

Abstract
Natural killer (NK) cells can spontaneously lyse certain virally infected and transformed cells. However, early in immune responses NK cells are further activated and recruited to tissue sites where they perform effector functions. This process is dependent on cytokines, but it is unclear if it is regulated by NK cell recognition of susceptible target cells. We show here that infiltration of activated NK cells into the peritoneal cavity in response to tumor cells is controlled by the tumor major histocompatibility complex (MHC) class I phenotype. Tumor cells lacking appropriate MHC class I expression induced NK cell infiltration, cytotoxic activation, and induction of transcription of interferon γ in NK cells. The induction of these responses was inhibited by restoration of tumor cell MHC class I expression. The NK cells responding to MHC class I–deficient tumor cells were ∼10 times as active as endogenous NK cells on a per cell basis. Although these effector cells showed a typical NK specificity in that they preferentially killed MHC class I–deficient cells, this specificity was even more distinct during induction of the intraperitoneal response. Observations are discussed in relation to a possible adaptive component of the NK response, i.e., recruitment/activation in response to challenges that only NK cells are able to neutralize.
Keywords: natural killer cell, MHC class I, activation, interferon γ, tumor
Introduction
Natural killer cells are of the lymphoid lineage, although they are distinct from T and B cells. They can rapidly reject tumor cells and bone marrow transplants without prior sensitization 1. The specificity of NK cells depend on an interplay between germ line–encoded inhibitory receptors, which are specific for MHC class I, and less characterized activating receptors 2 3 4. Cells with defects in their MHC class I expression are particularly susceptible to NK cell–mediated lysis, as they fail to cancel the lytic program through inhibitory receptors 2 3 5. NK cells can infiltrate both murine and human malignancies, and they are especially efficient in the rejection of tumors lacking host MHC class I molecules, including those with defects in transporter associated with antigen processing (TAP) or β2 microglobulin (β2m) genes 6 7.
In vivo NK cells can be recruited locally by inoculation of tumor cells 8 9 10 11 12 13, treatment with biological response modifiers 14 15, and viral infection 16 17 18. NK cell migration can be induced by several chemokines such as macrophage inflammatory protein 1α and IFN-γ–inducible protein 10 18 19 20 21, as well as TNF-α 13 15 21 and IL-2 22. Gene knockout mice deficient for certain of these cytokines show considerable defects in the migration of NK cells during local responses to tumors 13 and viruses 18. In addition, NK cells are activated to become more cytotoxic and to produce IFN-γ during the early phases of infections 16 17 18 23 24, and this may improve the chance for survival of the host 25 26 27. The activation of NK cells occurs in response to α/β IFNs or IL-12 23 24 28. Although NK cells may be activated during early immune responses by cytokines, they are typically regarded as an innate immune mechanism. It is thus unclear whether they can also be activated in response to abnormal cells with a phenotype that they are particularly competent to fight. Several previous studies have demonstrated infiltration of NK cells in the peritoneal cavity upon inoculation of tumor cells 12 13, but the specificity in the induction of this response is unclear. We set out to investigate this problem and particularly whether the induction was controlled by tumor MHC class I molecules, as NK cells detect the lack of MHC class I during the effector cell stage.
We found a low cytotoxic activity of resident intraperitoneal NK cells and a strong recruitment and activation of host NK cells after inoculation of tumor cells, provided that the tumor cells lacked critical host MHC class I molecules. In contrast, tumors with full MHC class I expression induced only weak responses in terms of both recruitment and activation of NK cells. At least two different mechanisms contributed to the response: an increase in NK cell number and a strongly augmented cytotoxic capacity per cell (compared with endogenous NK cells). The activated NK cells preferentially killed MHC class I–deficient target cells that had been used for activation, but the specificity for MHC class I–deficient cells was even more distinct in the activation step compared with the effector–target interaction. We also found that transcription of IFN-γ was induced in NK cells by MHC class I–deficient tumor cells but not by MHC class I transfectants. This NK activation pathway may increase early host resistance by amplification of the inflammatory response and bias the development of Th cells.
Materials and Methods
Mice.
All mice, except for SCID mice 29, were bred at the Department of Tumor Biology and at the Microbiology and Tumor Biology Center, Karolinska Institute. B6 mice are of the H-2b haplotype, and A × CBA mice are an F1 cross between A/Sn of the H-2a and CBA of the H-2Kk haplotype, respectively.
Cell Lines.
RMA is a Rauscher's virus–induced lymphoma cell line derived from B6 (H-2b), and RMA-S is a TAP-2–defective variant of RMA 30 31. RMA-S.mtp2 32 and RMA-S.Ham-2 33 are rat and mouse TAP-2 transfectants of RMA-S, respectively. YAC is a Moloney leukemia virus–induced lymphoma cell line from A/Sn (H-2a), and AH-2− is a β2m− variant of YAC. E49.3-β2m is a β2m+ transfectant of AH-2− 34.
Generation of In Vivo–activated NK Cells.
B6 or A × CBA mice were inoculated intraperitoneally with 106 or 107 cells resuspended in PBS, irradiated with 10,000 rads, or PBS alone. A × CBA F1 mice were used instead of parental A/Sn due to the low endogenous NK activity present in A/Sn. The tumor cells were for most experiments maintained as ascite cell lines in vivo but were also grown in vitro (the cell lines derived from YAC, used for in vivo injections, were always maintained as ascite lines). After 1–6 d, the mice were killed, and 2–3 ml of RPMI was injected twice into the intraperitoneal cavity to harvest peritoneal exudate cells (PECs). The cells were washed and analyzed for cytotoxic activity in a standard 4–5-h 51Cr-release assay or tested for expression of cell surface markers by FACS®.
mAbs, FACS® Analysis, FACS® Sorting, and Complement-mediated Depletion of Effector Cell Populations.
For flow cytometry, FITC- or PE-conjugated mAbs directed against various cell surface antigens were used. These were directed against NK1.1 (PK136), Ly49C (5E6), CD4 (L3T4), and CD8 (53-6.7). All conjugated antibodies were purchased from PharMingen. Before FACS® analysis (on a FACScan™; Becton Dickinson), cells were incubated with 0.5–1.0 μg of antibody for 30–60 min at 4°C. For inhibition of unspecific binding of fluoresceinated mAbs, the staining buffer contained 30% FCS and 5% mouse serum in hybridoma supernatant of 2G.4 specific for FcRIIγ (HB197; American Type Culture Collection). The analysis of 5E6 on NK1.1+ cells (see Fig. 4) was gated on forward scatter (FSC)low side scatter (SSC)low cells, due to high autofluorescence in FSChighSSChigh cells (Wright-Giemsa stain suggested that these may correspond to macrophages). NK1.1+ cells were always in the FSClowSSClow population. To generate pure NK1.1+ and NK1.1− populations, PECs were labeled with anti-NK1.1 mAb according to the procedure above and sorted on a FACScan™ (Becton Dickinson). For complement-mediated effector cell depletion, the cells were first incubated with an antisera against asialo-GM1 or mAbs against CD4 or CD8 for 30 min and subsequently incubated with rabbit complement diluted 1:8 for 75–90 min. These cells were subsequently used in a standard 51Cr-release assay.
Figure 4.

Strong increase in cytotoxic activity of NK1.1+ cells induced by intraperitoneal RMA-S inoculation. PECs from RMA-S–injected B6 (d–f) or control mice (a–c) were analyzed by FACS® (b and e) and tested for cytotoxic activity against 51Cr-labeled YAC-1 target cells (a and d) 3 d after tumor cell inoculation. A significant increase in NK1.1+ cell numbers is induced by RMA-S. In the right panels, PECs from control (c) or RMA-S–inoculated (f) mice were FACS® sorted into NK1.1+ and NK1.1− populations and tested for cytotoxic activity against YAC-1 target cells. A strong activation of cytotoxic activity, as measured per NK1.1+ cell, is induced by RMA-S.
Competitive PCR for Cytokine Transcripts.
Total RNA was obtained from single-cell suspensions by using acid guanidium thiocyanate and phenol–chloroform extraction 35, based on the rapid one-step procedure of Chomczynski and Sacchi 36. The aqueous phase was collected, and the RNA was further purified and concentrated by precipitation with isopropanol and ethanol. 2 mg of mRNA was denatured at 94°C for 5 min, reverse transcribed at 40°C for 45 min, and treated at 94°C for 5 min. The reverse transcription was carried out in a total of 40 μl with 7.5 μM dithiothreitol (GIBCO BRL), 0.5 mM nucleotides (dNTP; Pharmacia), 1 U/ml RNAsin (Promega Corp.), 5 mM random hexanucleotides (pd[N]6; Pharmacia), and 10 U/ml murine Moloney leukemia virus reverse transcriptase (GIBCO BRL).
Each cDNA sample was amplified in a competitive PCR assay 37. The PCR reaction was performed in 20 μl containing 0.2 mM dNTP, 25 mU/ml Taq polymerase (Perkin-Elmer Roche), 0.5 mM sense and antisense primers, cDNA, and 2 μl of competitor fragments of different lengths but with the same primer binding sequences as the target DNA. The competitors for each cytokine were diluted in a series of at least five threefold dilutions for every sample. A negative control containing no template, a competitor control, and cDNA controls were included in every assay. Primer sequences, annealing temperatures, and competitors for β-actin, IFN-γ, TNF-α, and IL-12 (p40) were the same as previously reported 38 39 40. The PCR products were loaded on a 2% agarose gel, electrophoresed, and photographed. The original concentration of the competitor was known in all cases. The concentrations of cDNA were calculated from cDNA and competitor bands with equal intensity. The cytokine mRNA concentrations were expressed as percentages of the β-actin mRNA concentration for each sample.
Immunoassay for IFN-α, -β, and -γ.
IFN-α and -β in serum was measured separately in dissociation-enhanced lanthanide fluoroimmunoassays (DELFIA) as described 41. In brief, microtiter plates were coated with sheep anti–IFN-α/β and incubated with samples or standards and then with mAbs to IFN-α or -β labeled with Europium lanthanide chelate. An enhancement solution was added, and fluorescence was measured in a DELFIA fluorometer. The values in Fig. 6 are displayed in international units, as determined by comparison with the signal generated by standard concentrations.
Figure 6.

Induction of IFN-γ but not IL-12 in NK1.1+ PECs by RMA-S cells. PECs from mice inoculated with either RMA-S or RMA-S.Ham-2 were FACS® sorted into NK1.1+ and NK1.1− populations and analyzed for IFN-γ (a) and IL-12 (p40) transcripts (b). Competitive PCR was used for the quantitation of transcripts (see Materials and Methods), and the intensity of the bands is displayed as percent compared with a β-actin standard. In c, the serum levels of IFN-α and IFN-β were measured using DELFIA (see Materials and Methods). Induction of IFN-γ transcription is observed in intraperitoneal NK1.1+ cells but not in NK1.1− cells and is dependent on tumor MHC class I expression.
The secretion of IFN-γ protein was determined by Quantikine ELISA (R & D Systems, Inc.). In brief, 2 × 106 PECs were plated in RPMI 1640 medium supplemented with 10% FCS in flat-bottomed 96-well plates. Supernatants were harvested after 20 h. PECs were plated alone and in the presence of RMA-S cells (2 × 105 cells per well) or IL-12 (50 IU/ml). The Quantikine ELISA is a sandwich ELISA, and the signal was quantified as compared with an IFN-γ standard.
Results
Induction of a Cytotoxic NK Cell Response by MHC Class I–deficient Lymphoma Cells.
To monitor the activity and specificity of NK cells during tumor cell rejection, we inoculated TAP-2–deficient mutant RMA-S cells intraperitoneally into syngeneic B6 mice. The mice were given irradiated tumor cells and killed 1–6 d after inoculation. PECs were obtained and tested for cytotoxicity against a panel of target cells. We found that PECs from RMA-S–inoculated B6 mice were highly cytotoxic against RMA-S (not shown) as well as YAC-1 cells ( Fig. 1 b), whereas the cytotoxic activity from BSS-injected control mice was low ( Fig. 1 b). The cytotoxic activity induced by RMA-S was detected after only 2 d, peaked by 3 d after inoculation ( Fig. 1 a), and was local; it could not be observed among spleen cells ( Fig. 1 b). The cytotoxicity was mediated by NK cells, as pretreatment of the effector cell population with anti-CD4 or anti-CD8 antibodies plus complement had no effect, whereas antiasialo-GM1 plus complement depleted the activity ( Fig. 1 c and data not shown). These data are in line with several previous studies showing migration of NK cells in response to tumor cells in the lung, liver, and peritoneal cavity (e.g., B16 melanoma [8, 9, 11], MCA 102 sarcoma [8], lung carcinomas [8, 12], MADB106 mammary carcinoma [9], and lymphomas including RMA-S cells [13]).
Figure 1.
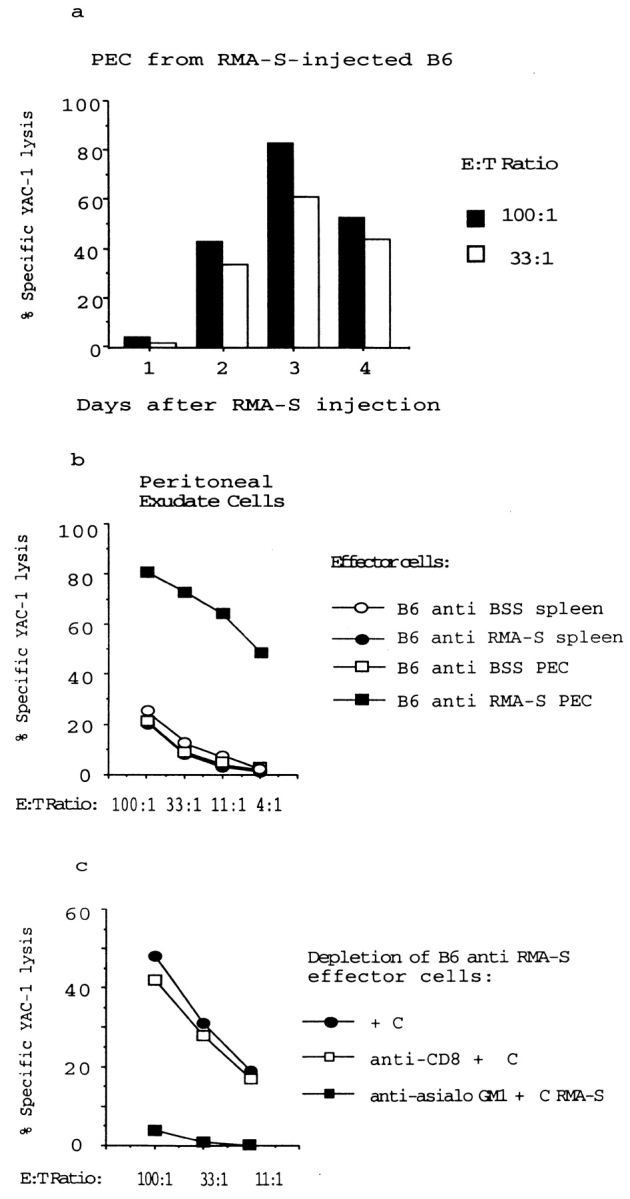
Intraperitoneal NK cell activity induced by RMA-S lymphoma cells. (a) Syngeneic B6 mice were inoculated with irradiated RMA-S cells, and the PECs were tested after 1–4 d against NK-susceptible, 51Cr-labeled YAC-1 target cells in a standard cytotoxicity assay. (b) Comparison of YAC-1 killing present in the peritoneal cavity and the spleen 3 d after intraperitoneal inoculation with irradiated RMA-S cells. (c) The cytotoxic PECs induced by RMA-S are asialo-GM1 +. PECs induced by RMA-S injection were stained with either anti-CD8 or antiasialo-GM1 antiserum, subjected to complement-mediated depletion, and subsequently tested in a 51Cr-release assay against YAC-1 target cells.
Induction of Cytotoxic NK Cell Responses in the Peritoneal Cavity Is Inhibited by Restoration of Tumor MHC Class I Expression.
RMA-S cells express low cell surface MHC class I due to a defect in TAP-2, and TAP-2 transfection of RMA-S cells restores cell surface MHC class I expression as well as resistance to NK cell–mediated lysis 7 30 31 32 33. Whereas RMA-S cells induced a strong intraperitoneal NK cell response, we found that wild-type RMA cells failed to do this ( Fig. 2, a–c). In addition, we found that TAP-2 transfection of RMA-S (RMA-S.mtp2) suppressed its ability to induce a cytotoxic NK response, demonstrating that the latter was indeed associated with the defect in antigen presentation of the challenging cells ( Fig. 2c and Fig. d).
Figure 2.

The induction of an intraperitoneal NK cell response is inhibited by restoration of tumor MHC class I expression. Irradiated tumor cells were inoculated intraperitoneally into syngeneic B6 mice, and PEC cells were tested on day 3 in a 51Cr-release assay against the target cells indicated. Stimulator cells used to inoculate syngeneic (H-2b) C57Bl/6 mice: (a) PBS control; (b) RMA; (c) RMA-S; and (d) RMA-S.mtp2. Each diagram represents a mean of at least five experiments.
A similar pattern of MHC class I–dependent activation was observed with a β2m-deficient variant of the YAC lymphoma called AH-2− and its β2m− transfectant, E49.3-β2m (all cells of A/Sn origin; reference 34). AH-2− induced a strong cytotoxic response intraperitoneally when inoculated into (A/Sn × CBA) F1 mice, whereas wild-type YAC and the β2m transfectant E49.3-β2m induced weaker responses, although they were stronger than the response induced by PBS (Table , top). The cytotoxic PECs induced by AH-2− were depleted by antiasialo-GM1 + complement, indicating that these effectors were NK cells (Table , top).
Table 1.
MHC Class I Specificity of NK Cell Induction in Allogeneic (H-2d) SCID Mice and (A × CBA) F1 Mice
| Target cell | In vivo stimulators of PECs from A × CBA F1 | |||
|---|---|---|---|---|
| BSS | YAC | AH-2− | E49.3-β2m | |
| AH-2− | 12, 0, 4 | 13, 7, 1 | 45, 28, 13 | 27, 13, 3 |
| AH-2− | AH-2− | |||
| + C′ | aA-GM1 + C′ | |||
| AH-2− | 39, 21, 9 | 1, 0, 2 | ||
| In vivo stimulators of C.B-17 SCID mice (H-2d) | ||||
| BSS | RMA | RMA-S | ||
| RMA | 6, 3, 3 | 29, 13, 6 | 42, 25, 14 | |
| RMA-S | 6, 6, 5 | 31, 19, 12 | 41, 28, 20 | |
| YAC-1 | 7, 4, 5 | 40, 21, 15 | 47, 31, 28 | |
We next tested if intraperitoneal responsiveness to tumor cells was dependent on the presence of functional T and B cells by tumor inoculation of C.B-17 (H-2d) SCID mice (lacking T and B cells; reference 29). We observed a moderate induction of NK cell activation after intraperitoneal inoculation of irradiated RMA cells into C.B-17 SCID, and a slightly stronger response to RMA-S inoculation (Table , bottom). We hypothesized that the response to wild-type H-2b RMA cells (unlike in B6 mice of the H-2b haplotype) was due to lack of host MHC class I molecules of the tumor. We therefore transfected RMA with the host MHC class I gene H-2Dd, and this resulted in a strong reduction of NK cell inducing capacity ( Fig. 3b and Fig. c). The intraperitoneal cytotoxic activity in RMA-Dd–inoculated mice was similar to that in BSS-injected control mice ( Fig. 3, a and c). This early induced response is therefore dependent on single host MHC class I alleles but independent of T and B cells.
Figure 3.

Regulation of NK cell activation in C.B-17 SCID mice by H-2Dd molecules. Irradiated (H-2b) tumor cells were injected intraperitoneally into allogeneic C.B-17 (H-2d) SCID mice, and PECs were tested on day 3 in a 51Cr-release assay against target cells indicated. Stimulator cells used to inoculate allogeneic (H-2d) C.B-17 SCID mice: (a) BSS control; (b) RMA; and (c) RMA-Dd. The MHC class I–specific induction of the response occurs independently of T and B cells.
The ability to induce an NK response in vivo in three different systems was thus linked to insufficient MHC class I expression in the tumor inoculate, as it was abrogated by transfection with three different genes involved in the class I pathway (TAP, heavy chain, or β2m), each in a cell line where the gene restored a self-phenotype in relation to the host. It should be noted that the NK cells induced by RMA-S showed a typical pattern of specificity when tested as effector cells (as they killed RMA-S more efficiently than RMA), but the MHC class I specificity was more distinct during induction of the NK cell response ( Fig. 2b–d). This was also observed for RMA and RMA-Dd when inoculated into C.B-17 SCID mice; these tumor cell lines had very different activation capability but were hardly distinguished at all when tested as target cells ( Fig. 3b and Fig. c). It should be noted that RMA and RMA-Dd cells have very different abilities to escape in vivo NK cell–mediated rejection when grafted as live tumors in vivo 42.
Induction of Strong NK Cell Activation but Not Blastogenesis by MHC Class I–deficient Tumor Cells.
Although NK cells are characterized by a constitutive cytotoxic activity, they can undergo cytotoxic activation and blastogenesis during early immune responses to viruses 43. We characterized the tumor cell–induced NK cell response in terms of NK cell numbers, phenotype, and activity of (FACS®-sorted) NK cells. MHC class I–expressing tumors (RMA and RMA-S.Ham-2) induced minor increases in the number of PECs, whereas RMA-S induced a two- to threefold increase in PECs (mean ± SD from 15 experiments: BSS, 7.5 ± 1.6 × 106; RMA, 7.8 ± 4.1 × 106; and RMA-S, 17.1 ± 7.1 × 106). FACS® analysis showed that, in addition, the PECs induced by RMA-S contained an increased proportion of NK cells, approximately threefold compared with untreated mice ( Fig. 4b and Fig. e). Among these NK cells, we did not find any indications of receptor-selective accumulation of NK cells (with the 5E6 epitope binding to Ly49C interacting with H-2Kb); the proportion of 5E6+ in the total NK1.1+ cells was roughly equal between control and RMA-S–inoculated mice ( Fig. 4b and Fig. e). Taken together, these observations show a local accumulation of NK cells; these may be 5–10 times as numerous in the peritoneal cavity in an animal inoculated with tumors of MHC class I–deficient phenotype.
The cytotoxic response induced by RMA-S revealed a more drastic increase than the induction in NK1.1+ cell numbers ( Fig. 4, a, b, d, and e). This may result from the activation of individual NK 1.1+ cells. To measure this, we FACS® sorted PECs from control and tumor cell–inoculated mice. In control mice, NK cell cytotoxic activity from both spleens and PECs was restricted to NK1.1+ cells and was comparatively low in PECs when compared with splenic NK1.1+ cells (approximately three- to fivefold reduced on a per cell basis; Fig. 5 a). In NK1.1+ cells sorted from RMA-S–inoculated mice, we found a strong cytotoxic activation compared with endogenous NK1.1+ cells, showing an ∼10-fold increase in cytotoxicity per NK1.1+ cell ( Fig. 4c and Fig. f, and Fig. 5 b). Also, among the PECs from RMA-S–inoculated mice, all of the cytotoxicity against YAC-1 cells was found in the NK1.1+ population. Furthermore, the cytotoxic activity of individual NK1.1+ cells was dependent on the MHC class I phenotype of the tumor, as TAP transfection of RMA-S inhibited its activating capacity ( Fig. 5 b).
Figure 5.

Cytotoxic activation of intraperitoneal NK1.1+ cells depends on the MHC class I expression of the tumor cells. PECs from untreated B6 (a) or tumor cell–injected B6 mice (b) were sorted by FACS® into NK1.1+ and NK1.1− populations and tested for cytotoxic activity against YAC-1 target cells. Note that the cytotoxic activity from all mice is restricted to the NK1.1+ population. (a) The cytotoxic activity of endogenous NK1.1+ cells from spleen and peritoneal cavity. NK1.1+ cells from the peritoneal cavity show comparatively low cytotoxic activity. (b) NK1.1+ cells from RMA-S–injected mice have a drastically induced cytotoxic activity compared with endogenous NK1.1+ cells from the intraperitoneal cavity and spleen (compare to Fig. 5 a). Activation of NK1.1+ cells is inhibited by TAP transfection of RMA-S. (c) Cell size of NK1.1+ cells in untreated (black line) or RMA-S–injected mice (gray line) versus poly I:C–induced NK1.1+ cells (stippled line). The mean FSC for spleen lymphocytes and RBCs in this experiment was 409 and 216, respectively. No difference in cell size was observed between RMA-S–activated and endogenous NK1.1+ cells.
NK cells induced by high serum levels of IFN-α/β during viral infection show augmented cytotoxicity combined with a blast-like phenotype 43. We observed a substantial population of large cells (FSChigh) in our peritoneal exudates induced by tumor cells. Therefore, control and RMA-S–inoculated mouse PECs were divided into one FSChighSSChigh and one FSClowSSClow population. The NK1.1+ cells were always present in the FSClowSSClow population, with a cell size not significantly different from that of endogenous NK1.1+ cells, which have low cytotoxic activity ( Fig. 5 a). These NK cells do not, therefore, resemble the blastoid NK cells induced by viral infection or by poly I:C ( Fig. 5 c). Wright-Giemsa stains of PECs indicated large proportions of monocytes and granulocytes in RMA-S–injected as well as control mice, which may explain the abundant FSChighSSChigh population of NK1.1− cells (data not shown). The observed NK1.1+ cells were TCR−, and the proportion of CD4+ and CD8+ cells was relatively unchanged at ∼5% of PECs (data not shown). In conclusion, the NK cell system is mobilized in response to target cells perceived as nonself, resulting in up to 100-fold total augmentation of NK cell activity locally in the peritoneal cavity. This is explained in part by the increase in NK1.1+ cell numbers and, in addition, by an ∼10-fold increase in activity per NK1.1+ cell.
Expression of Cytokine Genes in RMA-S–activated NK1.1+ Cells.
NK cells can influence the activation of Th1- and Th2-type CD4+ T cells through the production of IFN-γ 27. To test whether in vivo confrontation with MHC class I–deficient tumor cells can influence cytokine production of PECs, we prepared cDNA from FACS®-sorted NK1.1− and NK1.1+ cells of tumor-inoculated mice. The cDNA was amplified with cytokine-specific primers and used in a competitive PCR assay to quantify cytokine transcripts. β-Actin was amplified as a comparative standard 37. NK1.1+ cells from RMA-S–inoculated mice had high levels of transcripts for IFN-γ, whereas NK cells from mice inoculated with TAP-2–transfected RMA-S cells contained comparatively low levels, although they were above background ( Fig. 6 a). When amplifying cDNA from NK1.1− cells in parallel, the signal was always below the level of detection, indicating absence of IFN-γ transcription in NK1.1− cells. The induction of IFN-γ transcripts correlated with production of IFN-γ protein, but this occurred only in the presence of either RMA-S or IL-12 ( Fig. 7). NK cell recruitment in itself is therefore not sufficient for release of the IFN-γ protein. One major pathway for the induction of IFN-γ in NK cells is mediated through IL-12 secreted by macrophages, as observed during murine CMV infection 44 45. However, we did not detect any transcripts for the IL-12 p40 chain in either intraperitoneal NK1.1+ or NK1.1− cells, indicating that the induction of IFN-γ transcripts in NK1.1+ cells does not depend on local production of IL-12 ( Fig. 6 b). Attempts to amplify IL-12 transcripts from PECs from RMA-S–treated mice were also negative when performed 1 d after inoculation, and the same primers amplified IL-12 transcripts from control samples (data not shown). These data support recent data from viral infection models where IL-12 is required for NK cell production of IFN-γ protein 44 45 but also suggest that this requirement can be partially replaced by NK cell interaction with RMA-S. TNF-α is necessary for RMA-S–induced NK cell trafficking 13, and we did detect induction of TNF-α in NK cells from mice inoculated with RMA-S but not with RMA-S.Ham-2 (data not shown). This may suggest that NK cells themselves may mediate the attraction of other NK cells upon recognition of tumor cells when these tumor cells lack appropriate MHC class I expression. Furthermore, we detected increased levels of IFN-α and -β in serum from RMA-S–injected mice and lower but significant levels in RMA-S.Ham-2–inoculated mice ( Fig. 6 d). These levels correspond approximately to those found in mice chronically infected with LCMV but are much lower than those detected during the acute infection, which is also consistent with the lack of blastogenesis in these NK cells 46.
Figure 7.

Secretion of IFN-γ protein by RMA-S–induced PECs. Secretion of IFN-γ protein from untreated (unfilled bars) and RMA-S–induced (filled bars) PECs was measured by sandwich ELISA. 2 × 105 RMA-S cells or 50 IU of IL-12 was added for 20 h before measuring secretion. In the presence of medium only, the release of IFN-γ was <10 pg/ml. Secretion of IFN-γ protein is induced, but this requires the presence of either RMA-S or IL-12.
Discussion
Adaptive T cell–mediated immune responses against intracellular antigens include two related but distinct components: (a) a first step of recruitment/activation/expansion of effector cells in response to cognate interaction with cells expressing an MHC-presented antigen that the effector cell can eliminate, and (b) generation of memory, leading to a more rapid response upon reexposure to the same antigen. Our study is consistent with the notion that NK cells can also generate responses according to their specificity in the effector cell stage corresponding to the first step listed above. Tumor-infiltrating NK cells were recruited to the peritoneal cavity, acquired efficient cytotoxic capacity, and transcribed IFN-γ provided that the challenging tumor lacked appropriate MHC class I expression. Activation and recruitment of NK cells was inhibited by restoration of MHC class I expression in the tumor. Note also that the MHC-associated specificity was at least as distinct during activation of NK effector cells as during effector cell lysis of target cells. This is of particular relevance because a multitude of intracellular parasites (e.g., MCMV) as well as mutations in tumors cause defects in expression of MHC class I molecules 47 48 49. We have not been able to demonstrate the second component of adapted responses, a stronger memory response upon secondary tumor cell challenge (not shown).
NK cells are usually considered as part of the innate immune system, which can neutralize or eliminate pathogens or cells instantly without the need for activation. For example, the rejection of intravenously injected NK-sensitive tumor cells is rapid and occurs within hours 1 31 50. However, it is not excluded that an activation of NK cells occurs in response to a target and that this phase may be crucial at certain sites where the endogenous NK cell activity is low, as in the intraperitoneal NK cells studied here. The NK cell system shows considerable flexibility, and trafficking of NK cells to organs as well as induction of cytotoxicity occurs in response to many different stimuli, including tumor cell inoculation 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22. This has been studied with tumor cells of various origins (e.g., B16 melanoma [8, 9, 11], MCA 102 sarcoma [8], lung carcinomas [8, 12], MADB106 mammary carcinoma [9], and lymphomas including RMA-S [13]), but the role of NK cell specificity with respect to MHC class I during the induction of this response has not been studied. It was possible to establish this role in our study; note that activation was not only caused by cells that had been selected and mutagenized in vitro, but it could also be induced by MHC class I–expressing cells lacking a critical host MHC class I product (H-2Dd) and inhibited by transfection of the corresponding gene.
NK cells activated by IL-2 are highly lytic against many tumor targets and are interesting in relation to tumor immunotherapy. IL-2–activated NK cells can in some cases infiltrate solid tumors and induce regression of established lung and liver tumors 51 52 53. However, in many cases the primary tumor and metastases are not efficiently infiltrated by NK cells, leading to absence of an antitumor effect. We describe a phenotype resulting in both efficient infiltration and cytotoxic activation. A local decrease in MHC class I expression (e.g., by TAP inhibition by infected cell protein (ICP)47; reference 54) could possibly contribute to making this therapy more efficient.
Cells deficient for MHC class I expression have increased susceptibility for NK cells, but relatively little is known about the ligands that normally trigger NK cells. The NKp44 and NKp46 receptors that are involved in the triggering of human NK cells during lysis of tumor cells have recently been characterized 55 56. In addition, NKp46 has been cloned from mouse NK cells and would be one candidate for a triggering receptor that is required for the RMA-S–induced response 57. Many normal cell types are believed to express activatory NK cell ligands, and self-tolerance must then be maintained by the inhibitory signal provided by normal MHC class I expression. Target cell ligands that can trigger NK cell effector function range from adhesion molecules such as intercellular adhesion molecule 2 (when redistributed at the cell surface by Ezrin; reference 58) to costimulatory molecules expressed by APCs 59. Whereas deficient MHC class I expression was necessary for efficient NK cell activation in this study, it was not sufficient. We did not observe any NK cell activation by inoculation of irradiated β2m-deficient spleen cells, despite the fact that these cells have a very low MHC class I expression (data not shown). At the effector stage, NK cells can reject β2m-deficient hematopoietic precursors in the bone marrow and kill β2m-deficient Con A–activated T cell blasts in vitro 60 61. The latter may not, however, express enough activatory ligands to activate an NK cell response. Allogeneic cells may however possess such ligands, as rat spleen cells from certain strains activate CD8+ NK cells upon intraperitoneal inoculation 62. The molecular identity of the activatory ligands expressed by the cell lines tested in this paper have not been determined, but this system may serve as a model for the characterization of such ligands.
A number of viruses reduce MHC class I expression of infected host cells. This can occur either through a general cytopathic effect 63 or the synthesis of proteins that specifically interfere with processing in the MHC class I pathway 47. Interestingly, certain cytopathic viruses such as MCMV and murine hepatitis virus accumulate high numbers of cytotoxic NK/LGL at the site of infection, which is not observed with a noncytopathic LCMV strain 16. MCMV encodes genes that prevent normal antigen processing 64, and infection by MCMV induces local accumulation and cytotoxic activation of NK cells during early infection 17. The kinetics of NK cell activation induced by RMA-S is similar to that observed during viral infection, i.e., peaking after 3 d. In this process, antiviral NK cells undergo blastogenesis 43, and they thus accumulate at the site of viral replication as a result of both recruitment and proliferation. Activation by RMA-S inoculation was not associated with NK cell blastogenesis, supporting the conclusion that this activation pathway is different from the IFN-α/β–dependent virus induction pathway 45. It may also be different from the well characterized pathway based on macrophages secreting IL-12, leading to IFN-γ secretion by NK cells 28 44 45. In our system, RMA-S–induced recruitment in itself was not sufficient for secretion of IFN-γ protein, despite an induction of IFN-γ transcription. Secretion of IFN-γ protein was induced by RMA-S cells and more efficiently by IL-12. ( Fig. 7).
Several studies of early immune responses show the importance of activated IFN-γ–producing NK cells for protection of the host 27 28 65. IFN-γ produced by NK cells can activate macrophages; it may also be important in CD4+ T cell differentiation to the Th1 subset 27 66, and it is therefore possible that inoculates of MHC class I–deficient tumor cells could bias these pathways in vivo. For induction of IFN-γ transcription, we cannot exclude the possibility that NK cells may be activated indirectly by cytokines secreted by other cells such as macrophages, but we did not detect cytokine transcripts in NK1.1− cells for IFN-γ, TNF-α, or IL-12.
The RMA-S–induced peritoneal exudate contained a significant fraction of macrophages (as seen by Wright-Giemsa stains; data not shown), and it is possible that this mechanism may contribute to the development of an early inflammatory response. Indeed, NK cell–produced IFN-γ is reported as a T cell–independent pathway of macrophage activation 67. This pathway may be controlled by cells expressing MHC class I–specific inhibitory receptors, such as NK cells and potentially other cells of the immune system 68. This mechanism could operate in early responses against viruses that inhibit antigen processing (e.g., against HSV, expressing ICP47 that blocks TAP; reference 54). The simplest model to explain the induced NK cell response we observe is a direct interaction between RMA-S and NK cells in the peritoneal cavity. The interaction between cell surface–expressed molecules on RMA-S cells and NK cells that triggers cytotoxicity 55 56 57 may also trigger activation of augmented cytotoxic capacity and production of IFN-γ; other cytokines such as TNF-α 13, which attract new NK cells to the site, may also be secreted simultaneously. It was recently shown that NK cell infiltration as well as tumor rejection after intraperitoneal inoculation depends on TNF-α but not IL-12 13. In the situations where a complete self-phenotype is detected by inhibitory MHC class I receptors (e.g., in TAP-2–transfected RMA-S), all of the events above are canceled. Our data suggest that cells with deficient MHC class I expression may influence the outcome of an infection, and we may speculate about whether it is possible to use a cell with an MHC-deficient phenotype as an adjuvant for NK activation as well as for the induction of adaptive immune responses. In future studies, it will be important to delineate the activating receptor–ligand interactions underlying the type of induced NK activity studied here, as well as to understand the cellular mechanisms for local expansion of NK activity.
Acknowledgments
We thank M. Hagelin, M.L. Solberg, Marcello Toro, Birgitta Wester, and H. Morén for excellent technical assistance. We also thank all of the members of the Klas Kärre laboratory, Hans-Gustaf Ljunggren, and Benedict Chambers for interesting discussions.
This work was supported by grants from the Swedish Cancer Society, the Swedish Medical Research Council, Stiftelsen för Internationaliserandet av Högre Utbildning och Forskning (STINT), the Karolinska Institute, Stiftelsen Lars Hiertas Minne, Alex och Eva Wallströms minnesfond, the Göran Gustafsson Foundation, and Arbetsmarknadens Försäkringsaktiebolag.
Footnotes
Abbreviations used in this paper: DELFIA, dissociation-enhanced lanthanide fluoroimmunoassays; FSC, forward scatter; PECs, peritoneal exudate cells; SSC, side scatter; TAP, transporter associated with antigen processing.
References
- Trinchieri G. Biology of natural killer cells. Adv. Immunol. 1989;47:187–376 . doi: 10.1016/S0065-2776(08)60664-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanier L.L. NK cell receptors. Annu. Rev. Immunol. 1998;16:359–393 . doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- Moretta L., Ciccone E., Mingari M.C., Biassoni R., Moretta A. Human natural killer cellsorigin, clonality, specificity, and receptors. Adv. Immunol. 1994;55:341–380 . doi: 10.1016/s0065-2776(08)60513-1. [DOI] [PubMed] [Google Scholar]
- Fan Q.R., Mosyak L., Winter C.C., Wagtmann N., Long E.O., Wiley D.C. Structure of the inhibitory receptor for human natural killer cells resembles haematopoietic receptors. Nature. 1997;389:96–100 . doi: 10.1038/38028. [DOI] [PubMed] [Google Scholar]
- Ljunggren H.-G., Kärre K. In search of the missing selfMHC molecules and NK recognition. Immunol Today. 1990;11:237–244 . doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- Glas R., Sturmhöfel K., Hämmerling G.J., Kärre K., Ljunggren H.G. Restoration of a tumorigenic phenotype by β2-microglobulin transfection to EL-4 mutant cells. J. Exp. Med. 1992;175:843–846 . doi: 10.1084/jem.175.3.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franksson L., George E., Powis S., Butcher G., Howard J., Kärre K. Tumorigenicity conferred to lymphoma mutant by major histocompatibility complex–encoded transporter gene. J. Exp. Med. 1993;177:201–205 . doi: 10.1084/jem.177.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse P., Herberman R.B., Nannmark U., Johansson B.R., Hokland M., Wasserman K., Goldfarb R.H. Accumulation of adoptively transferred adherent, lymphokine-activated killer cells in murine metastasis. J. Exp. Med. 1991;174:479–488 . doi: 10.1084/jem.174.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink M.R., Palomba M.L., Basse P.H., Hiserodt J.C. In situ localization of 3.2.3+ natural killer cells in tissues from normal and tumor-bearing rats. Cancer Res. 1991;51:4931–4936 . [PubMed] [Google Scholar]
- Whiteside T.L., Herberman R.B. Extravasation of antitumor effector cells. Invasion Metastasis. 1992;12:128–146 . [PubMed] [Google Scholar]
- Fogler W.E., Volker K., McCormick K.L., Watanabe M., Ortaldo J.R., Wiltrout R.H. NK cell infiltration into lung, liver, and subcutaneous B16 melanoma is mediated by VCAM/VLA-4 interaction. J. Immunol. 1996;156:4707–4714 . [PubMed] [Google Scholar]
- Kurosawa S., Matsuzaki G., Harada M., Ando T., Nomoto K. Early appearance and activation of natural killer cells in tumor-infiltrating lymphoid cells during tumor development. Eur. J. Immunol. 1993;23:1029–1033 . doi: 10.1002/eji.1830230507. [DOI] [PubMed] [Google Scholar]
- Smyth M.J., Kelly J.M., Baxter A.G., Korner H., Sedgewick J.D. An essential role for tumor necrosis factor in natural killer cell–mediated tumor rejection in the peritoneum. J. Exp. Med. 1998;188:1611–1619 . doi: 10.1084/jem.188.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiltrout R.H., Mathieson B.J., Talmadge J.E., Reynolds C.W., Zhang S.R., Herberman R.B., Ortaldo J.R. Augmentation of organ-associated natural killer activity by biological response modifiers. Isolation and characterization of large granular lymphocytes from the liver. J. Exp. Med. 1984;160:1431–1449 . doi: 10.1084/jem.160.5.1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilaro A.M., Taub D.D., McCormick K.L., Williams H.M., Sayers T.J., Fogler W.E., Wiltrout R.H. TNF-α is a principal cytokine involved in the recruitment of NK cells to liver parenchyma. J. Immunol. 1994;153:333–342 . [PubMed] [Google Scholar]
- McIntyre K.W., Welsh R.M. Accumulation of natural killer and cytotoxic T large granular lymphocytes in the liver during virus infection. J. Exp. Med. 1986;164:1667–1681 . doi: 10.1084/jem.164.5.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natuk R.J., Welsh R.M. Accumulation and chemotaxis of natural killer/large granular lymphocytes at sites of virus replication. J. Immunol. 1987;138:877–883 . [PubMed] [Google Scholar]
- Salazar-Mather T.P., Orange J.S., Biron C.A. Early murine cytomegalovirus (MCMV) infection induces liver natural killer (NK) cell inflammation and protection through macrophage inflammatory protein 1α (MIP-1α)-dependent pathways. J. Exp. Med. 1998;187:1–14 . doi: 10.1084/jem.187.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allavena P., Bianchi G., Paganin C., Giardina G., Mantovani A. Regulation of adhesion and transendothelial migration of natural killer cells. Nat. Immun. 1996;15:107–116 . [PubMed] [Google Scholar]
- Taub D.D., Sayers T.J., Carter C.R., Ortaldo J.R. α and β chemokines induce NK cell migration and enhance NK cell mediated cytolysis. J. Immunol. 1995;155:3877–3888 . [PubMed] [Google Scholar]
- Maghazachi A.A. Tumor necrosis factor-α is chemokinetic for lymphokine-activated killer cellsregulation by cyclic adenosine monophosphate. J. Leukoc. Biol. 1991;49:302–308 . doi: 10.1002/jlb.49.3.302. [DOI] [PubMed] [Google Scholar]
- Lövik G., Vaage J.T., Naper C., Benestad H.B., Rolstad B. Recruitment of alloreactive natural killer cells to the rat peritoneum by a transfected cell line secreting rat recombinant interleukin-2. J. Immunol. Methods. 1995;179:59–69 . doi: 10.1016/0022-1759(94)00270-7. [DOI] [PubMed] [Google Scholar]
- Bancroft G.J. The role of natural killer cells in innate resistance to infection. Curr. Opin. Immunol. 1993;5:503–510 . doi: 10.1016/0952-7915(93)90030-v. [DOI] [PubMed] [Google Scholar]
- Biron C.A. Activation and function of natural killer cell responses during viral infections. Curr. Opin. Immunol. 1997;9:24–34 . doi: 10.1016/s0952-7915(97)80155-0. [DOI] [PubMed] [Google Scholar]
- Bukowski J.F., Warner J.F., Dennert G., Welsh R.M. Adoptive transfer studies demonstrating the antiviral effects of natural killer cells in vivo. J. Exp. Med. 1985;161:40–52 . doi: 10.1084/jem.161.1.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron C.A., Byron K.S., Sullivan J.L. Severe herpes virus infections in an adolescent without natural killer cells. N. Engl. J. Med. 1989;320:1731–1735 . doi: 10.1056/NEJM198906293202605. [DOI] [PubMed] [Google Scholar]
- Scharton T.M., Scott P. Natural killer cells are a source of interferon γ that drives differentiation of CD4+ T cell subsets and induces early resistance to Leishmania major in mice. J. Exp. Med. 1993;178:567–577 . doi: 10.1084/jem.178.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange J.S., Wang B., Terhorst C., Biron C.A. Requirement for natural killer cell–produced interferon γ in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 1995;182:1045–1056 . doi: 10.1084/jem.182.4.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosma M.J., Carroll A.M. The SCID mouse mutantdefinition, characterization and potential uses. Annu. Rev. Immunol. 1991;9:323–350 . doi: 10.1146/annurev.iy.09.040191.001543. [DOI] [PubMed] [Google Scholar]
- Kärre K., Ljunggren H.-G., Piontek G., Kiessling R. Selective rejection of H-2 deficient lymphoma variants suggests alternative immune defence strategy. Nature. 1986;319:675–678 . doi: 10.1038/319675a0. [DOI] [PubMed] [Google Scholar]
- Ljunggren H.G., Kärre K. Host resistance directed selectively against H-2–deficient lymphoma variants. Analysis of the mechanism. J. Exp. Med. 1985;162:1745–1759 . doi: 10.1084/jem.162.6.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis S.J., Townsend A.R., Deverson E.V., Bastin J., Butcher G.W., Howard J.C. Restoration of antigen presentation to the mutant cell line RMA-S by an MHC-linked transporter. Nature. 1991;354:528–531 . doi: 10.1038/354528a0. [DOI] [PubMed] [Google Scholar]
- Attaya M., Jameson S., Martinez C.K., Hermel E., Aldrich C., Forman J., Lindahl H.K.F., Bevan M.J., Monaco J.J. Ham-2 corrects the class I antigen processing defect in RMA-S cells. Nature. 1992;355:647–649 . doi: 10.1038/355647a0. [DOI] [PubMed] [Google Scholar]
- Ljunggren H.G., Sturmhöfel K., Wolpert E., Hämmerling G.J., Kärre K. Transfection of β2-microglobulin restores the IFN-mediated protection from natural killer cell lysis in YAC-1 lymphoma variants. J. Immunol. 1990;145:380–386 . [PubMed] [Google Scholar]
- Lagoo-Deenadayalan S., Lagoo A.S., Barber W.H., Hardy K.J. A standardized approach to PCR-based semiquantitation of multiple cytokine gene transcripts from small cell samples. Lymphokine Cytokine Res. 1993;12:59–67 . [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159 . doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Gilliland G., Perrin S., Blanchard K., Bunn H. Analysis of cytokine mRNA and DNAdetection and quantitation by competitive polymerase chain reaction. Proc. Natl. Acad. Sci. USA. 1990;87:2725–2729 . doi: 10.1073/pnas.87.7.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg M., Sporrong L., Persson I., Wigzell H., Örn A. Cytokine gene expression during infection of mice lacking CD4 and/or CD8 with Trypanosoma cruzi . Scand. J. Immunol. 1995;41:164–170 . doi: 10.1111/j.1365-3083.1995.tb03549.x. [DOI] [PubMed] [Google Scholar]
- Laucella S., Salcedo R., Castaños-Velez E., Riarte A., De Titto E., Patarroyo M., Örn A., Rottenberg M. Increased expression and secretion of ICAM-1 during experimental infection with Trypanosoma cruzi . Parasite Immunol. 1996;18:227–239 . doi: 10.1046/j.1365-3024.1996.d01-95.x. [DOI] [PubMed] [Google Scholar]
- Guevara-Mendoza O., Une C., Franceschi Carreira P., Örn A. Experimental infection of Balb/c mice with Leishmania panamensis and Leishmania Mexicanainduction of early IFN-γ but not IL-4 is associated with the development of cutaneous lesions. Scand. J. Immunol. 1997;46:35–40 . doi: 10.1046/j.1365-3083.1997.d01-96.x. [DOI] [PubMed] [Google Scholar]
- Eloranta M.-L., Sandberg K., Alm G.V. The interferon α/β responses of mice to herpes simplex virus studied at the blood and tissue level in vitro and in vivo. Scand. J. Immunol. 1996;43:356–360 . doi: 10.1046/j.1365-3083.1996.d01-62.x. [DOI] [PubMed] [Google Scholar]
- Glas R., Waldenström M., Höglund P., Klein G., Kärre K., Ljunggren H.G. Rejection of tumors in mice with severe combined immunodeficiency syndrome determined by the major histocompatibility complex class I expression on the graft. Cancer Res. 1995;55:1911–1916 . [PubMed] [Google Scholar]
- Biron C.A., Welsh R.M. Blastogenesis of natural killer cells during viral infection in vivo. J. Immunol. 1982;129:2788–2795 . [PubMed] [Google Scholar]
- Orange J.S., Biron C.A. An absolute and restricted requirement for IL-12 in natural killer cell IFN-gamma production and antiviral defense. Studies of natural killer and T cell responses in contrasting viral infections. J. Immunol. 1996;156:1138–1142 . [PubMed] [Google Scholar]
- Cousens L.P., Peterson R., Hsu S., Dorner A., Altman J.D., Ahmed R., Biron C.A. Two roads divergedinterferon α/β– and interleukin 12–mediated pathways in promoting T cell interferon γ responses during viral infection. J. Exp. Med. 1999;189:1315–1328 . doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukowski J.F., Biron C.A., Welsh R.M. Elevated natural killer cell-mediated cytotoxicity, plasma interferon, and tumor cell rejection in mice persistently infected with lymphocytic choriomeningitis virus. J. Immunol. 1983;131:991–996 . [PubMed] [Google Scholar]
- Ploegh H.L. Viral strategies of immune evasion. Science. 1998;280:248–253 . doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- Momburg F., Koch S. Selective loss of beta 2-microglobulin mRNA in human colon carcinoma. J. Exp. Med. 1989;169:309–314 . doi: 10.1084/jem.169.1.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo N.P., Esquivel F., Kawakami Y., Yewdell J.W., Mulé J.J., Rosenberg S.A., Bennink J.R. Identification of human cancers deficient in antigen processing. J. Exp. Med. 1993;177:265–272 . doi: 10.1084/jem.177.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund P., Glas R., Ohlen C., Ljunggren H.-G., Kärre K. Alteration of the natural killer repertoire in H-2 transgenic micespecificity of rapid lymphoma clearance determined by the H-2 phenotype of the target. J. Exp. Med. 1991;174:327–334 . doi: 10.1084/jem.174.2.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside T.L., Vujanovic N.L., Herberman R.B. Natural killer cells and tumor therapy. Curr. Top. Microbiol. Immunol. 1998;230:221–244 . doi: 10.1007/978-3-642-46859-9_13. [DOI] [PubMed] [Google Scholar]
- Schwarz R.E., Vujanovic N.L., Hiserodt J.C. Enhanced anti-metastatic activity of lymphokine-activated killer cells purified and expanded by their adherence to plastic. Cancer Res. 1989;49:1441–1446 . [PubMed] [Google Scholar]
- Yasumura S., Lin W.C., Hirabayashi H., Vujanovic N.L., Herberman R.B., Whiteside T.L. Immunotherapy of liver metastases of human gastric carcinoma with IL-2 activated natural killer cells. Cancer Res. 1994;54:3808–3816 . [PubMed] [Google Scholar]
- Hill A., Jugovic P., York I., Russ G., Bennink J., Yewdell J., Ploegh H., Johnson D. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1998;375:411–415 . doi: 10.1038/375411a0. [DOI] [PubMed] [Google Scholar]
- Vitale M., Bottino C., Sivori S., Sanseverino L., Castriconi R., Marcenaro E., Augugliaro R., Moretta L., Moretta A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex–restricted tumor cell lysis. J. Exp. Med. 1998;187:2065–2072 . doi: 10.1084/jem.187.12.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessino A., Sivori S., Bottino C., Malaspina A., Morelli L., Moretta L., Biassoni R., Moretta A. Molecular cloning of NKp46a novel member of the immunoglobulin superfamily involved in triggering of natural cytotoxicity. J. Exp. Med. 1998;188:953–960 . doi: 10.1084/jem.188.5.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biassoni R., Pessino A., Bottino C., Pende D., Moretta L., Moretta A. The murine homologue of the human NKp46, a triggering receptor involved in the induction of natural cytotoxicity. Eur. J. Immunol. 1999;29:1014–1020 . doi: 10.1002/(SICI)1521-4141(199903)29:03<1014::AID-IMMU1014>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Helander T., Carpen O., Turunen O., Kovanen P.E., Vaheri A., Timonen T. ICAM-2 redistributed by ezrin as a target for killer cells. Nature. 1996;382:265–268 . doi: 10.1038/382265a0. [DOI] [PubMed] [Google Scholar]
- Chambers B.J., Salcedo M., Ljunggren H.G. Triggering of a natural killer cells by the costimulatory molecule CD80 (B7-1) Immunity. 1996;5:311–317 . doi: 10.1016/s1074-7613(00)80257-5. [DOI] [PubMed] [Google Scholar]
- Bix M., Liao N.S., Ziljstra M., Loring J., Jaenisch R., Raulet D. Rejection of class I-MHC deficient haemopoietic cells by irradiated MHC-matched cells. Nature. 1991;349:329–331 . doi: 10.1038/349329a0. [DOI] [PubMed] [Google Scholar]
- Hoglund P., Ohlén C., Carbone E., Franksson L., Ljunggren H.G., Latour A., Koller B., Karre K. Recognition of beta 2-microglobulin-negative (beta 2m−) T-cell blasts by natural killer cells from normal but not from beta 2m− micenonresponsiveness controlled by beta 2m− bone marrow in chimeric mice. Proc. Natl. Acad. Sci. USA. 1991;88:10332–10336 . doi: 10.1073/pnas.88.22.10332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson P.O., Hansson J., Dohlsten M., Sjögren H.O., Hiserodt J.C., Hedlund G. In vivo induced allo-reactive natural killer cells. J. Immunol. 1992;149:1504–1509 . [PubMed] [Google Scholar]
- Brutkiewicz R.R., Klaus S.J., Welsh R.M. Window of vulnerability of vaccinia virus-infected cells to natural killer (NK) cell-mediated cytolysis correlates with enhanced NK cell triggering and is concomitant with a decrease in H-2 class I antigen expression. Nat. Immun. 1992;11:203–214 . [PubMed] [Google Scholar]
- Kleijnen M.F., Huppa J.B., Lucin P., Mukherjee S., Farrell H., Campell A.E., Koszinowski U.H., Hill A.B., Ploegh H.L. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:685–694 . doi: 10.1093/emboj/16.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay C.H., Welsh R.M. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J. Virol. 1997;71:267–275 . doi: 10.1128/jvi.71.1.267-275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constant S.L., Bottomly K. Induction of Th1 and Th2 CD4+ T cell responsesthe alternative approaches. Annu. Rev. Immunol. 1997;15:297–322 . doi: 10.1146/annurev.immunol.15.1.297. [DOI] [PubMed] [Google Scholar]
- Bancroft G.J., Schreiber R.D., Unanue E.R. Natural immunitya T-cell-independent pathway of macrophage activation, defined in the scid mouse. Immunol. Rev. 1991;124:5–24 . doi: 10.1111/j.1600-065x.1991.tb00613.x. [DOI] [PubMed] [Google Scholar]
- Cosman D., Fanger N., Borges L. Human cytomegalovirus, MHC class I and inhibitory signalling receptorsmore questions than answers. Immunol. Rev. 1999;168:177–185. doi: 10.1111/j.1600-065x.1999.tb01292.x. [DOI] [PubMed] [Google Scholar]