

Abstract
The c-Jun NH2-terminal kinases (JNKs) are a group of mitogen-activated protein (MAP) kinases that participate in signal transduction events mediating specific cellular functions. Activation of JNK is regulated by phosphorylation in response to cellular stress and inflammatory cytokines. Here, we demonstrate that JNK is regulated by a second, novel mechanism. Induction of Jnk gene expression is required in specific tissues before activation of this signaling pathway. The in vivo and in vitro ligation of the T cell receptor (TCR) leads to induction of JNK gene and protein expression. TCR signals are sufficient to induce JNK expression, whereas JNK phosphorylation also requires CD28-mediated costimulatory signals. Therefore, both expression and activation contribute to the regulation of the JNK pathway to ensure proper control during the course of an immune response.
Keywords: c-Jun NH2-terminal kinase, gene expression, T cells, phosphorylation, signal transduction
Introduction
Naive (precursor) CD4+ T cells recognize specific MHC–peptide complexes on APCs via the TCR complex. In addition to TCR-mediated signals, a costimulatory signal is provided at least partially by the ligation of CD28 expressed on T cells with B7 proteins on APCs 1. The combination of these two signals is necessary for full activation and clonal expansion of T cells. Engagement of the TCR and costimulatory molecules initiates multiple signal transduction cascades that lead to the activation of transcription factors and the expression of genes necessary for T cell responses.
The mitogen-activated protein (MAP) kinases have been implicated in the death, proliferation, and differentiation of mammalian cells 2. The extracellular signal–regulated kinases (ERKs), c-Jun NH2-terminal kinases (JNKs, also called stress-activated protein kinases [SAPKs]), and p38 MAP kinases constitute three distinct pathways within the MAP kinase family. Activation of each MAP kinase requires dual phosphorylation on threonine and tyrosine residues within the protein kinase subdomain VIII by MAP kinase kinases (MKKs) 3. Two MKKs that mediate JNK activation have been identified. MKK4, also known as SEK1, was initially identified as a component of the JNK signaling pathway that directly phosphorylates JNK 4 5 6. Disruption of the MKK4 gene in mice has shown that MKK4 is a physiological activator of JNK 7 8. MKK4 can also activate the p38 MAP kinase in vitro 4 5. More recently, the MKK7 protein kinase was identified and shown to specifically phosphorylate JNK 9 10 11 12. MKK4 and MKK7 are activated by MAP kinase kinase kinases, including members of the MEK kinase (MEKK) and the mixed-lineage kinase (MLK) groups of protein kinases 13. The presence of multiple upstream activators of JNK potentially offers a mechanism to achieve specific responses from different stimuli through the JNK pathway. Various forms of cellular stress and inflammatory cytokines activate the JNK pathway. Upon activation, JNK phosphorylates c-Jun, which heterodimerizes with Fos proteins to form the transcription factor activator protein 1 (AP-1). Other JNK substrates include JunD, activating transcription factor 2 (ATF2), Elk1, and nuclear factor of activated T cells 4 (NFAT4) 2 14.
Previous studies have implicated MAP kinase signaling pathways in the control of immune responses 2. The specific roles of MAP kinases in regulating T cell activation and/or differentiation are beginning to be understood. During the activation of T lymphocytes, MAP kinase pathways are triggered upon ligation of the TCR, although JNK but not ERK activation needs a costimulatory signal provided by CD28 15. A costimulatory signal is also required to activate transcription mediated by AP-1 16.
Three different Jnk genes (Jnk1, Jnk2, and Jnk3) are alternatively spliced to create 10 JNK protein kinases that differ in their interaction with transcription factors 17. Recent reports characterizing mice deficient of Jnk genes suggest that differential expression of these genes may influence the role of the JNK pathway in various tissues 18 19 20 21. Mice deficient of either Jnk1 or Jnk2 exhibit severe defects in T cell–mediated immune responses 18 19 20. CD4+ T cells from JNK1 knockout mice produce increased amounts of IL-4, IL-5, and IL-10, and preferentially develop a Th2 immune response 18. Impaired IFN-γ production and diminished Th1 responses have been observed in CD4+ T cells from mice lacking JNK2 19. These studies establish that the JNK1 and JNK2 protein kinases are essential for the normal function of T cells. In contrast, Jnk3 is expressed in the brain, testis, and heart, and Jnk3 knockout mice are defective in excitotoxic stress–induced neuronal apoptosis 21.
Due to the complex nature of signal transduction pathways, the JNK cascade requires strict control to ensure accurate cellular responses to specific stimuli. Here, we report a novel mechanism of JNK regulation in T cells. Expression of Jnk genes is induced in T cells upon activation. TCR-mediated signals are sufficient to induce JNK expression, whereas activation of JNK requires both ligation of the TCR and CD28-mediated costimulatory signals. The additional regulatory mechanism we have identified may play a role in maintaining proper control of JNK-mediated signals that are necessary for T cell activation.
Materials and Methods
Cell Purification.
Total CD4+ T cells were isolated from spleen and lymph nodes from wild-type mice by negative selection using anti-NK (NK1.1; PharMingen), anti-CD8 (PharMingen), and anti–MHC class II mAbs to deplete NK, CD8, and B cells, respectively 22 23. Total T cells were isolated using only anti-NK1.1 and anti–MHC class II mAbs.
Northern Blot Analysis.
Total RNA was extracted using the UltraSpec™ RNA isolation system (Biotex Laboratories, Inc.) as recommended by the manufacturer. Total RNA (10 μg) was analyzed by Northern blot as described previously 24. Specific cDNA probes for JNK1 and JNK2 were labeled with [γ-32P]dCTP using the Random Primer kit (Stratagene, Inc.). For analysis of Jnk tissue distribution ( Fig. 1 A), poly(A)+ RNA was isolated from different mouse tissues (Clontech) and analyzed as described.
Figure 1.




Delayed activation of JNK and AP-1 in resting CD4+ T cells. (A) Northern blot analysis of different mouse tissues for Jnk1 and Jnk2. A β-actin probe served as a control. (B) Analysis of JNK activity in CD4+ T cells stimulated with immobilized anti-CD3 (5 μg/ml) and soluble anti-CD28 (1 μg/ml) mAbs for short (left) or long (right) time periods. GST–c-Jun was used as the substrate. (C) Purified CD4+ T cells stimulated as in B were examined for JNK activity by intracellular staining using an antiphospho-JNK polyclonal antibody (P-JNK) followed by an FITC-conjugated secondary antibody. Background staining determined by intracellular staining of cells with the secondary antibody alone is represented by the dark vertical line. (D) Analysis of luciferase activity in CD4+ T cells purified from AP-1 luciferase reporter transgenic mice (references 16, 22) that were stimulated as in B for the indicated time periods. (E) Purified CD4+ T cells were stimulated as in B for the indicated time periods, and IL-2 production was measured.
Protein Kinase Assays.
Protein kinase assays were performed as described 25 26. T cell lysates were incubated in Triton lysis buffer with glutathione S-transferase (GST)–c-Jun immobilized on glutathione (GSH)-agarose beads. After 12 h at 4°C, the beads were washed extensively in lysis buffer followed by kinase assay buffer, and the activity of the bound JNK was detected by the addition of [γ-32P]ATP for 30 min at 30°C. The reaction products were resolved by SDS-PAGE, and the incorporation of [32P] phosphate was quantitated by PhosphorImager® analysis (Molecular Dynamics).
Intracellular Staining.
Analysis of JNK activity by intracellular staining was performed as directed by PharMingen. Cells were harvested, surface stained with a PE-conjugated anti-CD4 mAb, fixed in 4% paraformaldehyde, resuspended in permeabilization buffer (1% FCS, 0.1% sodium azide, and 0.1% saponin in PBS), and stained with an antiphospho-JNK polyclonal antibody (New England Biolabs), followed by staining with an FITC-conjugated anti–rabbit IgG (Caltag). Cells were then analyzed by flow cytometry. To examine JNK expression, cells were harvested, fixed in 4% paraformaldehyde, resuspended in permeabilization buffer, and stained with an anti-JNK mAb (PharMingen), followed by staining with an FITC-conjugated anti–mouse IgG (Jackson ImmunoResearch Laboratories). Cells were then surface stained with a PE-conjugated anti-CD4 mAb and analyzed by flow cytometry.
IL-2 Production.
IL-2 production was measured using the CTLL assay, as described previously 27. CTLL cells were incubated with conditioned media from cells of interest for 16–18 h and subsequently pulsed with [3H]thymidine for 6 h. CTLL cells were harvested, and IL-2 production was determined as a measurement of CTLL proliferation.
AP-1 Transcriptional Activity.
AP-1 transcriptional activity was determined by analysis of the luciferase activity in cell extracts from AP-1 luciferase reporter transgenic mice 16 22 using the Luciferase Assay kit (Promega Corp.).
Western Blot Analysis.
JNK protein levels were assayed by immunoblot analysis using an anti–human JNK mAb (PharMingen). Immune complexes were detected by enhanced chemiluminescence as instructed by the manufacturer (Kirkegaard & Perry).
Results and Discussion
We analyzed the tissue distribution of Jnk1 and Jnk2 genes by Northern blot and observed that both genes were widely expressed in several tissues ( Fig. 1 A). However, Jnk1 mRNA was absent and only a low amount of Jnk2 mRNA was detected in mouse spleen ( Fig. 1 A). Similarly, Jnk1 and Jnk2 mRNAs were not detected in lymph nodes (data not shown). The low levels of Jnk1 and Jnk2 gene expression in peripheral lymphoid organs suggested that the JNK signaling pathway was not functional in resting cells from these immune tissues.
Ligation of the TCR–CD3 complex in combination with costimulatory signals provided by CD28 activates JNK in cultured Jurkat T cells 15. Therefore, we performed in vitro studies of JNK activity in primary CD4+ T cells isolated from mouse spleen and lymph nodes. Stimulation with anti-CD3 and anti-CD28 mAbs for short periods of time (0–120 min) failed to increase JNK activity ( Fig. 1 B, left). However, increased JNK activity was observed in CD4+ T cells activated for longer time periods (24–48 h; Fig. 1 B, right). Similar results were obtained by flow cytometry using an antibody that specifically recognizes the activated (Thr and Tyr dual phosphorylated) form of JNK ( Fig. 1 C). In correlation with these results, other studies have reported only marginal JNK activity in primary T cells upon stimulation of the TCR in combination with costimulation for short periods of time 20 22 28.
c-Jun is a component of the AP-1 transcription factor and is phosphorylated by JNK at Ser-63 and Ser-73 within the transactivation domain 3. Therefore, we compared the time course of AP-1 activity with the delayed kinetics of JNK activity in stimulated T cells. AP-1 luciferase reporter transgenic mice 16 22 were used to monitor AP-1 transcriptional activity in primary CD4+ T cells. Stimulation with anti-CD3 and anti-CD28 mAbs for long periods of time (24–48 h) was necessary for induction of AP-1 transcriptional activity in primary CD4+ T cells ( Fig. 1 D). Similar results were obtained using CD4+ T cells activated with concanavalin A in the presence of APCs 22. Together, these data indicate that both JNK and AP-1 display delayed kinetics of increased activity during stimulation of primary resting CD4+ T cells.
The AP-1 regulatory element present in the IL-2 promoter has been shown to be important for early transcription of the IL-2 gene in cultured Jurkat T cells 29. However, analysis of IL-2 production by primary mouse CD4+ T cells showed that IL-2 expression occurred before the detection of increased JNK and AP-1 activity ( Fig. 1 E), suggesting that AP-1 and JNK are not required for the initial production of IL-2. The lack of a requirement of JNK and AP-1 for IL-2 expression is consistent with the observation that normal levels of IL-2 are produced by naive CD4+ T cells deficient of either Jnk1 or Jnk2 when stimulated through the TCR in the presence of costimulation 18 19 20. The role of JNK in the regulation of IL-2 production by other T cell populations (e.g., memory CD4+ T cells) remains to be determined.
What is the mechanism that accounts for the delayed increase in JNK activity detected in stimulated primary CD4+ T cells? One possible explanation relates to the observation that only low amounts of Jnk1 and Jnk2 gene expression were detected in lymphoid tissues ( Fig. 1 A). Increased expression of JNK may be required before increased JNK activity is observed. To test this hypothesis, we examined Jnk expression by splenocytes stimulated with either PMA plus ionomycin or with anti-CD3 mAb by Northern blot analysis ( Fig. 2 A). Both Jnk1 and Jnk2 mRNAs were increased in activated cells compared with unstimulated cells ( Fig. 2 A). To determine whether Jnk gene expression could be induced in specific lymphoid populations, we examined Jnk1 and Jnk2 mRNA in T and B cells isolated from spleen and lymph nodes. No Jnk1 mRNA was detected in either resting B cells or T cells by Northern blot analysis; in T cells, a low amount of Jnk2 mRNA could be observed ( Fig. 2 B, left). Interestingly, PMA and ionomycin stimulation greatly increased Jnk1 and Jnk2 mRNA in both T and B cells ( Fig. 2 B, left). Unlike T cells, both Jnk1 and Jnk2 mRNAs were detected in thymocytes ( Fig. 2 B, left). These data indicate that Jnk gene expression is inducible in lymphoid cells in vitro. To determine whether this regulation also occurred during an immune response in vivo, we examined Jnk gene expression in T cells from mice immunized with staphylococcal enterotoxin B (SEB). A large increase in Jnk mRNA was detected in T cells from SEB-injected mice compared with control mice ( Fig. 2 C). Thus, Jnk gene expression was increased after antigen stimulation in vivo.
Figure 2.
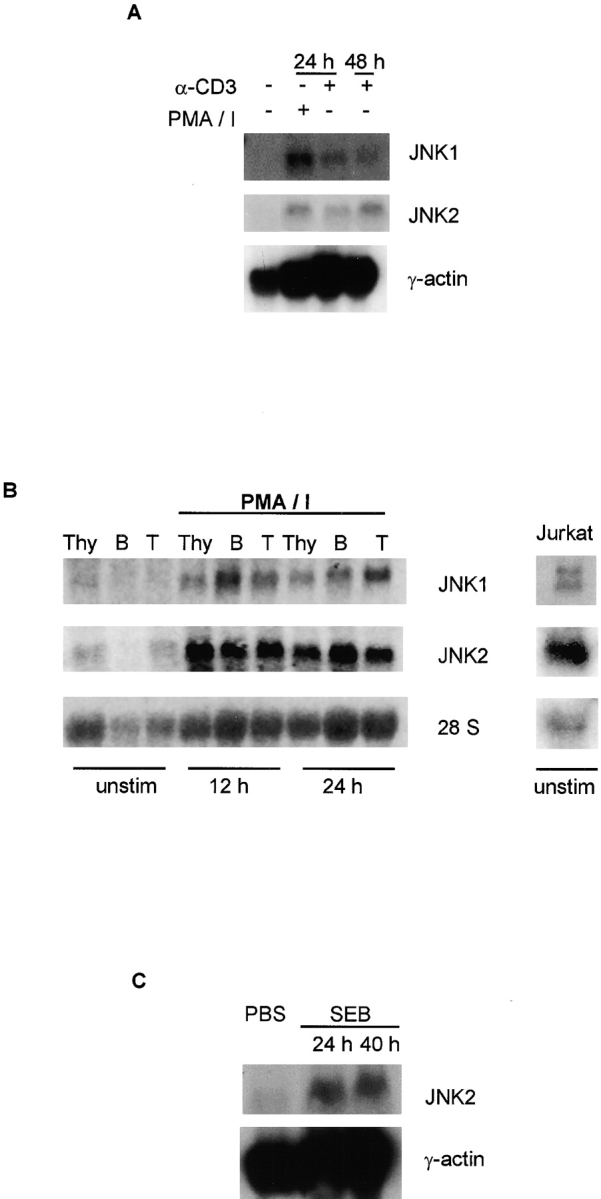

JNK expression in lymphoid tissues. (A) Northern blot analysis of Jnk expression in spleen cells stimulated with PMA (5 ng/ml) and ionomycin (250 ng/ml) (PMA/I) or with soluble anti-CD3 mAb (1 μg/ml) for the indicated time periods. A γ-actin probe served as a control. (B) Total RNA was isolated from unstimulated or PMA and ionomycin (PMA/I) stimulated thymocytes (Thy), B cells (B), and T cells (T), or from unstimulated Jurkat cells. Jnk expression was analyzed by Northern blot using mouse (left) or human (right) Jnk1 and Jnk2 cDNA probes. 28S ribosomal RNA expression served as a control. (C) Northern blot analysis of T cells purified from mice injected intraperitoneally with either PBS or SEB (50 ng in 200 μl PBS) for the indicated time periods. A γ-actin probe served as a control. (D) Western blot analysis of JNK from T cells stimulated with PMA and ionomycin (PMA/I) for the indicated time periods (left), or from unstimulated Jurkat cells (right). (E) JNK expression in CD4+ T cells stimulated with PMA and ionomycin for the indicated time periods was determined by intracellular staining using an anti-JNK mAb followed by an FITC-conjugated secondary antibody. Background staining determined by intracellular staining of cells with the secondary antibody alone is represented by the vertical line.
To test whether the increased Jnk mRNA in activated T cells correlated with increased JNK protein, we examined JNK1 and JNK2 expression by immunoblot analysis of CD4+ T cells. JNK1 was not detected, and only a low amount of JNK2 was present in unstimulated cells ( Fig. 2 D, left). However, both the 55- and 46-kD forms of JNK1 and JNK2 were strongly increased upon stimulation with PMA and ionomycin ( Fig. 2 D, left). We also examined the expression of JNK in activated CD4+ T cells by flow cytometry using an anti-JNK mAb that recognizes both JNK1 and JNK2. This analysis confirmed that JNK expression by CD4+ T cells increased after stimulation with PMA and ionomycin ( Fig. 2 E). Together, these data demonstrate that JNK is present only in low amounts in primary T cells, but increased JNK expression occurs after T cell activation. Furthermore, examination of MAP kinase kinase expression by Northern blot analysis indicated that Mkk4 and Mkk7 mRNAs were also increased in activated T cells (data not shown), suggesting that increased gene expression may be important not only for JNK, but also for upstream components of the JNK signaling pathway in activated T cells.
In contrast to the delayed kinetics of JNK activation observed in primary CD4+ T cells ( Fig. 1 B), JNK was reported to be rapidly activated in cultured Jurkat T cells after treatment with anti-CD3 and anti-CD28 mAbs 15. This rapid activation of JNK suggests that, unlike primary T cells, the cultured Jurkat cells may express JNK1 and JNK2 before stimulation. Therefore, we examined JNK expression by unstimulated Jurkat cells. High amounts of both Jnk1 and Jnk2 mRNAs ( Fig. 2 B, right) and JNK1 and JNK2 proteins ( Fig. 2 D, right) were detected in Jurkat cells. The presence of JNK1 and JNK2 in unstimulated Jurkat cells may account for the difference in the time course of increased JNK activity between Jurkat cells (early) and primary mouse T cells (delayed).
T cells require two signals for activation. The first signal is provided by the TCR, and the second costimulatory signal is mediated by CD28. A TCR signal is sufficient to activate the ERK MAP kinase pathway, whereas an additional costimulatory signal mediated by CD28 is required to increase JNK activity in Jurkat cells 15. A CD28 costimulatory signal is also required for activation of AP-1 in naive CD4+ T cells 16, but not in effector Th2 cells 22. Therefore, we investigated the involvement of costimulation by CD28 in the regulation of JNK expression by T cells. Purified CD4+ T cells were stimulated with anti-CD3 mAb alone or in combination with anti-CD28 mAb. Stimulation with anti-CD3 mAb in the absence of additional costimulatory signals was sufficient to increase expression of Jnk1 and Jnk2 mRNA ( Fig. 3 A). IL-2 production was detected only after treatment with anti-CD3 mAb together with anti-CD28 mAb, excluding the possibility that cells cultured with anti-CD3 mAb alone contained a source of costimulation (data not shown). These data indicate that a TCR signal alone was sufficient to increase Jnk expression by CD4+ T cells.
Figure 3.

Signaling requirement for JNK expression and activation. (A) Jnk expression was analyzed by Northern blot in CD4+ T cells stimulated with immobilized anti-CD3 mAb (5 μg/ml) (Y-CD3) alone or in combination with soluble anti-CD28 mAb (1 μg/ml) for the indicated time periods. 28S ribosomal RNA served as a control. (B) Northern blot analysis of Jnk expression in splenocytes stimulated with soluble anti-CD3 mAb (1 μg/ml) alone or in the presence of CTLA4-Ig (15 μg/ml). A γ-actin probe served as a control. (C) Western blot analysis of JNK in CD4+ T cells stimulated with immobilized anti-CD3 mAb (Y-CD3) alone or in combination with soluble anti-CD28 mAb for the indicated time periods. (D) Analysis of JNK activity in CD4+ T cells stimulated with immobilized anti-CD3 mAb (Y-CD3) alone or in combination with soluble anti-CD28 mAb for 48 h. GST–c-Jun was used as the substrate.
To test whether interference with CD28-mediated signals could modify Jnk expression, we examined the effect of soluble CTLA4-Ig, which can bind the CD28 ligands B7.1 and B7.2 expressed by APCs 30. Jnk expression was detected in total spleen cells, which contain APCs in addition to T cells, when stimulated with anti-CD3 mAb ( Fig. 3 B). However, when costimulation was blocked with CTL antigen–Ig (CTLA4-Ig), the increased expression of Jnk was unaffected ( Fig. 3 B). These data confirm the conclusion that increased Jnk gene expression by stimulated T cells was independent of costimulation.
Immunoblot analysis demonstrated that the amounts of JNK1 and JNK2 were increased after treatment of CD4+ T cells with anti-CD3 mAb alone, indicating that TCR signals were sufficient for increased expression of JNK protein ( Fig. 3 C). In contrast, both anti-CD3 and anti-CD28 mAbs were required for JNK activation ( Fig. 3 D), as reported in a previous study of Jurkat T cells 15. Together, these data suggest that the two modes of JNK regulation observed in primary T cells (gene expression and protein phosphorylation) differ in their signaling requirements. Signals induced upon TCR activation were sufficient for increased JNK expression, whereas activation of JNK by phosphorylation required a costimulatory signal mediated by CD28 in addition to TCR signaling.
The combination of TCR and costimulatory signals triggers the proliferation and differentiation of primary CD4+ T cells into effector Th1 and Th2 cells, which can rapidly secrete high levels of specific cytokines during an immune response. We have previously shown that, unlike primary CD4+ T cells, effector Th1 and Th2 cells contain high levels of JNK mRNA and protein 19; consequently, the JNK signaling pathway is rapidly activated (30 min) in Th1 cells in response to antigen stimulation 19. In correlation, reduced IFN-γ production by Th1 cells is observed in JNK2-deficient mice 19, whereas mice lacking JNK1 exhibit predominant Th2 responses 18. Therefore, we propose that regulation of JNK gene expression constitutes a mechanism to ensure that JNK activation is coordinated with specific cellular events required during immune responses.
The constitutive expression of JNK in thymocytes but not in mature T cells ( Fig. 2) suggested that suppression of Jnk gene expression may be required before the migration of thymocytes to the peripheral immune system. In the thymus, several cell populations can be identified based on cell surface markers and represent different stages of maturation. Immature CD4+CD8+ double-positive thymocytes downregulate either CD4 or CD8 to become mature single CD4+ or single CD8+ thymocytes, which can then leave the thymus and enter the spleen or lymph nodes. We examined the expression of JNK in CD4+CD8+ double-positive thymocytes, CD4+ single-positive thymocytes, and CD4+ T cells isolated from spleen and lymph nodes. Interestingly, Jnk1 and Jnk2 mRNAs were detected in both immature CD4+CD8+ thymocytes and mature CD4+ thymocytes, but not in peripheral CD4+ T cells ( Fig. 4 A). Examination of JNK expression by flow cytometry indicated that JNK was present in both CD4+CD8+ and CD4+ thymocytes, but was not detected in peripheral CD4+ T cells ( Fig. 4 B). These data suggest that the CD4+ thymocytes and peripheral CD4+ T cells can respond differentially to specific stimuli. A large fraction of CD4+ thymocytes undergoes apoptosis (negative selection) in response to signals mediated by TCR and CD28 31. In contrast, the combination of these two signals results in the activation of peripheral CD4+ T cells. Thus, the absence of JNK in peripheral resting CD4+ T cells may prevent death of these cells during the early phase of antigen stimulation. Analysis of JNK expression in two previously described subsets of CD4+ thymocytes (HSAhigh and HSAlow [31]) suggests that downregulation of JNK expression occurs within the thymus (Weiss, L., and M. Rincón, unpublished data).
Figure 4.

JNK expression in the thymus. (A) CD4+CD8+ double-positive (DP) and CD4+ single-positive thymocyte populations were obtained by FACS® after staining the cells with anti-CD4 and anti-CD8 mAbs. Peripheral CD4+ T cells were purified from lymph nodes and spleens. Total RNA was isolated and examined by Northern blot analysis. (B) JNK expression in CD4+CD8+ double-positive thymocytes (DP) and CD4+ single-positive thymocytes (SP CD4+) or purified peripheral CD4+ T cells was determined by intracellular staining using an anti-JNK mAb followed by an FITC-conjugated secondary antibody (thick histograms). Background staining determined by intracellular staining of cells with the secondary antibody alone (thin histograms).
The regulation of JNK expression represents a novel mechanism for controlling MAP kinase signaling in mammalian cells. This form of regulation may contribute to the control of MAP kinase signaling in plants 32 and yeast 33, suggesting that it is a functional mechanism conserved among organisms. It is likely that this mechanism contributes to the regulation of other MAP kinase pathways in differentiated mammalian cells. Our results indicate that JNK expression is strictly regulated during maturation and activation of T cells to provide coordination between the JNK signaling pathway and the immune response.
Acknowledgments
We thank C. Charland for expert flow cytometry analysis, and C. Merritt for technical assistance.
This work was supported by National Institutes of Health grant R29 AI42138 (to M. Rincón), and National Cancer Institute grants CA58396 and CA72009 (to R.J. Davis). R.J. Davis and R.A. Flavell are Investigators of the Howard Hughes Medical Institute.
Footnotes
D.D. Yang's present address is Lilly Research Laboratory, Eli Lilly and Co., Indianapolis, IN 46285.
Abbreviations used in this paper: AP-1, activator protein 1; ERK, extracellular signal–regulated kinase; GST, glutathione S-transferase; JNK, c-Jun NH2-terminal kinase; MAP, mitogen-activated protein; MKK, MAP kinase kinase; SAPK, stress-activated protein kinase; SEB, staphylococcal enterotoxin B.
References
- Linsley P.S., Ledbetter J.A. The role of the CD28 receptor during T cell responses. Annu. Rev. Immunol. 1993;11:191–212 . doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- Ip Y.T., Davis R.J. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr. Opin. Cell Biol. 1998;10:205–219 . doi: 10.1016/s0955-0674(98)80143-9. [DOI] [PubMed] [Google Scholar]
- Whitmarsh A.J., Davis R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996;17:2360–2371 . doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- Dérijard B., Rainjeaud J., Barret T., Wu I.-H., Han J., Ulevitch R.J., Davis R.J. Independent human MAP kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:683–685 . doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- Lin A., Minden A., Martinetto H., Claret F.-X., Lange-Carter C., Mercurio F., Johnson G.L., Karin M. Identification of a dual specificity kinase that activates the Jun kinases and p38-Mpk2. Science. 1995;268:286–290 . doi: 10.1126/science.7716521. [DOI] [PubMed] [Google Scholar]
- Sanchez I., Hughes R.T., Mayer B.J., Yee K., Woodgett J.R., Avruch J., Kyriakis J.M., Zon L.I. Role of SAP/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature. 1994;372:794–798 . doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- Nishina H., Bachmann M., Oliveira-dos-Santos A.J., Odermatt B., Wakeham A., Shahinian A., Takimoto H., Bernstein A., Mak T.M., Woodgett J.R. Impaired CD28-mediated interleukin 2 production and proliferation in stress kinase SAPK/ERK1 kinase (SEK1)/mitogen-activated protein kinase kinase 4 (MKK4)-deficient T lymphocytes. J. Exp. Med. 1997;186:941–953 . doi: 10.1084/jem.186.6.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D., Tournier C., Wysk M., Lu H.-T., Jie X., Davis R.J., Flavell R.A. Targeted disruption of the MKK4 gene causes embryonic death inhibition of c-Jun NH2 terminal kinase activation, and defects in AP-1 transcriptional activity. Proc. Natl. Acad. Sci. USA. 1997;94:3004–3009 . doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland P.M., Suzanne M., Campbell J.S., Noselli S., Cooper J.A. MKK7 is a stress-activated mitogen-activated protein kinase kinase functionally related to hemipterous. J. Biol. Chem. 1997;272:24994–24998 . doi: 10.1074/jbc.272.40.24994. [DOI] [PubMed] [Google Scholar]
- Lu X., Nemoto S., Lin A. Identification of c-Jun NH2-terminal protein kinase (JNK)-activating kinase 2 as an activator of JNK but not p38. J. Biol. Chem. 1997;272:24751–24754 . doi: 10.1074/jbc.272.40.24751. [DOI] [PubMed] [Google Scholar]
- Moriguchi T., Toyoshima F., Masuyama N., Hanafusa H., Gotoh Y., Nishida E. A novel SAPK/JNK kinase, MKK7, stimulated by TNFα and cellular stresses. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:7045–7053 . doi: 10.1093/emboj/16.23.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournier C., Whitmarsh A.J., Cavanagh J., Barrett T., Davis R.J. Mitogen-activated protein kinase kinase 7 is an activator of the c-Jun NH2-terminal kinase. Proc. Natl. Acad. Sci. USA. 1997;94:7337–7342 . doi: 10.1073/pnas.94.14.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger G.R., Gerwins P., Widmann C., Jarpe M., Johnson G. MEKKs, GCKs, MLKs, PAKs, TAKs and Tplsupstream regulators of the c-Jun amino-terminal kinase? Curr. Opin. Genet. Dev. 1997;7:67–74 . doi: 10.1016/s0959-437x(97)80111-6. [DOI] [PubMed] [Google Scholar]
- Minden A., Karin M. Regulation and function of the JNK subgroup of MAP kinases. Biochim. Biophys. Acta. 1997;1333:F85–F104 . doi: 10.1016/s0304-419x(97)00018-8. [DOI] [PubMed] [Google Scholar]
- Su B., Jacinto E., Hibi M., Kallunki T., Karin M., Ben-Neriah Y. JNK is involved in signal integration during costimulation of T lymphocytes. Cell. 1994;77:727–736 . doi: 10.1016/0092-8674(94)90056-6. [DOI] [PubMed] [Google Scholar]
- Rincón M., Flavell R.A. AP-1 transcriptional activity requires both T-cell receptor-mediated and co-stimulatory signals in primary T lymphocytes. EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:4370–4381 . doi: 10.1002/j.1460-2075.1994.tb06757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Barret T., Whitmarsh A.J., Cavanagh J., Sluss H.K., Dérijard B., Davis R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2760–2770 . [PMC free article] [PubMed] [Google Scholar]
- Dong C., Yang D.D., Wysk M., Whitmarsh A.J., Davis R.J., Flavell R.A. Defective T cell differentiation in the absence of Jnk1. Science. 1998;282:2092–2095 . doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Conze D., Whitmarsh A.J., Barret T., Davis R.J., Rincón M., Flavell R.A. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585 . doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- Sabapathy K., Hu Y., Kallunki T., Schreiber M., David J.-P., Jochum W., Wagner E.F., Karin M. JNK2 is required for efficient T-cell activation and apoptosis but not for normal lymphocyte development. Curr. Biol. 1999;9:116–125 . doi: 10.1016/s0960-9822(99)80065-7. [DOI] [PubMed] [Google Scholar]
- Yang D.D., Kuan C.-Y., Whitmarsh A.J., Rincón M., Zheng T.S., Davis R.J., Rakic P., Flavell R.A. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870 . doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Rincón M., Dérijard B., Chow C.-W., Davis R.J., Flavell R.A. Reprogramming the signaling requirement for AP-1 (activator protein-1) activation during differentiation of precursor CD4+ T cells to effector Th1 and Th2 cells. Genes Funct. 1997;1:51–68 . doi: 10.1046/j.1365-4624.1997.00007.x. [DOI] [PubMed] [Google Scholar]
- Kamogawa Y., Minasi L.-A.E., Carding S., Bottomly K., Flavell R.A. The relationship of IL-4 and IFNγ-producing T cells studied by lineage ablation of IL-4-producing cells. Cell. 1993;75:985–995 . doi: 10.1016/0092-8674(93)90542-x. [DOI] [PubMed] [Google Scholar]
- Rincón M., Tugores A., López-Rivas A., Silva A., Alonso M., de Lándazuri M.O., López-Botet M. Prostaglandin E2 and the increase of intracellular cAMP inhibit the expression of interleukin 2 receptors in human T cells. Eur. J. Immunol. 1988;18:1791–1796 . doi: 10.1002/eji.1830181121. [DOI] [PubMed] [Google Scholar]
- Dérijard B., Hibi M., Wu I.-H., Barret T., Su B., Deng T., Karin M., Davis R.J. JNK1a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–1037 . doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- Raingeaud J., Gupta S., Roger J., Dickens M., Han J., Ulevitch R.J., Davis R.J. Pro-inflammatory cytokines and environmental stress cause p38 MAP kinase activation by dual phophorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426 . doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- Gillis S., Ferm M.N., Smith K.A. T cell growth factorparameters of production and quantitative microassay for activity. J. Immunol. 1978;120:1109–1113 . [PubMed] [Google Scholar]
- Swat W., Fujikawa K., Ganiatsas S., Yang D.D., Xavier R.J., Harris N.L., Davidson L., Ferrini R., Davis R.J., Labow M.A. SEK1/MKK4 is required for maintenance of a normal peripheral lymphoid compartment but not for T lymphocyte development. Immunity. 1998;8:625–634 . doi: 10.1016/s1074-7613(00)80567-1. [DOI] [PubMed] [Google Scholar]
- Durand D.B., Shaw J.P., Bush M.R., Replogle R.E., Belageje R., Crabtree G.R. Characterization of antigen receptor response elements within the interleukin 2 enhancer. Mol. Cell. Biol. 1988;8:1715–1724 . doi: 10.1128/mcb.8.4.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson C. Distinct roles for the costimulatory ligands B7.1 and B7.2 in T helper cell differentiation. Cell. 1995;81:979–982 . doi: 10.1016/s0092-8674(05)80001-7. [DOI] [PubMed] [Google Scholar]
- Kishimoto H., Sprent J. Negative selection in the thymus includes semimature T cells. J. Exp. Med. 1997;185:263–271 . doi: 10.1084/jem.185.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirt H. Multiple roles of MAP kinases in signal transduction in plants. Trends Plant Sci. 1997;2:1–7 . [Google Scholar]
- Krisak L., Strich R., Winters S., Hall J.P., Mallory M.J., Kreitzer D., Tuan R.S., Winter E. SMK1, a developmentally regulated MAP kinase, is required for spore wall assembly in Saccharomyces cerevisiae . Genes Dev. 1994;8:2151–2161. doi: 10.1101/gad.8.18.2151. [DOI] [PubMed] [Google Scholar]