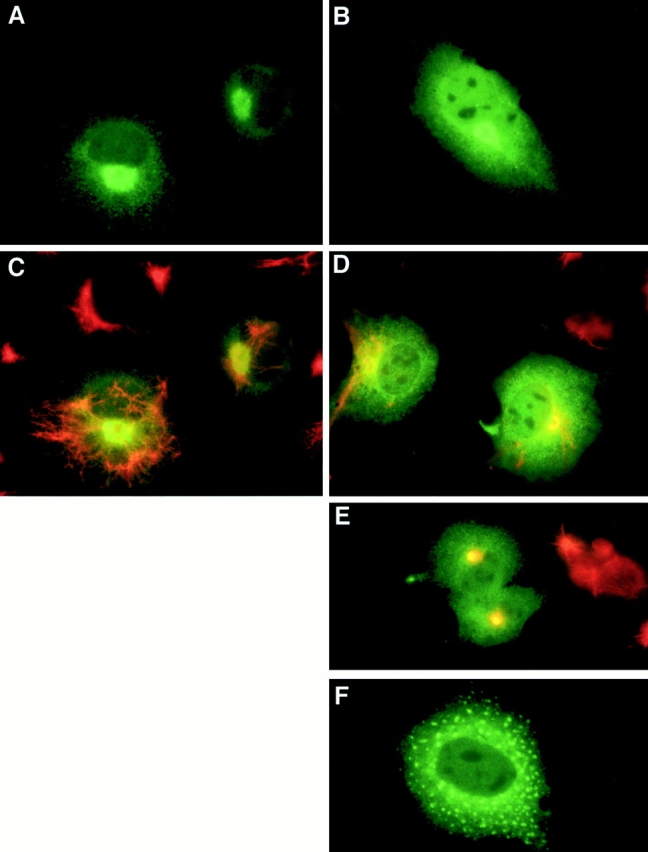
SMA is dislocated from the ER and accumulates in vimentin-ringed aggresomes. Cells transfected with LEN (A and C) or SMA (B and D–F) were incubated without (A–D and F) or with (E) 10 μg/ml ALLN overnight. Cells were processed for immunofluorescence with anti–human kappa antibody only (green, A, B, and F), or double labeled with the same antibody and anti–vimentin antibody (red, C–E). LEN shows reticular and Golgi staining typical of a secreted protein (A and C); SMA (B, D, and E) exhibits diffuse staining of the cytosol and nucleus, plus concentrated staining of a perinuclear aggresome. In LEN-transfected cells (C), vimentin is distributed throughout the cytoplasm; in SMA-transfected cells (D and E), the network of filaments collapses, concentrating around the aggresome. In SMA-transfected cells treated with nocodazole (F), the aggresomes are dispersed.
Figure 6.

Hsp70 promotes SMA solubility and degradation. Cells transfected with SMA alone (open bars) or cotransfected with Hsp70 (filled bars) were incubated overnight in the absence (−) or presence (+) of 10 μg/ml ALLN and processed for immunofluorescence with anti–kappa antibody. SMA-expressing cells were scored for the presence of aggresomes (n = 100 cells counted for each condition). (B) Cells were transfected with SMA alone (−), or with SMA plus one of the following: BiP, T19G BiP, Hsp70, or VSV G. Soluble (S) and insoluble (P) fractions were analyzed by Western blotting with anti–kappa antibody (top). The same blot was reprobed with anti–raf antibody (bottom). When coexpressed with Hsp70, more SMA partitions into the soluble fraction. (C) Cells were transfected as in B, incubated overnight in the presence of 10 μg/ml ALLN, and analyzed as above. Here, two times more insoluble than soluble material was loaded. Short exposure shows an increase in soluble SMA expressed with chaperones; Hsp70 coexpression also leads to a decrease in insoluble SMA (top). Long exposure of the same blot shows that the insoluble SMA ladder is present when SMA is expressed alone, or with BiP, but not when Hsp70 is coexpressed with SMA (bottom). (D) Cells transfected with SMA (lane 1), SMA plus Hsp70 (2 and 4), Hsp70 (3), or SMA plus SGK (5) were treated overnight with 10 μg/ml ALLN. They were metabolically labeled with 35S-methionine for 3 h, also in the presence of ALLN. Soluble fractions were immunoprecipitated with anti–human kappa antibody and protein-A beads (1–3 and 5) or protein-A beads alone (4). Hsp70 specifically coprecipitates with SMA when they are coexpressed. (E) Cells transfected with SMA plus HA-Hsp70 (lanes 1 and 4), SMA plus HA-SGK (2 and 5), or mock transfected (3 and 6) were treated overnight with 10 μg/ml ALLN. Soluble fractions were immunoprecipitated with anti–kappa antibody. Soluble lysate (1–3) and precipitated material (4–6) were analyzed by Western blotting with anti–HA antibody. Only HA-Hsp 70 coprecipitates with SMA.
Figure 8.

Inhibition of aggresome formation in the presence of TAT-TISS peptide. Transfected cells were treated overnight with 10 μg/ml ALLN and 100 μM peptide and processed for immunofluorescence with anti–kappa antibody. (A) Representative cells are shown for each treatment. (B) SMA-expressing cells were scored for the presence of aggresomes (n = 80–100 cells counted for each condition).
Similar structures, termed aggresomes, were observed in cells where the degradation of cystic fibrosis transmembrane conductance regulator (CFTR) was blocked. These aggresomes were shown to be surrounded by vimentin filaments and to require intact microtubules to form (Johnston et al. 1998). To further characterize the SMA-induced inclusion bodies, transfected cells were double labeled with anti–kappa and –vimentin antibodies. In LEN-expressing cells, the vimentin filaments formed a dense, filamentous network that extended to the periphery of the cells (Fig. 4 C). In cells expressing SMA, the vimentin distribution was quite different, having collapsed into a perinuclear location coincident with the inclusion body. This collapse of the vimentin filaments was observed in cells overexpressing SMA regardless of ALLN treatment, although the presence of the drug intensified the effect (Fig. 4D and Fig. E).
Treatment of SMA-expressing cells with nocodazole completely abrogated inclusion body formation (Fig. 4 F). Instead, multiple smaller aggregates were found dispersed throughout the cytosol. In contrast to the ALLN-induced aggresomes of CFTR (Johnston et al. 1998), even pre-existing SMA aggresomes were disrupted and dispersed by nocodazole treatment. Thus, intact microtubules play a critical role in the generation and maintenance of aggresomes.
Electron microscopy of SMA-expressing cells treated with ALLN revealed a large area (2–4 μm in diameter) near the nucleus, containing electron-dense clusters of material (Fig. 5 A). The appearance of this area was dependent on SMA expression; neither nontransfected nor LEN-expressing cells treated with ALLN exhibited this type of structure. These areas were, for the most part, devoid of other organelles and were surrounded by intermediate filaments (Fig. 5 B). Hence, the inclusions formed by cells in response to the overexpression of SMA have the hallmarks of aggresomes (Johnston et al. 1998).
Figure 5.

Ultrastructure of aggresomes. SMA-transfected cells were treated overnight with 10 μg/ml ALLN and processed for electron microscopy. (A) A low magnification view showing a cell with a perinuclear zone excluding other organelles and containing electron-dense clusters of material (arrows). N, nucleus. Bar, 4 μm. (B) High magnification view showing an aggresome composed of aggregated protein (electron dense material, white stars) surrounded by filamentous structures (arrowheads), consistent with vimentin intermediate filaments. Bar, 0.5 μm.
Coexpression of the Cytosolic Chaperone Hsp70 Inhibits SMA Aggregation
Recently, we showed that purified, recombinant Hsp70 blocked fibril formation by recombinant variable domains in vitro (Davis et al. 2000). BiP, the ER homologue of Hsp70, also prevented in vitro fibril formation, but was less efficient than Hsp70. Moreover, BiP suppressed aggregation in vivo. In particular, a mutant BiP that is unable to release substrate (T19G BiP) caused a larger pool of SMA to be retained within the ER and decreased the frequency of aggresome formation (Davis et al. 2000). Therefore, we asked whether coexpression of Hsp70 with SMA would decrease the frequency of aggresome formation (Fig. 6 A). Without ALLN treatment, 30% of the COS cells contained aggresomes as assayed by immunofluorescence. Addition of ALLN increased the proportion to 75%, but coexpression of Hsp70 dramatically decreased the frequency of aggresomes to 12% whether or not ALLN was present.
To further investigate the effects of Hsp70 and BiP on the aggregation of SMA in vivo, we used a biochemical assay. The experiments in Fig. 6 demonstrate that forced interaction with these chaperones improves the fate of SMA. When BiP or T19G BiP were coexpressed with SMA, a modest effect on the pool size of soluble LC was observed, as compared with cells transfected with SMA alone. This effect was variable in the absence (Fig. 6 B, top), but consistently observed in the presence (C, top), of ALLN. However, coexpression of either of these chaperones had no effect on the size of the insoluble SMA pool (Fig. 6 C, top).
Overexpression of cytosolic Hsp70, however, had a dramatic effect on SMA aggregation, both quantitatively and qualitatively. The yield of insoluble SMA was reduced three- to eightfold, while that of soluble SMA was proportionately increased (Fig. 6B and Fig. C, top). In addition, longer exposure of the same blot showed a decrease in the insoluble ladder of SMA only when Hsp70 was coexpressed (Fig. 6 C, bottom). To demonstrate direct interaction between Hsp70 and SMA, soluble fractions from metabolically labeled cells were immunoprecipitated with anti–kappa antibody and analyzed with reducing SDS-PAGE (Fig. 6 D). In cells cotransfected with Hsp70 and SMA, a 70-kD band was specifically coimmunoprecipitated with SMA (Fig. 6 D, lane 2). By contrast, overexpression of an irrelevant cytosolic protein, SGK, did not result in coprecipitation of that protein with SMA (lane 5). To verify that the coprecipitating band was indeed Hsp70, Western blotting was performed on anti–kappa immunoprecipitates from cotransfected cells (Fig. 6 E). The HA-tagged Hsp70 was detected by anti–HA antibody only in soluble lysates (Fig. 6 E, lane 1), and anti–kappa precipitates (lane 4) from cells cotransfected with SMA and HA-Hsp70. The HA-tagged SGK, although present in the lysate, was not found in the precipitated samples (compare lane 2 with 5). We conclude that Hsp70 interacts directly with SMA in the cytosol to decrease aggregation and aggresome formation, and improve targeting for degradation by the proteasome.
Together with our previous work (Davis et al. 2000), these experiments show that the fate of misfolded, amyloidogenic LC was altered by increased interactions with Hsp70 family chaperones on either side of the ER membrane: more SMA was retained in the ER, less was aggregated, and more of it remained in a soluble state, whether in the lumen of the ER or in the cytosol. Since the majority of SMA at steady state is in the dislocated, cytosolic pool, BiP would be expected to have less of an effect than Hsp70 when the total cellular protein is analyzed, and this is indeed what we observed.
An Hsp70-binding Peptide Derived from the LC Sequence Inhibits SMA Aggregation In Vivo
The activity of Hsp70 family proteins in the in vitro fiber-forming assay is due to their peptide-binding activity, because inclusion of a specific, VL-derived BiP- and Hsp70-binding peptide, FTLTISS (amino acids 71–77), blocked the polymerization of SMA fibrils (Davis et al. 1999, Davis et al. 2000). Therefore, we asked whether this peptide also inhibits SMA aggregation in vivo (Fig. 7). To optimize delivery of the peptide to all cellular compartments, it was synthesized with the 11-mer sequence from the HIV TAT protein at the NH2 terminus (Gius et al. 1999; Fig. 7 A). This TAT peptide permits the transduction of denatured proteins across cell membranes rapidly and efficiently in an energy- and receptor-independent fashion (Schwarze et al. 1999). In addition to the test peptide, TAT-TISS, another TAT-fusion was employed as a specificity control. This peptide, TAT-PASS, contains four amino acid substitutions (Fig. 7 A) and does not inhibit fibril formation in vitro (Davis et al. 2000). SMA-transfected cells were incubated overnight in the presence of increasing concentrations of ALLN and 50 μM of each peptide (Fig. 7 B). In the range of 1–10 μg/ml ALLN, there was a progressive increase in the amount of SMA found in the soluble fraction on a per cell basis (Fig. 7 B, top, N). Inclusion of the TAT-TISS peptide dramatically reduced the amount of SMA recovered at all ALLN concentrations tested (Fig. 7 B, top, T). In contrast, the TAT-PASS peptide had no effect (Fig. 7 B, top, N and P). Incubation of the same blots with anti–raf antibody demonstrated that equal cell equivalents were loaded across the gel (Fig. 7 B, bottom).
Figure 7.

A VL-derived peptide inhibits SMA aggregation in the cell. (A) Schematic representation of VL (numbers indicate amino acid residues; CDR1-3 indicates the portions of the sequence that contribute to the antigen-binding site), including the location and sequence of the BiP- and Hsp70-binding peptide (TISS). This peptide was synthesized as a fusion with the HIV-derived 11-mer TAT peptide to permit diffusion across all cell membranes. A control peptide that does not bind BiP (PASS), because of four substitutions, was also fused to TAT in similar fashion. (B) SMA-transfected cells were incubated overnight in the presence of the indicated concentrations of ALLN and 50 μM peptide, as indicated. Equal cell equivalents from the soluble fractions were loaded on an SDS gel and analyzed by Western blotting with anti–kappa antibody (top). N, no peptide added; T, TAT-TISS peptide added; P, TAT-PASS peptide added. Steady state levels of SMA are decreased in the presence of TAT-TISS peptide. The same blot was reprobed with anti–raf antibody (bottom). (C) Quantification of SMA in the soluble and insoluble fractions. SMA-transfected cells were incubated overnight in the presence of 10 μg/ml ALLN plus the indicated concentrations of either the Hsp70-binding peptide (TISS) or the control peptide (PASS). Soluble (unfilled bars) and insoluble (filled bars) fractions were analyzed by Western blotting with anti–kappa antibody and quantified. Note the loss of SMA with increasing TISS peptide concentration, particularly from the detergent-insoluble fractions. (D) Long exposure of a blot prepared as in C, but with twofold more insoluble than soluble material loaded, shows that with increasing TISS peptide there is a diminution of the ladder of SMA molecules.
We also examined the effect of different concentrations of peptide on SMA after treatment with 10 μg/ml ALLN. As shown in Fig. 7C and Fig. D, the TAT-TISS peptide decreased the yield of SMA in the insoluble fractions much more than in the detergent-soluble fractions. The magnitude of the decrease was from 4- to 10-fold (n = 3), in a peptide concentration-dependent fashion, whereas the TAT-PASS peptide had only a marginal effect even at the highest concentration used. As observed with coexpression of Hsp70, upon addition of the TAT-TISS peptide, the ladder of SMA was diminished (Fig. 7 D). This indicated that these molecules were being maintained in a soluble form long enough to be kept off the aggregation pathway and were degraded by the proteasome.
Lastly, we asked whether the decrease in steady state level of SMA in the presence of TAT-TISS peptide correlated with a decrease in the frequency of aggresome formation, by scoring anti–kappa stained cells (Fig. 8 A). Roughly 30% of untreated cells exhibited aggresomes, and this number increased to ∼65% upon addition of ALLN (Fig. 8 B). Transduction of TAT-TISS decreased the number of aggresomes by more than half, to 25%, about the same as in untreated cells, whereas addition of TAT-PASS peptide had no significant effect (Fig. 8 B). Hence, the large decrease in insoluble SMA observed in the presence of TAT-TISS peptide coincides with a drop in aggresome formation.
Discussion
This study shows that when an amyloidogenic LC fails to fold properly, it is dislocated out of the ER to the cytosol, where two linked coping mechanisms come into play. Much of the LC is targeted for degradation by proteasomes, but a considerable fraction aggregates intracellularly into one large inclusion body. The two alternative pathways are linked because inhibition of degradation increases aggregation and, most remarkably, introduction of a specific peptide into the cell inhibits aggregation and increases the efficiency of LC degradation. Thus, the two pathways seem to be in a kinetic competition.
Despite the fact that SMA was isolated from an extracellular fibrillar aggregate, it is not secreted and is disposed of quite efficiently. Even when overexpressed, as in our transient expression system, the majority of SMA molecules are degraded efficiently, disappearing from the soluble lysate with a half-time of <1 h. In parallel, a large fraction of cells exhibit a SMA-containing inclusion body. As a result of both mechanisms, steady state levels of SMA are only barely detectable when cells are lysed with nonionic detergents. Hence, our data show that COS cells have sufficient quality control capacity to deal with a heavy load of nonsecreted, highly aggregation-prone protein. They also show that a soluble secretory protein is disposed of via the same dislocation and proteasomal degradation pathway as misfolded proteins that span the ER membrane.
The inclusion bodies formed by SMA have the properties of aggresomes as defined by Johnston et al. 1998. As shown by both light and electron microscopy, there is usually only one large, perinuclear inclusion per cell; the ultrastructure of the dense material is similar to that of CFTR-containing aggresomes; the inclusion body does not appear to be bound by membrane, but does appear to be ringed by filaments that include microtubules and intermediate filaments (Johnston et al. 1998). The aggresomes are distinct from Russell bodies, which are intracellular inclusion bodies consisting of ER-derived vesicles. Russell bodies appear when intact Ig is expressed at high levels and in cells expressing germline heavy chain μ sequences (Matthews 1983; Tarlinton et al. 1992).
Given recent evidence that a large fraction of nascent chains is never fully folded and is therefore subject to proteasomal degradation (Schubert et al. 2000), it should be noted that our data show SMA to be efficiently translated and even translocated into the ER lumen. When degradation is inhibited, the total amount of full-length SMA is of the same order of magnitude as that of full-length wild-type LC, except that much of it is found in a detergent-insoluble form. Thus, there is no evidence for translational control mechanisms downregulating SMA expression. Second, although our polyclonal antibodies recognize multiple epitopes in the κ chain, only full-length molecules were detectable by metabolic labeling or immunoblotting. Third, the ability to affect the intracellular distribution of SMA via prolonged interaction with a nonreleasing version of BiP (Davis et al. 2000) shows that at least a large fraction of molecules are translocated into the ER. Almost all of these molecules, however, never fold properly and are evidently dislocated into the cytosol for disposal.
The severe folding defect of SMA is rather surprising and was not predicted from the sequence of the protein; most of the somatic mutations relative to the germline sequence are localized within the antigen-binding loops, and none are found in the constant domain. Previous work with a number of murine LC mutants showed that, in general, mutations in the variable domain only affect the folding of this domain while allowing folding of the constant domain (Hendershot et al. 1996; Skowronek et al. 1998; Aviel, S., J. Melnick, T. Gidalevitz, F.J. Stevens, J.L. Dul, and Y. Argon, submitted for publication). The failure of SMA to oxidize even its constant domain is therefore unusual and indicates that VL and CL folding is coupled. Studies are in progress to determine whether this property is due to the particular sequence of SMA or to differences between murine and human LC.
Surprisingly, immunofluorescence staining shows that SMA is also transported into the nucleus, where it is evenly distributed, although it is excluded from the nucleoli (Fig. 4). This pattern is not observed with other misfolded proteins that are degraded via the ERAD system. Active proteasomes have been shown (using GFP labeling) to translocate slowly from the cytosol to the nucleus (Reits et al. 1997). We therefore hypothesize that full-length SMA, or a large fragment thereof, is associated with proteasomes for a considerable time before degradation, and in this way migrates into the nucleus. If so, this prolonged association may reflect the rate-limiting step in the degradation pathway and may explain the reason for the kinetic competition between proteasome degradation and aggresome formation.
Hsp70 chaperones are instrumental in dictating the fate of SMA at several levels. First, our data are consistent with previously published work on the role of BiP in binding and “selecting” inappropriately folded LC, both in the lumen of the ER and at the translocon itself (Hamman et al. 1998; Gething 1999). BiP binds to LC mainly via its VL domain; this interaction is transient and likely repetitive (Hendershot et al. 1996; Davis et al. 1999). The presence of a nonreleasing BiP mutant (or high levels of wild-type BiP) “traps” SMA molecules in the ER lumen that would otherwise be quickly dislocated to the cytosol. Second, the data show that once dislocated, interaction of SMA with cytosolic Hsp70 (and/or Hsc70) improves the efficiency of degradation. Overexpression of Hsp70 decreased the insoluble pool of SMA that accumulated in the presence of proteasome inhibitors. In the absence of proteasome inhibitors, however, the level of endogenous Hsp70 was sufficient to support degradation of most SMA LCs. This supports the idea of kinetic competition between degradation and aggregation: when there is sufficient Hsp70 present to keep SMA soluble, more of it gets degraded, but if the level of Hsp70 is inadequate, then more SMA aggregates. Thus, SMA is a type of proteasomal substrate whose degradation is improved by chaperone interactions, as shown in vitro with other proteins by Bercovich et al. 1997.
Our data can be incorporated into the following model (Fig. 9). SMA fails to progress along a productive folding pathway and hence both its domains remain in the reduced state in vivo. The presence of the highly unstable κ4 protein is detected by BiP, presumably during or soon after its translocation across the ER membrane. Binding to BiP prevents SMA aggregation in the lumen and facilitates its dislocation back to the cytosol. Once there, binding to Hsp70 (or related chaperones) serves to maintain SMA in a degradation-competent state so that it can be ubiquitinated and rapidly targeted to proteasomes. At the same time, Hsp70 inhibits the tendency of SMA to aggregate in the cytosol, thus regulating the balance between degradation and aggregation. The inability to fold exposes (at least) the two major peptides in each of the two β sheets of the V domain that are good sites for binding of Hsp70 family chaperones. Continued exposure of these sites enables associations first with BiP (within the ER), and then with Hsp70 (in the cytosol). The FTLTISS peptide, which is effective in reducing intracellular aggregation, has the sequence of one of these two major sites, and, importantly, the same features that are required for its antiaggregation activity are necessary for its Hsp70 binding activity (Davis et al. 1999). We envisage the peptide interacting with the same amino acids in the hydrophobic core of the V domain normally occupied in the folded molecule by the endogenous FTLTISS peptide. In this way, the peptide acts as a surrogate chaperone, inhibiting aggregation and promoting degradation. This suggests the possibility of new treatment modalities using rationally designed peptides to suppress aggregation.
Figure 9.

Model of competing pathways of SMA degradation and aggregation in vivo. Binding to BiP in the ER, or Hsp70 or TAT-TISS peptide in the cytosol, helps prevent SMA from aggregating. Ubiquitination (Ub) targets soluble SMA for degradation by proteasomes. ALLN inhibits degradation, thus driving more SMA into insoluble aggresomes. Dotted line indicates that a small portion of insoluble SMA is ubiquitinated; otherwise, the size of the arrows is not meant to indicate efficiency of the step.
If our data with SMA holds true for other amyloidogenic LCs, then how do amyloids form? In our view, both arms of the cellular quality control system, degradation and aggresome formation, explain the low frequency of LC amyloidosis and provide a mechanism for fibrillogenesis. Amyloid LCs are always somatically mutated, and the frequency of destabilizing mutations is much higher than the frequency of expressed amyloidogenic LCs (Stevens and Argon 1999). This implies that there is selection against such mutants. The efficient recognition of unstable LC by BiP (and possibly other ER molecules), combined with the dislocation and degradation pathways, enable a B cell to dispose of the undesired LC and perhaps to express a new LC, through a process known as “receptor editing” (Gay et al. 1993; Retter and Nemazee 1998). Normally, there is sufficient degradative capacity, but when it is overloaded, aggresomes are formed as a generalized cellular response to the stress of protein aggregation. We infer from the high fraction of cells that spontaneously form aggresomes that mere overexpression of an amyloidogenic LC is sufficient to induce aggresome formation. The increase in aggresome formation that occurs when proteasome activity is inhibited further suggests that there is a balance between the two disposal pathways. It is not clear whether SMA molecules in the aggresome are still a viable substrate for degradation (Johnston et al. 1998). If so, the aggresome may serve as a substrate “holding station” for proteasomes. Either way, aggresomes may be highly relevant for extracellular amyloid formation, because their induction may lead to cell death, releasing large quantities of aggregated LC into the extracellular space. When a large clone of cells undergoes such cell death, sufficient material may be released to initiate fibrillogenesis.
Acknowledgments
We thank Dr. R. Morimoto for a gift of the human Hsp70 clone, Dr. J. Burkhardt for antibodies and advice with immunofluorescence, Dr. S. Conzen for the HA-SGK construct, and Dr. G. Reddy, from The University of Chicago Peptide Synthesis Facility, for the synthesis of peptides. We also thank the members of our laboratories for helpful discussions, T. Gidalevitz and S. Vogen for a critical reading of the manuscript, and J. Voss for administrative help.
This work was supported by grants from the National Institutes of Health (AI-30178 to Y. Argon and AG-18001 to F.J. Stevens and Y. Argon) and from the American Cancer Society, Illinois Division (to J.L. Dul). D.P. Davis was supported in part by a National Research Service Award fellowship and by a Baron fellowship.
Footnotes
Abbreviations used in this paper: ALLN, N-Acetyl-Leu-Leu-Norleu-Al; CFTR, cystic fibrosis transmembrane conductance regulator; HA, hemagglutinin; LC, light chain; SGK, serum and glucocorticoid-inducible kinase; VL, variable domain of light chain.
References
- Abravaya K., Myers M.P., Murphy S.P., Morimoto R.I. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 1992;6:1153–1164. doi: 10.1101/gad.6.7.1153. [DOI] [PubMed] [Google Scholar]
- Bercovich B., Stancovski I., Mayer A., Blumenfeld N., Laszlo A., Schwartz A.L., Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem. 1997;272:9002–9010. doi: 10.1074/jbc.272.14.9002. [DOI] [PubMed] [Google Scholar]
- Bole D.G., Hendershot L.M., Kearney J.F. Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol. 1986;102:1558–1566. doi: 10.1083/jcb.102.5.1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky J.L., McCracken A.A. ER protein quality control and proteasome-mediated protein degradation. Semin. Cell Dev. Biol. 1999;10:507–513. doi: 10.1006/scdb.1999.0321. [DOI] [PubMed] [Google Scholar]
- Burkhardt J.K., Wiebel F.A., Hester S., Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J. Exp. Med. 1993;178:1845–1856. doi: 10.1084/jem.178.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis D.P., Khurana R., Meredith S., Stevens F.J., Argon Y. Mapping the major interaction between BiP and immunoglobulin light chains to sites within the variable domain. J. Immunol. 1999;163:3842–3850. [PubMed] [Google Scholar]
- Davis D.P., Raffen R., Dul J.L., Vogen S.M., Williamson E.K., Stevens F.J., Argon Y. Inhibition of amyloid fiber assembly by both BiP and its target peptide. Immunity. 2000;13:433–442. doi: 10.1016/s1074-7613(00)00043-1. [DOI] [PubMed] [Google Scholar]
- Denoroy L., Deret S., Aucouturier P. Overrepresentation of the V kappa IV subgroup in light chain deposition disease. Immunol. Lett. 1994;42:63–66. doi: 10.1016/0165-2478(94)90036-1. [DOI] [PubMed] [Google Scholar]
- Dul J.L., Burrone O.R., Argon Y. A conditional secretory mutant in an Ig L chain is caused by replacement of tyrosine/phenylalanine 87 with histidine. J. Immunol. 1992;149:1927–1933. [PubMed] [Google Scholar]
- Gardner A.M., Aviel S., Argon Y. Rapid degradation of an unassembled immunoglobulin light chain is mediated by a serine protease and occurs in a pre-Golgi compartment. J. Biol. Chem. 1993;268:25940–25947. [PubMed] [Google Scholar]
- Gay D., Saunders T., Camper S., Weigert M. Receptor editingan approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething M.-J., McCammon K., Sambrook J. Expression of wild-type and mutant forms of influenza hemagglutininthe role of folding in intracellular transport. Cell. 1986;46:939–950. doi: 10.1016/0092-8674(86)90076-0. [DOI] [PubMed] [Google Scholar]
- Gething M.J. Role and regulation of the ER chaperone BiP. Semin. Cell Dev. Biol. 1999;10:465–472. doi: 10.1006/scdb.1999.0318. [DOI] [PubMed] [Google Scholar]
- Gething M.J., Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Gius D.R., Ezhevsky S.A., Becker-Hapak M., Nagahara H., Wei M.C., Dowdy S.F. Transduced p16INK4a peptides inhibit hypophosphorylation of the retinoblastoma protein and cell cycle progression prior to activation of Cdk2 complexes in late G1. Cancer Res. 1999;59:2577–2580. [PubMed] [Google Scholar]
- Hamman B.D., Hendershot L.M., Johnson A.E. BiP maintains the permeability barrier of the ER membrane by sealing the lumenal end of the translocon pore before and early in translocation. Cell. 1998;92:747–758. doi: 10.1016/s0092-8674(00)81403-8. [DOI] [PubMed] [Google Scholar]
- Hammond C., Helenius A. Quality control in the secretory pathway. Curr. Opin. Cell Biol. 1995;7:523–529. doi: 10.1016/0955-0674(95)80009-3. [DOI] [PubMed] [Google Scholar]
- Hendershot L., Wei J., Gaut J., Melnick J., Aviel S., Argon Y. Inhibition of immunoglobulin folding and secretion by dominant negative BiP ATPase mutants. Proc. Natl. Acad. Sci. USA. 1996;93:5269–5274. doi: 10.1073/pnas.93.11.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieter P.A., Max E.E., Seidman J.G., Maizel J.V., Jr., Leder P. Cloned human and mouse kappa immunoglobulin constant and J region genes conserve homology in functional segments. Cell. 1980;22:197–207. doi: 10.1016/0092-8674(80)90168-3. [DOI] [PubMed] [Google Scholar]
- Ishihara T., Takahashi M., Koga M., Yokota T., Yamashita Y., Uchino F. Amyloid fibril formation in the rough endoplasmic reticulum of plasma cells from a patient with localized A lambda amyloidosis. Lab. Invest. 1991;64:265–271. [PubMed] [Google Scholar]
- Johnston J.A., Ward C.L., Kopito R.R. Aggresomesa cellular response to misfolded proteins. J. Cell Biol. 1998;143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly J.W., Colon W., Lai Z., Lashuel H.A., McCulloch J., McCutchen S.L., Miroy G.J., Peterson S.A. Transthyretin quaternary and tertiary structural changes facilitate misassembly into amyloid. Adv. Prot. Chem. 1997;50:161–181. doi: 10.1016/s0065-3233(08)60321-6. [DOI] [PubMed] [Google Scholar]
- Kjeldsberg C.R., Eyre H.J., Totzke H. Evidence for intracellular amyloid formation in myeloma. Blood. 1977;50:493–504. [PubMed] [Google Scholar]
- Ma J., Kearney J.F., Hendershot L.M. Association of transport-defective light chains with immunoglobulin heavy chain binding protein. Mol. Immunol. 1990;27:623–630. doi: 10.1016/0161-5890(90)90004-j. [DOI] [PubMed] [Google Scholar]
- Matthews J.B. The immunoglobulin nature of Russell bodies. Br. J. Exp. Pathol. 1983;64:331–335. [PMC free article] [PubMed] [Google Scholar]
- Melnick J., Dul J.L., Argon Y. Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature. 1994;370:373–375. doi: 10.1038/370373a0. [DOI] [PubMed] [Google Scholar]
- Mosmann T.R., Williamson A.R. Structural mutations in a mouse immunoglobulin light chain resulting in failure to be secreted. Cell. 1980;20:283–292. doi: 10.1016/0092-8674(80)90614-5. [DOI] [PubMed] [Google Scholar]
- Perutz M. Polar zipperstheir role in human disease. Prot. Sci. 1994;3:1629–1637. doi: 10.1002/pro.5560031002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffen R., Dieckman L.J., Szpunar M., Wunschl C., Pokkuluri P.R., Dave P., Wilkins Stevens P., Cai X., Schiffer M., Stevens F.J. Physicochemical consequences of amino acid variations that contribute to fibril formation by immunoglobulin light chains. Prot. Sci. 1999;8:509–517. doi: 10.1110/ps.8.3.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reits E.A.J., Benham A.M., Plougastel B., Neefjes J., Trowsdale J. Dynamics of proteasome distribution in living cells. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6087–6094. doi: 10.1093/emboj/16.20.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Retter M.W., Nemazee D. Receptor editing occurs frequently during normal B cell development. J. Exp. Med. 1998;188:1231–1238. doi: 10.1084/jem.188.7.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U., Anton L.C., Gibbs J., Norbury C.C., Yewdell J.W., Bennink J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Schwarze S.R., Ho A., Vocero-Akbani A., Dowdy S.F. In vivo protein transductiondelivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Shapiro A.L., Scharff M.D., Maizel J.V., Uhr J.W. Synthesis of excess light chains of gamma globulin by rabbit lymph node cells. Nature. 1966;211:243–245. doi: 10.1038/211243a0. [DOI] [PubMed] [Google Scholar]
- Skowronek M.H., Hendershot L.M., Hass I.G. The variable domain of nonassembled Ig light chains determines both their half-life and binding to the chaperone BiP. Proc. Natl. Acad. Sci. USA. 1998;95:1574–1578. doi: 10.1073/pnas.95.4.1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon A., Frangione B., Franklin E.C. Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V lambda VI subgroup of human light chains with amyloidosis AL (lambda) J. Clin. Invest. 1982;70:453–460. doi: 10.1172/JCI110635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens F.J., Argon Y. Pathogenic light chains and the B-cell repertoire. Immunol. Today. 1999;20:451–457. doi: 10.1016/s0167-5699(99)01502-9. [DOI] [PubMed] [Google Scholar]
- Tarlinton D., Forster I., Rajewsky K. An explanation for the defect in secretion of IgM Mott cells and their predominant occurrence in the Ly-1 B cell compartment. Eur. J. Immunol. 1992;22:531–539. doi: 10.1002/eji.1830220236. [DOI] [PubMed] [Google Scholar]
- Werner E.D., Brodsky J.L., McCracken A.A. Proteasome-dependent endoplasmic reticulum-associated protein degradationan unconventional route to a familiar fate. Proc. Natl. Acad. Sci. USA. 1996;93:13797–13801. doi: 10.1073/pnas.93.24.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermark P., Araki S., Benson M.D., Cohen A.S., Frangione B., Masters C.L., Saraiva M.J., Sipe J.D., Husby G., Kyle R.A., Selkoe D. Nomenclature of amyloid fibril proteins. Report from the meeting of the International Nomenclature Committee on Amyloidosis, August 8–9, 1998. Amyloid. 1999;6:63–66. doi: 10.3109/13506129908993290. [DOI] [PubMed] [Google Scholar]
- Wilkins Stevens P., Raffen R., Hanson D.K., Deng Y.L., Berrios-Hammond M., Westholm F.A., Murphy C., Eulitz M., Wetzel R., Solomon A. Recombinant immunoglobulin variable domains generated from synthetic genes provide a system for in vitro characterization of light-chain amyloid proteins. Prot. Sci. 1995;4:421–432. doi: 10.1002/pro.5560040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi H.Y., Orr H.T. Spinocerebellar ataxia type 1. Semin. Cell Biol. 1995;6:29–35. doi: 10.1016/1043-4682(95)90012-8. [DOI] [PubMed] [Google Scholar]