

Abstract
Helicobacter pylori colonizes the gastric epithelium of ∼50% of the world's population and plays a causative role in the development of gastric and duodenal ulcers. H. pylori is phagocytosed by mononuclear phagocytes, but the internalized bacteria are not killed and the reasons for this host defense defect are unclear. We now show using immunofluorescence and electron microscopy that H. pylori employs an unusual mechanism to avoid phagocytic killing: delayed entry followed by homotypic phagosome fusion. Unopsonized type I H. pylori bound readily to macrophages and were internalized into actin-rich phagosomes after a lag of ∼4 min. Although early (10 min) phagosomes contained single bacilli, H. pylori phagosomes coalesced over the next ∼2 h. The resulting “megasomes” contained multiple viable organisms and were stable for 24 h. Phagosome–phagosome fusion required bacterial protein synthesis and intact host microtubules, and both chloramphenicol and nocodazole increased killing of intracellular H. pylori. Type II strains of H. pylori are less virulent and lack the cag pathogenicity island. In contrast to type I strains, type II H. pylori were rapidly ingested and killed by macrophages and did not stimulate megasome formation. Collectively, our data suggest that megasome formation is an important feature of H. pylori pathogenesis.
Keywords: phagocytosis, phagosome maturation, pathogenicity island, phagocytic killing, Yersinia enterocolitica
Introduction
Helicobacter pylori (Hp) is a microaerophilic gram-negative bacterium that colonizes the gastric epithelium of at least 50% of the world's population and plays a causative role in the development of chronic gastritis, gastric and duodenal ulcers, and gastric adenocarcinoma 1 2. One hallmark of Hp is its persistence in spite of the host immune response, and colonization of the stomach is associated with bacterial replication followed by chronic inflammation. The immune response to Hp is characterized by the production of IgG and secretory IgA, the synthesis and release of proinflammatory cytokines such as IL-6, IL-8, GM-CSF, and TNF-α, and the recruitment of neutrophils, mononuclear phagocytes, and lymphocytes to the gastric mucosa 1 2 3 4. Phagocyte recruitment correlates directly with the ability of bacteria to induce gastritis, and it is thought that both bacterial products and host inflammatory mediators contribute to the subsequent tissue damage 1 2 3. To date, strains of Hp have been divided into two groups 4 5 6 7. Type I strains contain the cag pathogenicity island (PAI) and secrete a vacuolating cytotoxin (VacA). In contrast, type II strains lack the cag PAI and synthesize small amounts of inactive VacA. Type I strains are more prevalent in persons with ulcer disease and induce more inflammation and tissue damage than do the less virulent type II strains 1 2 3 8.
The available data suggest that bacterial persistence can be explained in part by the failure of Hp to be killed by professional phagocytes. Phagocytosis occurs in vivo, particularly at sites of tissue damage such as ulcer margins and in regions of metaplasia 9 10 11 12. Unopsonized Hp bind readily to mononuclear phagocytes and neutrophils, and organisms are internalized into close-fitting phagosomes 13 14 15 16 17. Importantly, however, only about half of the ingested bacilli are killed 16 17 18. The reasons for this host defense defect are unknown, and Hp–phagocyte interactions are only beginning to be explored at the molecular level. As phagocytosis is an essential component of the innate immune response, it is likely that the failure of macrophages and neutrophils to eliminate ingested Hp contributes significantly to bacterial persistence.
Many pathogenic microorganisms evade killing in macrophages by preventing phagosome–lysosome fusion. For example, Listeria monocytogenes and Shigella flexneri escape from the phagosome and replicate in the cytosol (for review see reference 19). Legionella pneumophilia resides in a compartment that does not fuse with endosomes or lysosomes 19, and phagosomes containing pathogenic mycobacteria fuse with endosomes but do not acidify 19 20 21. In contrast, pathogens such as Coxiella survive and replicate in acidic phagolysosomes 19. Thus far, the Hp phagosome has not been characterized. To this end, we used immunofluorescence and electron microscopy (EM) to monitor ingestion of Hp by human and murine macrophages. We now show that phagocytosis of virulent type I Hp (but not less virulent type II organisms) exhibits two unusual features. First, actin polymerization and phagosome formation are delayed until several minutes after bacterial attachment to macrophages. Second, Hp phagosomes undergo extensive clustering and fusion during the first few hours after bacterial ingestion. Organisms inside these “megasomes” remained viable for at least 24 h. Collectively, our data support the hypothesis that intracellular Hp reside in a protected niche 16 22.
Materials and Methods
Materials.
Trypticase soy agar, Brain heart infusion broth, Luria-Bertani broth (LB), and LB agar were obtained from Difco Labs., Inc. Pyrogen-free tissue culture reagents Dulbecco's (D)PBS, RPMI, Hepes–RPMI, MEMα, DMEM, l-glutamine, and penicillin/streptomycin were from BioWhittaker, Inc. FBS was from HyClone or GIBCO BRL. Round glass coverslips (12 mm) were from Fisher Scientific Co. PMA was from LC Labs. Live-Dead BacLight Bacterial Viability Kit, rhodamine–phalloidin, FITC–phalloidin, acridine orange, and 4′,6-diamidino-2-phenylindole hydrochloride (DAPI) were from Molecular Probes, Inc. Polyclonal (p)Abs to Hp (YVS-6601) were from Accurate Chemical and Science Corp. Mouse mAbs to β-tubulin (1111876) were obtained from Boehringer Mannheim. Affinity-purified tetramethylrhodamine isothiocyanate (TRITC)- or FITC-conjugated donkey anti–rabbit IgG F(ab′)2 and FITC-conjugated goat anti–mouse IgG+IgM were from Jackson ImmunoResearch Labs., Inc. Mouse mAbs to talin (8d4) and other reagents were obtained from Sigma Chemical Co.
Bacterial Strains and Culture Conditions.
Type I (11637 [type strain] and 60190) and type II (Tx30a) Hp were obtained from the American Type Culture Collection (ATCC). Hp were grown on trypticase soy agar, pH 6, containing 5% sheep blood (Remel) under microaerophilic conditions (5% O2, 10% CO2, 85% N2) and 98% humidity in a Heraeus trigas incubator (Heraeus Instruments) at 37°C. Fresh plates were started from glycerol stocks each week and passaged after 48 h. Where indicated, liquid cultures of Hp were grown in Brain heart infusion broth containing 10% heat-inactivated (HI) FBS. Avirulent plasmid-cured Yersinia enterocolitica (Ye) 8081c 23 was grown overnight in LB or on LB agar at 30°C. Bacteria from plates or liquid cultures were washed with DPBS, and concentrations were estimated using OD600 = 0.1 as 108 Hp or 5 × 107 Ye. Viability of washed bacteria was determined by vital staining using the Live-Dead BacLight Bacterial Viability kit according to the manufacturer's instructions. Cultures with <90% viable organisms were discarded. For experiments using dead organisms, Hp were killed by heating to 65°C for 10 min.
Macrophage Isolation and Culture.
Resident macrophages were harvested by peritoneal lavage from female CD-1 (ICR) mice (Charles River Labs.; references 24 and 25) and plated in MEMα supplemented with 10% HI FBS, 1% l-glutamine, 100 U/ml penicillin G, and 100 μg/ml streptomycin. After 2 h at 37°C, lymphocytes were removed by washing, and adherent macrophages were incubated overnight at 37°C in antibiotic-free medium before use. The murine macrophage cell line J774a.1 (ATCC TIB-67) and U-937 human histiocytic lymphoma cells (ATCC CRL-1593.2) were grown in DMEM or RPMI supplemented with HI FBS, l-glutamine, penicillin, and streptomycin as indicated above. U-937 cells were differentiated into adherent macrophage-like cells in medium containing 20 nM PMA for 48–72 h 26 before use. Human monocyte-derived macrophages (MDMs) were obtained by culturing PBMCs in teflon wells for 5 d at 37°C in RPMI containing 10% HI autologous serum 27 and were a gift from Dr. Richard Fawcett (University of Iowa). MDMs were purified by adherence to collagen-coated coverslips. All macrophages were incubated for at least 16 h in antibiotic-free medium before contact with bacteria.
Synchronized Phagocytosis and Phagocytic Killing.
Bacteria were dispersed in Hepes–RPMI containing 10% HI FBS at a ratio of 25 bacteria/macrophage unless indicated otherwise, and phagocytosis was synchronized by centrifugation. Hp and Ye were bound to macrophages by a 3-min, 12°C spin at 600 g, and phagocytosis was initiated by rapid warming to 37°C after a DPBS wash. Attachment indices (bacteria per 100 macrophages), phagocytic indices (phagosomes per 100 macrophages), and bacterial viability after 0–60 min at 37°C was determined using vital staining. Macrophages were washed three times with DPBS (to remove any unbound organisms), permeabilized with 1 mg/ml saponin (5 min, 25°C), and stained with BacLight reagents diluted in DPBS. Green (viable) and red (dead) cell-associated bacteria were counted using fluorescence microscopy. At least 300 consecutive macrophages were counted per experiment in triplicate samples. Parallel staining of unpermeabilized macrophages determined that the fraction of cell-associated bacteria that was not ingested was <5% at 30 min. Phagocytic killing at later time points (2–24 h) was determined using the gentamicin-CFU method 28. In brief, uningested organisms were killed by adding 100 μg/ml gentamicin (BioWhittaker, Inc.) to the culture medium 1 h after initiation of phagocytosis. After 60 min in gentamicin, macrophages were washed and returned to antibiotic-free medium at 37°C. 2–24 h after initiation of phagocytosis, diluted macrophage lysates were plated in triplicate for determination of CFU.
Immunofluorescence Microscopy.
For immunofluorescence microscopy (IFM), cells were plated on uncoated acid-washed glass coverslips in 35-mm dishes (peritoneal macrophages, J774, or PMA-treated U-937) or coverslips coated with collagen (MDMs) to achieve 1–2 × 105 cells per 12-mm coverslip. After the desired treatments, macrophages were fixed in 10% buffered formalin (Sigma Chemical Co.) and permeabilized in −20°C acetone 24 25. Fixed cells were blocked overnight at 4°C in DPBS containing 0.5 mg/ml NaN3, 5 mg/ml BSA, and 10% horse serum (blocking buffer) and then incubated sequentially with primary and secondary Abs or fluorescent phalloidins, washed, and mounted using mowiol as we previously described 24 25. Staining with phalloidins detects F-actin on forming phagosomes and correlates with particle ingestion 24 25. Reagents were diluted in blocking buffer as follows: anti-Hp pAb, 1:3,000; FITC–phalloidin, 1:300; rhodamine–phalloidin, 1:500; anti–β-tubulin mAb, 1:50; antitalin mAb, 1:200; and all secondary Abs, 1:100 or 1:200. Specificity of staining was verified by omission of primary Abs and by the use of mouse, rabbit, or rat isotype control Abs (Zymed Labs., Inc.). Immunofluorescence was viewed using a Zeiss Axioplan2 photomicroscope (Carl Zeiss, Inc.). Cells were photographed using Kodak Ektachrome ASA 400 color slide film (Eastman Kodak Co.). Composite images were generated using Adobe Photoshop 3.0 (Adobe Systems, Inc.).
Transmission Electron Microscopy.
Macrophages (8 × 105 per glass coverslip) were incubated with Hp at a ratio of 100 bacteria/cell, and phagocytosis was synchronized using centrifugation as described above. Gentamicin treatment was performed as described above except that the drug was added 2 h after initiation of phagocytosis. After 10 min–24 h at 37°C, infected macrophages were fixed in 2.5% glutaraldehyde/0.1 M sodium cacodylate buffer, and samples were processed for transmission (T)EM essentially as previously described 29 30. Samples were examined using a Hitachi H-7000 transmission electron microscope in the University of Iowa Central Microscopy Research Facility. To quantify phagosome–phagosome fusion, the number of bacilli in each macrophage section and the number of bacilli per phagosome were counted in samples from one to three independent experiments. For each sample containing strain 11637, at least 50 sections and 200 phagosomes were scored. For strain Tx30a, a minimum of 46 sections and 100 phagosomes was scored.
Quantitation of Coalesced Phagosomes Using Fluorescence Microscopy.
Macrophages were infected with bacteria and processed for IFM as indicated above. Fixed and permeabilized cells on triplicate coverslips were stained with DAPI (1:1,000 dilution) to detect Ye or with anti-Hp pAbs and FITC- or TRITC-conjugated secondary Abs. The number of phagosomes in ≥200 macrophages per coverslip was counted using fluorescence and phase optics. Phagosomes with apparent diameters greater than three bacilli were scored as megasomes, and all others were scored as single phagosomes. For each experiment, cell-associated bacterial aggregates (originating from bacterial stock suspensions) were enumerated in samples fixed immediately after the centrifugation step or after 5 min at 37°C, and these values were subtracted from the data shown.
Other Methods.
To depolymerize microtubules (MTs), macrophages were pretreated with 2 μg/ml nocodazole or 2 μg/ml colcemid for 10 min at 37°C before addition of Hp and initiation of phagocytosis. Depolymerization was confirmed by staining nocodazole- or colcemid-treated cells with mAbs to β-tubulin 31. After 20 h in medium containing nocodazole or colcemid, 10–15% of the macrophages had detached from the culture surface. Therefore, for overnight time points, both adherent cells and macrophages in the culture medium were collected for determination of CFU. To inhibit protein synthesis, macrophages or Hp were preincubated for 30 min at 37°C in Hepes–RPMI containing 100 μg/ml cycloheximide or 30–100 μg/ml chloramphenicol, and inhibitors were maintained in the culture medium during and after phagocytosis. Protein synthesis was quantified by labeling triplicate cultures of 3 × 105 macrophages or 6 × 108 Hp with 15 μCi/ml [35S]methionine–cysteine Express Protein Labeling Mix (43.5 TBq/mmol; New England Nuclear) in methionine/cyteine-free RPMI (GIBCO BRL) in the presence or absence of chloramphenicol and cycloheximide for 2 h at 37°C. Incorporation of radioactivity into proteins was quantified by precipitation with 10% TCA and liquid scintillation counting 32. Protein concentrations were determined using the BCA Protein Assay Kit (Pierce Chemical Co.). Assessment of Hp uptake using the double-immunofluorescence method was performed using anti-Hp pAbs essentially as previously described 33.
Results
Type I Hp Are Ingested by Macrophages yet Resist Phagocytic Killing.
Previous studies have shown that unopsonized Hp is inefficiently killed by human phagocytes 16 17 18. Similarly, we found that Hp 11637 or 60190 remained viable in resting mouse peritoneal macrophages or J774 cells 24 h after ingestion, whereas avirulent Ye 8081c did not ( Fig. 1). The ability of pathogens such as Mycobacterium tuberculosis 19 21 to disrupt phagosome maturation and phagosome–lysosome fusion is thought to be an important aspect of bacterial virulence and survival. However, the mechanism by which type I strains of Hp resist phagocytic killing is unknown. In this study, we used IFM and TEM to characterize the Hp phagosome.
Figure 1.

Phagocytic killing of Hp by murine macrophages is impaired. Peritoneal macrophages were incubated with Hp 11637 or Ye 8081c at a ratio of 1:25, and phagocytosis was synchronized using centrifugation. As indicated in Materials and Methods, bacterial viability before gentamicin treatment (15 min and 1 h) was determined by vital staining of permeabilized macrophages, whereas viability at later times (2–24 h) was determined by plating macrophage lysates for CFU. Data shown are the average ± SD of three independent experiments. Note that the y-axis is a log scale. Comparable data were obtained using Hp 60190 and J774 cells (not shown). Similar killing curves were generated using 5–100 bacteria per phagocyte (not shown). Mφ, macrophage.
Phagosome morphology and the kinetics of bacterial ingestion were followed using FITC– or rhodamine–phalloidin and fluorescence microscopy ( Fig. 2). As neither Ye nor Hp bound to macrophages at 4°C, phagocytosis was synchronized using centrifugation. Ye, which binds to β1 integrins 34, was rapidly internalized into close-fitting conventional phagosomes ( Fig. 2). The kinetics of Ye uptake were similar to those we obtained previously for zymosan, IgG beads, and complement-opsonized particles 24 25. Hp 11637 and 60190 were also detected in close-fitting phagosomes. However, unlike with Ye or other particles, both actin rearrangements beneath attached Hp and phagosome formation were significantly delayed relative to bacterial binding ( Fig. 2). Thus, most Ye were phagocytosed within 0.5–1 min, whereas actin polymerization in the vicinity of bound Hp was negligible until 3–4 min and peaked at 5–7 min. Ingestion kinetics similar to those shown in Fig. 2 (i.e., a delay with virulent Hp) were also obtained using Abs to the cytoskeletal protein talin or using the double-immunofluorescence method (data not shown). Moreover, in contrast to viable organisms, ingestion of dead Hp resembled Ye and occurred without a lag; 81.3 ± 7.0% of cell-associated heat-killed Hp were in actin-positive phagosomes after 1 min at 37°C (n = 3). Neither the receptor that mediates phagocytosis of Hp nor the accompanying signaling events have been elucidated. Nevertheless, it is tempting to speculate that delayed uptake of Hp may reflect alterations of the internalization process that are important for bacterial survival in macrophages.
Figure 2.
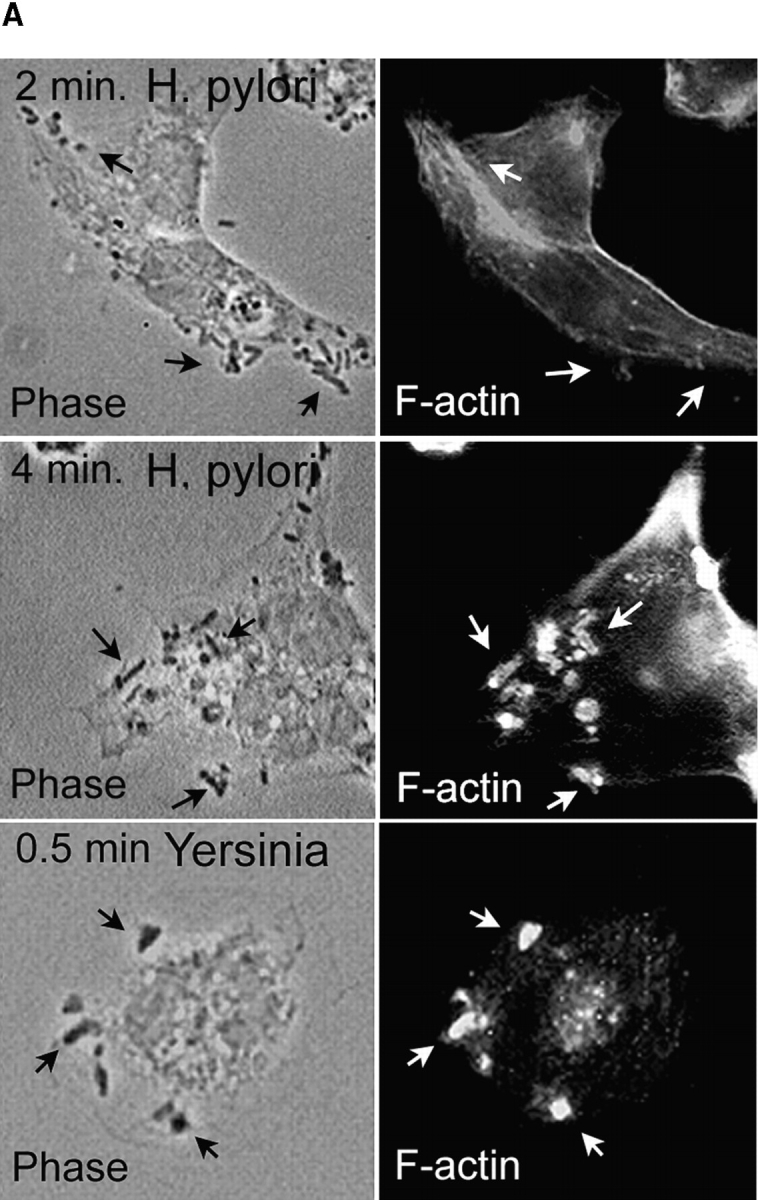

Ingestion of Hp by macrophages is delayed relative to bacterial binding. Phagocytosis of Hp 11637 or Ye by peritoneal macrophages was synchronized using centrifugation. After incubation at 37°C for the indicated times, forming phagosomes were detected by staining fixed and permeabilized macrophages with FITC– or rhodamine–phalloidin, and cell-associated organisms were detected using phase contrast microscopy. (A) Representative forming phagosomes containing Hp or Ye. Left column, phase contrast; Right column, F-actin. Forming phagosomes containing Ye were abundant after 0.5 min at 37°C (arrows, bottom panels). Actin rearrangements were not detected beneath bound Hp after 2 min at 37°C (arrows, top panels); however, numerous Hp phagosomes were detected after 4 min at 37°C (arrows, center panels). (B) Kinetics of phagosome formation and bacterial ingestion. Adherent macrophages ingested Hp or Ye for 0–15 min at 37°C before processing for IFM. F-actin was detected as in A, and the results are expressed as the percentage of actin-positive cell-associated bacteria over time. The total number of cell-associated bacteria per 100 macrophages did not change significantly over the time course of the experiment: 800 ± 87 Hp and 771 ± 106 Ye at 1 min, and 882 ± 91 Hp and 800 ± 92 Ye at 15 min. Data shown are the average ± SD from three independent experiments conducted in triplicate. At least 300 bacteria were scored per sample per time. Comparable data were obtained using J774 cells or Hp 60190 (not shown).
Hp Phagosomes Rapidly Coalesce into Structures Containing Multiple Organisms.
Further characterization of the Hp phagosome demonstrated that phagosomes containing virulent Hp underwent extensive clustering followed by homotypic fusion. In initial experiments using light microscopy (LM), we found that shortly after bacterial uptake Hp, phagosomes appeared to cluster in the cytoplasm as if they were actively migrating toward one another ( Fig. 3 A). Over the next ∼2 h, phagosome clustering continued and was followed by the appearance of larger phagosomes containing multiple viable organisms ( Fig. 3a and Fig. b). These structures were stable for at least 24 h ( Fig. 3 A). Due to their unusual morphology, we have named these large Hp phagosomes “megasomes.” As judged by IFM, we found that megasomes formed similarly in peritoneal macrophages, MDMs, J774 cells ( Fig. 3 B), and PMA-treated U-937 cells (data not shown). After 2 h at 37°C, the percentage of infected macrophages containing at least one megasome was 65.5 ± 10.4 (n = 4). Due to the ∼6 h doubling time of Hp, it is unlikely that bacterial replication contributed to initial megasome formation. However, we cannot exclude a role for Hp replication in megasome maintenance (i.e., at times >6 h). The shape of the killing curve shown in Fig. 1 indicates that Hp remains viable yet may not replicate inside macrophages. Importantly, it is unlikely that megasomes contained Hp aggregates. In contrast to organisms such as M. tuberculosis, viable Hp are not prone to aggregation (Allen, L., unpublished observation). Moreover, the frequency of cell-associated aggregates was quantified for each experiment and did not exceed 2.1% for Hp or 3.2% for Ye.
Figure 3.
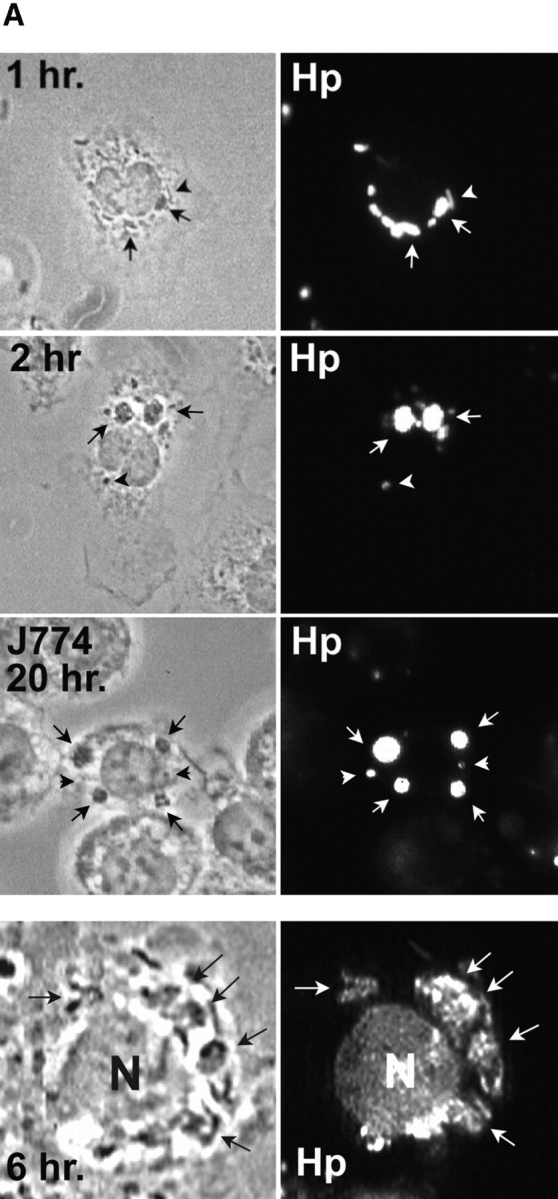

Hp phagosomes rapidly coalesce inside macrophages. Phagocytosis of Hp by adherent macrophages was synchronized as described above. After 1–20 h at 37°C, samples were processed for IFM. (A) Top panels, appearance of Hp megasomes 1–20 h after initiation of phagocytosis of Hp 60190. Peritoneal macrophages, 1–2 h panels; J774 cells, 20 h panels. Fixed and permeabilized cells were stained with pAbs to Hp and secondary Abs conjugated to FITC or TRITC. Left panels, phase contrast; right panels, Hp phagosomes. Arrowheads, small/conventional phagosomes; arrows, Hp megasomes. Note that 2 h megasomes are larger than 1 h megasomes. Bottom panels: 6 h after ingestion of Hp 11637, live macrophages were permeabilized with saponin and stained with BacLight reagents. Hp emitted green fluorescence, demonstrating that these bacteria were viable inside megasomes (right panel, greyscale image of emitted green fluorescence; left panel, phase contrast). Arrows, Hp; N, macrophage nucleus. (B) Time course of megasome formation. Peritoneal macrophages (PM) or human MDMs ingested Hp 11637 or 60190 for the indicated times. Fixed-permeabilized cells were stained with Abs to Hp as described above. Phagosomes were scored as small/conventional or large/megasomes as described in Materials and Methods. The graph indicates the number of megasomes as a percentage of total Hp phagosomes. Data shown are the average ± SD of three independent experiments conducted in triplicate (PMs) or duplicate (MDMs). nd, not determined.
We next used TEM to assess the structure of Hp megasomes at higher resolution. Ultrastructural analysis confirmed that Hp was phagocytosed in a conventional manner and that 93% of early (10 min) phagosomes contained single organisms (Table and Fig. 4A and Fig. B). The remaining 10 min phagosomes appeared to contain two to three organisms each ( Fig. 5). Importantly, our EM data confirmed that megasomes were large phagosomes containing multiple intact Hp ( Fig. 4C–F). Moreover, the results of quantitation experiments demonstrated that megasomes increased in both size and number over time ( Fig. 4 and Fig. 5 and Table ), consistent with the hypothesis that phagosome–phagosome fusion was progressive. After 30 min, the average megasome contained two to three Hp (range, 2–9), and by 24 h the average megasome contained ∼10 Hp (range, 2–50). The extent of phagosome–phagosome fusion is illustrated by the fact that 87–90% of Hp were inside megasomes after 24 h (Table ). Although the vast majority of Hp were inside megasomes after 6–24 h, we did detect many phagosomes containing single Hp in these samples (Table ). Importantly, however, these single phagosomes were usually in close proximity to a megasome ( Fig. 4D and Fig. E). Therefore, some or all of these “single” phagosomes may have been connected to the adjacent megasome in a plane of the macrophage that was not examined. Similar data were obtained for peritoneal macrophages and J774 cells (Table ) and are in good agreement with the results of our LM experiments ( Fig. 3 B). That more megasomes were detected by TEM than IFM likely reflects both the limited resolution of the light microscope and the fact that only very large phagosomes were scored as megasomes for the data shown in Fig. 3 B. Taken together, our data suggest that Hp utilizes an unusual mechanism to avoid phagocytic killing that involves both delayed entry and homotypic phagosome fusion.
Table 1.
Quantitation of Phagosome Size
| Time | Sections | Size* | Percent of total phagosomes | Percent of total Hp |
|---|---|---|---|---|
| P.Mφ | ||||
| 10 min | 54 | single | 93.1 | 85.0 |
| mega. | 6.9 | 15.0 | ||
| 30 min | 60 | single | 70.7 | 45.9 |
| mega. | 29.3 | 54.1 | ||
| 2 h | 58 | single | 76.9 | 45.8 |
| mega. | 23.1 | 54.2 | ||
| 24 h | 52 | single | 43.9 | 9.4 |
| mega. | 56.1 | 90.6 | ||
| J774 | ||||
| 30 min | 56 | single | 68.0 | 34.5 |
| mega. | 32.0 | 65.5 | ||
| 2 h | 57 | single | 75.9 | 41.1 |
| mega. | 24.1 | 58.9 | ||
| 6 h | 53 | single | 69.0 | 24.2 |
| mega. | 31.0 | 75.8 | ||
| 24 h | 54 | single | 51.6 | 12.6 |
| mega. | 48.4 | 87.4 |
Peritoneal macrophages or J774 cells ingested H. pylori 11637 for the indicated times, and for each condition at least 200 phagosomes were scored. Data shown are the number of single phagosomes (single) and megasomes (mega.) as a percentage of total phagosomes, and the distribution of Hp between the two types of phagosomes (total Hp).
P.Mφ, peritoneal macrophage.
* Single phagosomes contain one bacterium; mega. phagosomes contain two or more organisms.
Figure 4.
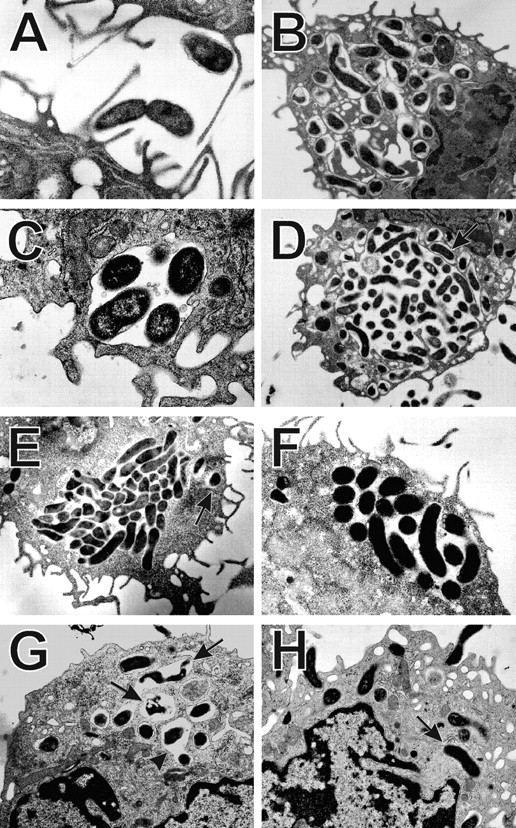
Ultrastructure of Hp phagosomes in macrophages. Peritoneal macrophages (A–D, G, H) or J774 cells (E and F) were infected with Hp 11637 (A–F) or Tx30a (G and H) at a ratio of 100 bacteria/macrophage. At various times after bacterial ingestion, samples were processed for TEM as described in Materials and Methods. (A) Ingestion of Hp. (B) 10 min phagosomes containing single bacteria. (C) 30 min megasome. (D and F) 24 h megasomes. (E) 6 h megasome in a J774 cell. Arrows in D and E indicate single phagosomes adjacent to megasomes. (G and H) 30 min phagosomes containing Tx30a. These type II Hp were found in conventional phagosomes (H, arrows), and some of the organisms appeared degraded (G, arrows). Arrowhead in G indicates a rare Tx30a phagosome that might contain two organisms. Magnifications: A and C, 20,000; B, 10,000; D, E, G, and H, 7,000; F, 12,000.
Figure 5.
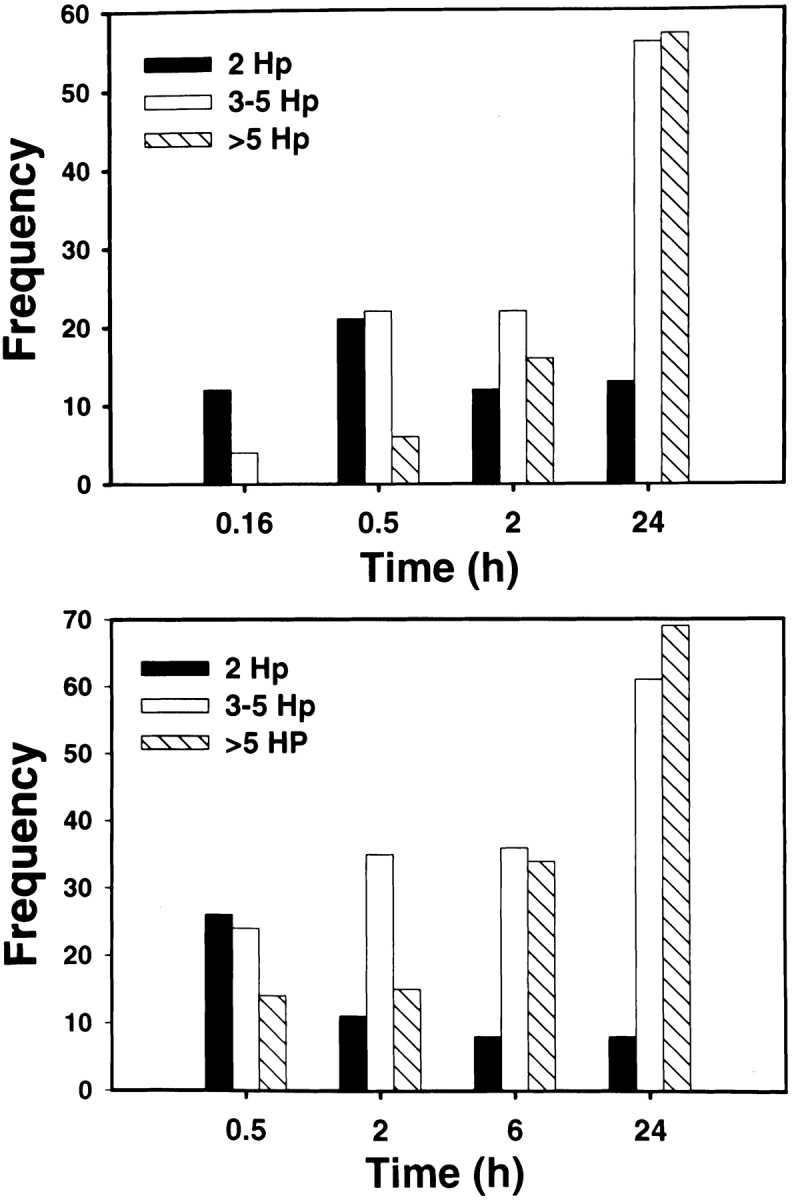
Megasomes increase in size and number over time. Peritoneal macrophages (top panel) or J774 cells (bottom panel) ingested Hp 11637 for the indicated times before processing for TEM. To assess whether phagosome–phagosome fusion was progressive, both the total number of megasomes (Table ) and the number of bacteria per megasome (this figure) was scored in cell sections as described in Materials and Methods. “Frequency” indicates the number of megasomes containing 2 Hp (black bars), 3–5 Hp (white bars), or >5 Hp (hatched bars) at each time point. Phagosomes containing single Hp are not shown in this figure.
Megasome Formation Requires Bacterial Protein Synthesis and Intact MTs.
Intracellular organelle transport is mediated by the cytoskeleton, and previous studies have shown that phagosomes move bidirectionally along microtubular tracts in macrophages 35 36. As clustering and fusion of Hp phagosomes appeared to be highly dynamic, we explored the role of MTs in this process. MTs were not essential for Hp phagocytosis, and the rate of Hp ingestion was not altered in nocodazole-treated cells ( Fig. 6). In contrast, we found that depolymerization of macrophage MTs with either nocodazole or colcemid inhibited megasome formation by ∼90% ( Fig. 7 A), and the few megasomes that did form were reduced in size (compare Fig. 7 B to Fig. 5, Fig. 2 h time point). Moreover, in the absence of intact MTs, 99% of ingested Hp were killed ( Fig. 7 C). However, neither drug impaired Hp viability in the absence of macrophages (data not shown). Efficient phagocytic killing of Hp in the absence of MTs was somewhat surprising, as MT-destabilizing drugs can reduce killing of some microorganisms via their ability to block phagosome–lysosome fusion 37. Nevertheless, we found that Ye was also killed by nocodazole-treated macrophages (2 logs killing at 3 h in the presence of nocodazole vs. 1.5 logs killing in the absence of the drug, n = 3). These data suggest that nonlysosomal killing mechanisms were bacteriocidal under these conditions. Although the exact nature of the Hp phagosome remains undefined, our data indicate that megasome formation is important for Hp survival in macrophages.
Figure 6.
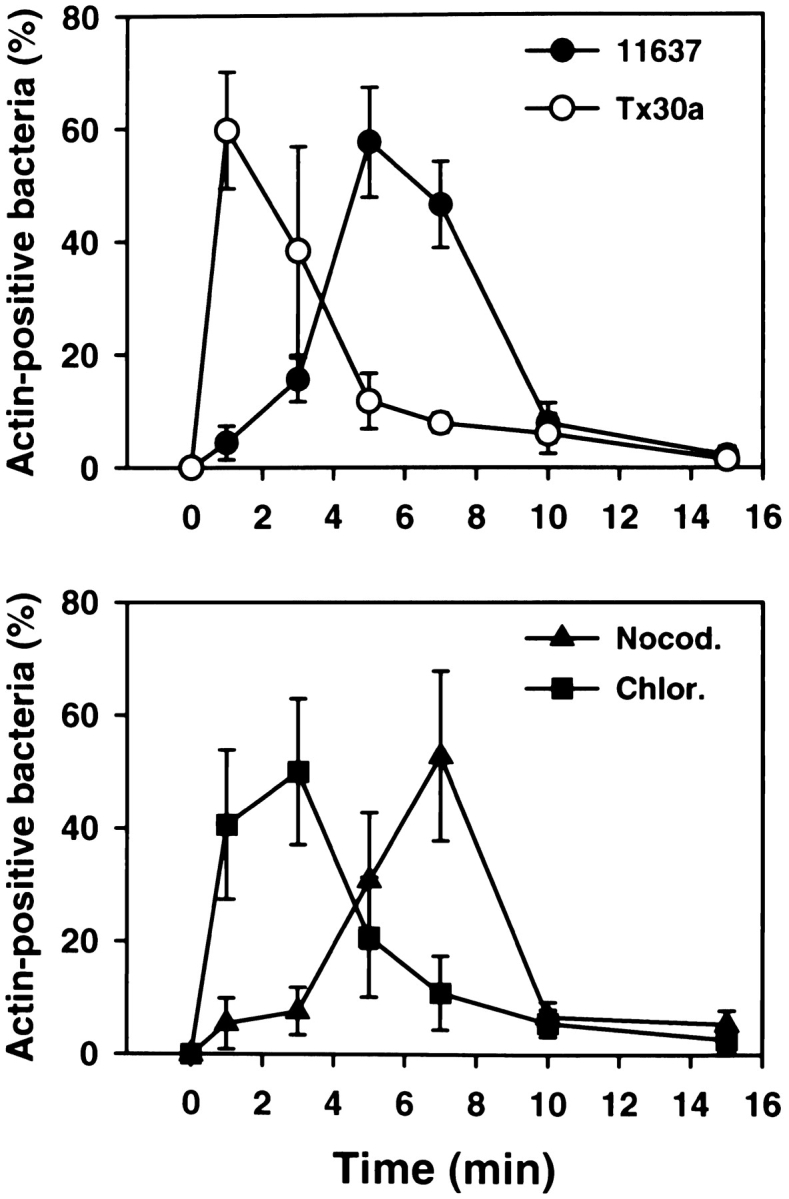
Kinetics of phagocytosis of Hp 11637 and Tx30a. Top panel: peritoneal macrophages were infected with Hp 11637 or Tx30a, and the kinetics of phagocytosis were followed in fixed and permeabilized cells double-stained with Abs to Hp and FITC–phalloidin. Results are expressed as described for Fig. 2 B. In contrast to the wild-type strain, phagocytosis of Tx30a was not significantly delayed relative to bacterial binding. Data shown are the average ± SD of three (11637) or five (Tx30a) independent experiments conducted in triplicate. Bottom panel: effect of nocodazole (Nocod.) and chloramphenicol (Chlor.) on the rate of phagocytosis of Hp 11637. Phagocytosis assays were performed in the presence of 2 μg/ml nocodazole or 100 μg/ml chloramphenicol as described above. Inhibition of bacterial protein synthesis, but not depolymerization of MTs, increased the rate of phagocytosis of Hp 11637. Data shown are the average ± SD of two (nocodazole) or four (chloramphenicol) independent experiments run in triplicate.
Figure 7.



Nocodazole and chloramphenicol inhibit megasome formation and increase intracellular killing of Hp. Peritoneal macrophages and Hp were treated with 2 μg/ml nocodazole, 30–100 μg/ml chloramphenicol, or 100 μg/ml cycloheximide as described in Materials and Methods. Megasome formation and megasome size was scored using LM and TEM. Phagocytic killing was assayed as described above. (A) Nocodazole and chloramphenicol inhibit megasome formation. Macrophages and Hp were treated with nocodazole (Nocod.), chloramphenicol (Clr.), or cycloheximide (Chx.) as indicated, and samples were fixed-processed for LM and TEM 2 h after initiation of phagocytosis. The graphs show the number of megasomes as a percentage of all Hp phagosomes. Top panel: megasomes were scored in fixed and permeabilized cells using LM. Data shown are the average ± SD of three to six independent experiments performed in triplicate. Bottom panel, effect of 100 μg/ml chloramphenicol and 2 μg/ml nocodazole on megasome formation as judged by TEM. Con., control. (B) Effect of 2 μg/ml nocodazole and 100 μg/ml chloramphenicol on megasome size. Macrophages were fixed and processed for TEM 2 h after initiation of phagocytosis. “Frequency” indicates the number of megasomes containing 2 Hp (black bars), 3–5 Hp (white bars), or >5 Hp (hatched bars). The size of 2 h megasomes in control macrophages is shown in Fig. 5. (C) Macrophages and Hp 11637 were left untreated or incubated with 2 μg/ml nocodazole or 100 μg/ml chloramphenicol, and phagocytic killing was measured after 0.5–20 h. Data shown are the average ± SD of four independent experiments.
We next examined whether eukaryotic or bacterial protein synthesis played a role in megasome formation. Hp and macrophages were pretreated with chloramphenicol or cycloheximide for 30 min at 37°C before initiation of phagocytosis, and inhibitors remained in the culture medium for the duration of each experiment. As shown in Fig. 7 A, we found that the bacterial protein synthesis inhibitor chloramphenicol reduced phagosome–phagosome fusion in a dose-dependent manner: megasome formation was reduced 16 and ∼72% by 30 and 100 μg/ml chloramphenicol, respectively. On the other hand, megasome formation did not change significantly in the presence of the eukaryotic protein synthesis inhibitor cycloheximide ( Fig. 7 A). These data suggest that clustering and fusion of Hp phagosomes was induced by metabolically active Hp yet was independent of macrophage protein synthesis. Consistent with this hypothesis, we found that heat-killed Hp did not induce megasome formation, and 98.1 ± 1.2% of these bacteria were inside single phagosomes (n = 5). [35S]methionine labeling experiments demonstrated the efficacy and specificity of the inhibitors used: 100 μg/ml of cycloheximide inhibited macrophage protein synthesis 79.8 ± 7.7% and Hp protein synthesis 1.0 ± 4.9%; 30 μg/ml of chloramphenicol inhibited macrophage protein synthesis 19.6 ± 18.0% and Hp protein synthesis 46.2 ± 15.0%; and 100 μg/ml of chloramphenicol inhibited macrophage protein synthesis 19.5 ± 1.1% and Hp protein synthesis 69.8 ± 6.7% (n = 3).
Next, we determined if bacterial protein synthesis was required for Hp uptake or intracellular killing. In addition to its effects on protein synthesis, chloramphenicol is bacteriostatic for most microorganisms 38 39. We found that the OD600 of Hp liquid cultures declined by 13 ± 4% (n = 3) after 20 h in medium containing 100 μg/ml of chloramphenicol, whereas the optical density of control cultures increased threefold. Although chloramphenicol was not toxic to Hp in the absence of macrophages, this drug dramatically increased phagocytic killing such that only ∼1% of ingested organisms survived ( Fig. 7 B). In contrast to the MT-destabilizing agents discussed above, chloramphenicol also affected the rate of Hp entry ( Fig. 6), and maximum phagocytosis occurred within 1–3 min of bacterial binding. Collectively, these data indicate that only metabolically active Hp exhibited delayed phagocytosis followed by megasome formation. The data support the hypothesis that Hp modifies its environment in macrophages and indicate a role for megasome formation in Hp persistence.
Type II Hp Are Rapidly Ingested and Killed by Macrophages and Are Not Found in Megasomes.
Type II strains of Hp lack the cag PAI in the chromosome, produce a nontoxic form of VacA, and are thought to be less virulent 1 2 3. However, the interaction of type II Hp with macrophages has not been explored. Therefore, we determined if Hp Tx30a 5 was more susceptible to phagocytic killing than were 11637 or 60190 and if these bacteria were found in megasomes. Using both LM and TEM, we found that unopsonized Tx30a readily bound to macrophages, and adherent bacilli were internalized into conventional close-fitting phagosomes ( Fig. 4G and Fig. H). Phagocytosis of Tx30a was rapid, and actin-positive phagosomes were numerous after 1–3 min at 37°C ( Fig. 6). In this regard, uptake of Tx30a resembled that of chloramphenicol-treated 11637 ( Fig. 6). In contrast to type I Hp, phagosomes containing Tx30a did not coalesce ( Fig. 4G and Fig. H, and Fig. 8 A). As judged by IFM, megasome formation was reduced by 75 and 72% 0.5 and 2 h after ingestion, respectively ( Fig. 8 A). Similarly, ultrastructural analysis of 102 phagosomes in sections from 46 macrophages demonstrated that 94% of Tx30a phagosomes appeared to contain single bacilli ( Fig. 4G and Fig. H). Three Tx30a phagosomes may have contained two organisms each ( Fig. 4 G, arrowhead). However, it was unclear whether these three organelles contained pairs of bacilli or whether there was a sectioning artifact. Larger vacuoles were not observed. Significantly, we also found that Tx30a was more susceptible to phagocytic killing than wild-type Hp. Damaged Tx30a were detected inside phagosomes as early as 30 min after ingestion ( Fig. 4 G, arrows), and by 20 h, 99% of ingested organisms were killed ( Fig. 8 B). These data suggest that the ability to induce megasome formation is a unique feature of virulent Hp and support the hypothesis that both delayed entry and megasome formation are important for Hp survival in macrophages.
Figure 8.


Tx30a does not induce megasome formation and is efficiently killed. Peritoneal macrophages were infected with Hp 11637 or Tx30a as described above. (A) After 0.5 or 2 h of phagocytosis at 37°C, fixed and permeabilized cells were stained with pAb to Hp and secondary Abs conjugated to TRITC. Megasome formation was scored using immunofluorescence and phase contrast microscopy. The graph indicates the number of megasomes as a percentage of total Hp phagosomes. Data shown are the average ± SD of three independent experiments conducted in triplicate. (B) Phagocytic killing of Tx30a and 11637 was determined as described for Fig. 1. Initial average phagocytic indices were 735 ± 20 and 670 ± 26 for 11637 and Tx30a, respectively. 20 h after initiation of phagocytosis, 99.5 ± 0.4% of ingested Tx30a were killed. Data shown are the average ± SD of four independent experiments. Similar data were obtained using J774 cells (data not shown).
Discussion
Hp has infected at least half of the world's population. In spite of the prevalence of this bacterium and the significant morbidity associated with ulcer disease, our understanding of the pathogenesis of Hp infection is in its infancy. Hp colonizes the mucus layer of the stomach, and acidic pH triggers bacterial invasion of the epithelium 40 41 42 43. Both secreted bacterial factors and invasion are cytotoxic 41 44 45 46 47 48, and cell death allows Hp to colonize exposed basement membranes and facilitates contact with macrophages 49 50 51 52 53. Although multiple studies have shown that Hp is not readily killed by professional phagocytes, the mechanism of bacterial survival has remained obscure. We now show that phagocytosis and intracellular trafficking of unopsonized Hp by macrophages is characterized by two unusual features. First, Hp phagocytosis is delayed for several minutes relative to other particles tested in this paper and previous work 24 25. Second, ingestion is followed by homotypic phagosome fusion. The resulting phagosomes are stable, contain multiple viable bacilli, and are named megasomes to indicate their large size. We found that phagosome–phagosome fusion required host cell MTs and was restricted to metabolically active type I strains of Hp. By contrast, type II strains of Hp, which are associated with asymptomatic infection rather than invasive ulcer disease, were ingested without a lag, did not induce megasome formation, and did not persist inside macrophages. Collectively, our data indicate that phagosome–phagosome fusion is required for Hp survival in macrophages and as such suggest that megasome formation is an important aspect of Hp virulence.
Phagocytosis is driven by receptor–ligand interactions that induce localized actin polymerization 54. Using EM, we demonstrated that Hp was internalized into conventional phagosomes. Importantly, however, our synchronized phagocytosis assay revealed that localized actin polymerization and Hp internalization were delayed until several minutes after bacterial attachment. To our knowledge, delayed phagocytosis has not previously been described for other microorganisms, and we have shown that phagocytosis via mannose receptors 24, Fc receptors, or complement receptors in activated cells 25 is rapid and occurs with kinetics similar to those shown here after Ye engagement of β1 integrin receptors. Although Chmiela et al. 55 demonstrated that sialic acid–dependent and –independent adhesins support Hp binding to macrophages, the receptor that mediates phagocytosis of these organisms has not been defined. In addition, the mechanism by which Hp alters its ingestion is unknown. Nonetheless, it is tempting to speculate that type I Hp may actively impede one or more of the signaling pathways activated during phagocytosis.
Three lines of evidence support our hypothesis that Hp actively modulates its ingestion by macrophages. First, dead Hp were ingested without a lag. Second, pretreatment of organisms with chloramphenicol shortened the lag between binding and phagocytosis such that maximum ingestion occurred after 1–3 rather than 5–7 min. Third, ingestion of Tx30a resembled that of chloramphenicol-treated 11637. Inhibition of bacterial protein synthesis may deplete short-lived proteins from the outer membranes of type I organisms, and some or all of these proteins may be lacking in type II strains. These data are consistent with a model in which type I and II Hp engage different phagocytic receptors. Indeed, this may also be the case for neutrophils, as type II organisms bind poorly to these cells, whereas type I strains are efficiently ingested (Allen, L., unpublished observation). In vivo, Hp persists at a site of inflammation, and in this milieu Hp-phagocyte interactions may be modulated by serum opsonins. Therefore, we are currently exploring whether complement proteins or IgG alter the rate of phagocytosis of Hp and subsequent megasome formation.
The most striking finding of this study is that phagosomes containing single Hp underwent homotypic fusion. Under normal circumstances, the interaction of phagosomes with one another and with other organelles is tightly regulated, and it is well documented that phagosomes containing inert particles do not undergo homotypic fusion 19. Therefore, the ability of Hp to induce rapid and extensive phagosome–phagosome fusion is significant. In this regard, Hp may most closely resemble Chlamydia trachomatis. Phagosomes containing C. trachomatis elementary bodies fuse with each other to generate an inclusion 56 57, and formation of both megasomes and inclusions requires bacterial protein synthesis (reference 56 and this study). Nevertheless, formation of these two types of organelles can be distinguished by the fact that megasomes are present within 30 min of Hp uptake, whereas generation of inclusions occurs after several hours 56.
Three lines of evidence suggest that megasome formation plays a key role in Hp persistence in macrophages. First, dead organisms were not found in megasomes. Second, treatments that inhibited megasome formation (nocodazole, chloramphenicol) also promoted phagocytic killing. Third, Tx30a was unable to induce megasome formation and did not survive in macrophages. Although megasomes have not been characterized at the molecular level, virulence factors that favor Hp survival in the gastric lumen might also allow bacteria to persistence in an acidic endosomal compartment in macrophages. Moreover, our observation that phagosome–phagosome fusion is induced by type I but not type II organisms suggests a role for VacA and/or the cag PAI in this process.
VacA is a “vacuolating cytotoxin” secreted by all type I strains of Hp. VacA is activated by exposure to acidic pH and, in epithelial cells, induces the formation of dilated acidic vacuoles that are derived from late endosomal membranes 58 59 60. These vacuoles accumulate rab 7 and lamp-1 yet exclude mannose-6-phosphate receptors and lysosomal hydrolases such as cathepsin D. Although the vacA gene is present in type II Hp, the gene sequence differs from type I strains and the protein synthesized appears to be inactive 6. Both vacuolation and megasome formation require intact MTs (reference 60 and this study), and we found that 24 h megasomes are acidic, as judged by their accumulation of acridine orange (reference 61 and Allen, L., unpublished data). Thus, it is tempting to speculate that VacA may be secreted and activated inside phagosomes and thereby promote megasome formation. We are currently investigating whether purified VacA can induce fusion of phagosomes containing latex beads and whether other types of phagosomes merge with the megasome compartment in infected macrophages. Importantly, any role of VacA in phagosome–phagosome fusion does not exclude a role for other Hp proteins. The cag PAI encodes a type IV secretion system 62, and a similar secretion system is required for modification of the L. pneumophilia phagosome in macrophages 62. Factors secreted by the Hp type IV organelle are uncharacterized. Nevertheless, bacterial attachment and secretion are required to induce IL-8 secretion from epithelial cells 62, and this system may also play a role in delaying phagocytosis of Hp or in megasome formation.
A growing body of evidence indicates that some species of “extracellular” bacteria can infect host cells and survive for prolonged periods in intracellular locales. With regard to phagocytes, we show here that type I Hp survives for 24 h in human and murine macrophages. Similarly, group B streptococci remain viable inside macrophages for 24–48 h 63, and Campylobacter jejuni survives inside macrophages for 6–7 d 64. It has been suggested that intracellular organisms are hidden from the immune response and therefore reside in protected niches 16 64. However, the vacuoles occupied by these organisms are at best only partially characterized 63 64. In all cases examined thus far, the ability of bacteria to invade host cells appears to be restricted to certain strains of organisms (such as clinical isolates) and is further modulated by bacterial growth conditions (in vitro) or local signaling events (in vivo) 63 64 65 66. For example, we found that Hp grown at pH 6 induces more megasomes than do bacteria grown at neutral pH (Allen, L., manuscript in preparation). Significantly, for Hp, C. jejuni, and group B streptococci, invasion and persistence in macrophages occurs in the apparent absence of bacterial replication (references 63 and 64 and this study). In fact, it may be that the ability to replicate inside cells, rather than the ability to invade and survive, is the feature that distinguishes some “intracellular” and “extracellular” pathogens. Nevertheless, Hp does not survive indefinitely inside cells, and lysis of infected macrophages has been observed at later times (Allen, L., unpublished observation).
We have shown that Tx30a is efficiently ingested and killed by macrophages in vitro. Nevertheless, infections with type II organisms are not cleared by the host 1 67. This may be explained in part by the fact that type II Hp appear to have only limited contact with phagocytes in vivo. Tx30a and related strains lack the cag PAI and synthesize a nontoxic form of VacA. Consequently, these organisms induce only modest amounts of inflammation, and phagocyte recruitment to the stomach is vastly reduced 1 4 68. Because the available data suggest that Hp–phagocyte contact often occurs at sites of tissue damage 10 11 12 17, the low toxicity of type II Hp may allow these bacteria to persist in the mucus layer and thereby avoid contact with macrophages.
Taken together, the results of this study demonstrate that type I Hp are ingested by an unusual mechanism that involves delayed entry followed by phagosome–phagosome fusion. To our knowledge, this mechanism is unique to Hp. Moreover, our results demonstrate for the first time that “extracellular” bacteria can alter the intraphagosomal environment in macrophages. Antibiotic resistance of Hp and treatment failure is a rapidly growing problem 22 69 70. A clear understanding of how all types of Hp avoid elimination by the host immune response is an essential first step in the development of novel treatments for ulcer disease.
Acknowledgments
The authors thank Dr. Richard Fawcett for the generous gift of MDMs and Mr. Joe Reardon at the VA Medical Center for assistance with figure preparation. We also acknowledge the use of the University of Iowa Central Microscopy Research Facility.
This work was supported in part by funds from the University of Iowa Biosciences Initiative and Central Investment Fund for Research Enhancement (CIFRE) to L. Allen and by funds from the National Institutes of Health (AI33004) to L.S. Schlesinger.
Footnotes
Abbreviations used in this paper: EM, electron microscopy; HI, heat-inactivated; Hp, Helicobacter pylori; IFM, immunofluorescence microscopy; LB, Luria-Bertani broth; LM, light microscopy; MDMs, monocyte-derived macrophages; MT, microtubule; p, polyclonal; PAI, pathogenicity island; TEM, transmission electron microscopy; Ye, Yersinia enterocolitica.
References
- Blaser M.J., Parsonnet J. Parasitism by the “slow” bacterium Helicobacter pylori leads to altered gastric homeostasis and neoplasia. J. Clin. Invest. 1994;94:4–8 . doi: 10.1172/JCI117336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czinn S.J., Nedrud J.G. Immunopathology of Helicobacter pylori infection and disease. Springer Semin. Immunopathol. 1997;18:495–513 . doi: 10.1007/BF00824055. [DOI] [PubMed] [Google Scholar]
- Blaser M.J. Helicobacter pylorimicrobiology of a ‘slow’ bacterial infection. Trends Microbiol. 1993;1:255–260 . doi: 10.1016/0966-842x(93)90047-u. [DOI] [PubMed] [Google Scholar]
- Telford J.L., Covacci A., Rappuoli R., Ghiara P. Immunobiology of Helicobacter pylori infection. Curr. Opin. Immunol. 1997;9:498–503 . doi: 10.1016/s0952-7915(97)80101-x. [DOI] [PubMed] [Google Scholar]
- Xiang Z., Censini S., Bayeli P.F., Telford J.L., Figura N., Rappuoli R., Covacci A. Analysis of expression of CagA and VacA virulence factors in 43 strains of Helicobacter pylori reveals that clinical isolates can be divided into two major types and that CagA is not necessary for expression of the vacuolating cytotoxin. Infect. Immun. 1995;63:94–98 . doi: 10.1128/iai.63.1.94-98.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton J.C., Cao P., Peek R.M., Jr., Tummuru M.K., Blaser M.J., Cover T.L. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori . J. Biol. Chem. 1995;270:17771–17777 . doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]
- Forsyth M.H., Atherton J.C., Blaser M.J., Cover T.L. Heterogeneity in levels of vacuolating cytotoxin gene (vacA) transcription among Helicobacter pylori isolates. Infect. Immun. 1998;66:3088–3094 . doi: 10.1128/iai.66.7.3088-3094.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E.D. Consequences of attachment of Helicobacter pylori to gastric cells. Biomed. Pharmacother. 1997;51:5–12 . doi: 10.1016/s0753-3322(97)87073-4. [DOI] [PubMed] [Google Scholar]
- Hessey S.J., Spencer J., Wyatt J.I., Sobala G., Rathbone B.J., Axon A.T.R., Dixon M.F. Bacterial adhesion and disease activity in Helicobacter associated chronic gastritis. Gut. 1990;31:134–138 . doi: 10.1136/gut.31.2.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori S., Tsutsumi Y. Helicobacter pylori infection in gastric xanthomasimmunohistochemical analysis of 145 lesions. Pathol. Int. 1996;46:589–593 . doi: 10.1111/j.1440-1827.1996.tb03658.x. [DOI] [PubMed] [Google Scholar]
- Steer H.W. Ultrastructure of Campylobacter pylori in vivo. In: Rathbone B.J., Heatley R.V., editors. Campylobacter pylori and Gastroduodenal Disease. Blackwell Scientific Publishers; Oxford, UK : 1989. pp. 146–154. [Google Scholar]
- Asaka M., Kato M., Kudo M., Meguro T., Kimura T., Miyazaki T., Inoue K. The role of Helicobacter pylori in peptic ulcer disease Gastroenterol. Jpn. 28Suppl.1993. 163 167 [DOI] [PubMed] [Google Scholar]
- Chmiela M., Ljungh A., Rudnicka W., Wadstrom T. Phagocytosis of Helicobacter pylori bacteria differing in the heparan sulfate binding by human polymorphonuclear leukocytes. Int. J. Med. Microbiol. Virol. Parasitol. Infect. Dis. 1996;283:346–350 . doi: 10.1016/s0934-8840(96)80070-3. [DOI] [PubMed] [Google Scholar]
- Teneberg S., Miller-Podraza H., Lampert H.C., Evans D.J., Jr., Evans D.G., Danielsson D., Karlsson K.A. Carbohydrate binding specificity of the neutrophil-activating protein of Helicobacter pylori . J. Biol. Chem. 1997;272:19067–19071 . doi: 10.1074/jbc.272.30.19067. [DOI] [PubMed] [Google Scholar]
- Chmiela M., Lelwala-Guruge J., Wadstrom T. Interaction of cells of Helicobacter pylori with human polymorphonuclear leukocytespossible role of hemagglutinins. FEMS Immunol. Med. Microbiol. 1994;9:41–48 . doi: 10.1111/j.1574-695X.1994.tb00472.x. [DOI] [PubMed] [Google Scholar]
- Andersen L.P., Blom J., Nielsen H. Survival and ultrastructural changes of Helicobacter pylori after phagocytosis by human polymorphonuclear phagocytes and monocytes. APMIS. 1993;101:61–72 . [PubMed] [Google Scholar]
- Kist M., Spiegelhalder C., Moriki T., Schaefer H.E. Interaction of Helicobacter pylori (strain 151) and Campylobacter coli with human peripheral polymorphonuclear granulocytes. Int. J. Med. Microbiol. Virol. Parasitol. Infect. Dis. 1993;280:58–72 . doi: 10.1016/s0934-8840(11)80941-2. [DOI] [PubMed] [Google Scholar]
- Pruul H., Lee P.C., Goodwin C.S., McDonald P.J. Interaction of Campylobacter pyloridis with human immune defence mechanisms. J. Med. Microbiol. 1987;23:233–238 . doi: 10.1099/00222615-23-3-233. [DOI] [PubMed] [Google Scholar]
- Joiner K.A. Membrane-protein traffic in pathogen-infected cells. J. Clin. Invest. 1997;99:1814–1817 . doi: 10.1172/JCI119347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgill-Koszycki S., Schlesinger P.H., Chakraborty P., Haddix P.L., Collins H.L., Fok A.K., Allen R.D., Gluck S.L., Heuser J., Russel D.G. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science. 1994;263:678–681 . doi: 10.1126/science.8303277. [DOI] [PubMed] [Google Scholar]
- Clemens D.L., Horwitz M.A. Characterization of the Mycobacterium tuberculosis phagosome, and evidence that phagosomal maturation is inhibited. J. Exp. Med. 1995;181:257–270 . doi: 10.1084/jem.181.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engstrand L., Graham D., Scheynius A., Genta R.M., El-Zaatari F. Is the sanctuary where Helicobacter pylori avoids antibacterial treatment intracellular? Am. J. Clin. Pathol. 1997;108:504–509 . doi: 10.1093/ajcp/108.5.504. [DOI] [PubMed] [Google Scholar]
- Kinder S.A., Badger J.L., Bryant G.O., Pepe J.C., Miller V.L. Cloning of the YenI restriction endonuclease and methyltransferase from Yersinia enterocolitica serotype O8 and construction of a transformable R-M+ mutant. Gene. 1993;136:271–275 . doi: 10.1016/0378-1119(93)90478-l. [DOI] [PubMed] [Google Scholar]
- Allen L.H., Aderem A. A role for MARCKS, the alpha isozyme of protein kinase C and myosin I in zymosan phagocytosis by macrophages. J. Exp. Med. 1995;182:829–840 . doi: 10.1084/jem.182.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen L.A., Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor–mediated phagocytosis in macrophages. J. Exp. Med. 1996;184:627–637 . doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minta J.O., Pambrun L. In vitro induction of cytologic and functional differentiation of the immature human monocyte-like cell line U-937 with phorbol myristate acetate. Am. J. Pathol. 1985;119:111–126 . [PMC free article] [PubMed] [Google Scholar]
- Kusner D.J., Hall C.F., Schlesinger L.S. Activation of phospholipase D is tightly coupled to the phagocytosis of Mycobacterium tuberculosis or opsonized zymosan by human macrophages. J. Exp. Med. 1996;184:585–595 . doi: 10.1084/jem.184.2.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsinghorst E.A. Measurement of invasion by gentamicin resistance. Methods Enzymol. 1994;236:405–420 . doi: 10.1016/0076-6879(94)36030-8. [DOI] [PubMed] [Google Scholar]
- Schlesinger L.S., Bellinger-Kawahara C.G., Payne N.R., Horwitz M.A. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol. 1990;144:2771–2780 . [PubMed] [Google Scholar]
- Horwitz M.A. Characterization of avirulent mutant Legionella pneumophilia that survive but do not multiply within human monocytes. J. Exp. Med. 1987;166:1310–1328 . doi: 10.1084/jem.166.5.1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen L.A., Aderem A. Protein kinase C regulates MARCKS cycling between the plasma membrane and lysosomes in fibroblasts. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:1109–1121 . doi: 10.1002/j.1460-2075.1995.tb07094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen L.A., Raetz C.R.H. Partial phenotypic suppression of a peroxisome-deficient animal cell mutant treated with aminoglycoside G418. J. Biol. Chem. 1992;267:13191–13199 . [PubMed] [Google Scholar]
- Dziwish L., Hessemann J., Kremer B. The microscopic double immunofluorescence technique, a method for quantitative differentiation between extra- and intracellularly located bacteria in isolated polymorphonuclear granulocytes. Med. Microbiol. Immunol. 1988;177:101–107 . doi: 10.1007/BF00189531. [DOI] [PubMed] [Google Scholar]
- Pierson D. Mechanisms of Yersinia entry into mammalian cells. In: Miller V.L., editor. Molecular Genetics of Bacterial Pathogenesis. ASM Press; Washington, DC : 1994. pp. 235–247. [Google Scholar]
- Blocker A., Severin F.F., Burkhardt J.K., Bingham J.B., Yu H., Olivo J.C., Schroer T.A., Hymann A.A., Griffiths G. Molecular requirements for bi-directional movement of phagosomes along microtubules. J. Cell Biol. 1997;137:113–129 . doi: 10.1083/jcb.137.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blocker A., Griffiths G., Olivo J.C., Hymann A.A., Severin F.F. A role for microtubule dynamics in phagosome movement. J. Cell Science. 1998;111:303–312 . doi: 10.1242/jcs.111.3.303. [DOI] [PubMed] [Google Scholar]
- Desjardins M., Huber L.A., Parton R.G., Griffiths G. Biogenesis of phagolysosomes proceeds through sequential series of interactions with the endocytic apparatus. J. Cell Biol. 1994;124:677–688 . doi: 10.1083/jcb.124.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder H.M., Jr., Osier C., Maderazo E.G. Chloramphenicola review of its use in clinical practice. Rev. Infect. Dis. 1981;3:479–491 . doi: 10.1093/clinids/3.3.479. [DOI] [PubMed] [Google Scholar]
- Garrett E.R., Won C.M. Kinetics and mechanisms of drug interaction on microorganisms. XVII. Bacteriocidal effects of penicillin, kanamycin, and rifampin with and without organism pretreatment with bacteriostatic chloramphenicol, tetracycline, and novobiocin. J. Pharm. Sci. 1973;62:1666–1673 . doi: 10.1002/jps.2600621018. [DOI] [PubMed] [Google Scholar]
- Corthesy-Theulaz I., Porta N., Pringault E., Racine L., Bogdanova A., Kraehenbuhl J.P., Blum A.L. Adhesion of Helicobacter pylori to polarized T84 human intestinal monolayers is pH dependent. Infect. Immun. 1996;64:3827–3832 . doi: 10.1128/iai.64.9.3827-3832.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S.M., Uhl J.R., Cockerill F.R., III. Assessment of invasion frequencies of cultured HEp-2 cells using clinical isolates of Helicobacter pylori using an acridine orange assay. J. Clin. Pathol. 1998;51:127–133 . doi: 10.1136/jcp.51.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesca M., Borgia S., Hoffman P., Lingwood C.A. Acidic pH changes receptor binding specificity of Helicobacter pyloria binary adhesion model in which surface heat shock (stress) proteins mediate sulfatide recognition in gastric colonization. Infect. Immun. 1996;64:2643–2648 . doi: 10.1128/iai.64.7.2643-2648.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huesca M., Goodwin A., Bhagwansingh A., Hoffman P., Lingwood C.A. Characterization of an acidic-pH-inducible stress protein (hsp70), a putative sulfatide binding adhesin from Helicobacter pylori . Infect. Immun. 1998;66:4061–4067 . doi: 10.1128/iai.66.9.4061-4067.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci V., Sommi P., Fiocca R., Romano M., Solcia E., Ventura U. Helicobacter pylori vacuolating cytotoxin accumulates within the endosomal-vacuolar compartment of cultured gastric cells and potentiates the vacuolating activity of ammonia. J. Pathol. 1997;183:453–459 . doi: 10.1002/(SICI)1096-9896(199712)183:4<453::AID-PATH950>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Papini E., Satin B., de Bernard M., Molinari M., Arico B., Galli C., Telford J.R., Rappuoli R., Montecucco C. Action site and cellular effects of cytotoxin VacA produced by Helicobacter pylori . Folia Microbiol. 1998;43:279–284 . doi: 10.1007/BF02818613. [DOI] [PubMed] [Google Scholar]
- Cover T.L. The vacuolating cytotoxin of Helicobacter pylori . Mol. Microbiol. 1996;20:241–246 . doi: 10.1111/j.1365-2958.1996.tb02612.x. [DOI] [PubMed] [Google Scholar]
- Fiocca R., Luinetti O., Villani L., Chiaravalli A.M., Capella C., Solicia E. Epithelial cytotoxicity, immune responses, and inflammatory components of Helicobacter pylori gastritis. Scand. J. Gastroenterol. Suppl. 1994;29:11–21 . [PubMed] [Google Scholar]
- el-Shoura S.M. Helicobacter pyloriI. Ultrastructural sequences of adherence, attachment, and penetration into the gastric mucosa. Ultrastruct. Pathol. 1995;19:323–333 . doi: 10.3109/01913129509064237. [DOI] [PubMed] [Google Scholar]
- Valkonen K.H., Wadstrom T., Moran A.P. Identification of the N-acetylneuraminyllactose-specific laminin-binding protein of Helicobacter pylori . Infect. Immun. 1997;65:916–923 . doi: 10.1128/iai.65.3.916-923.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall B.J. Virulence and pathogenicity of Helicobacter pylori . J. Gastroenterol. Hepatol. 1991;6:121–124 . doi: 10.1111/j.1440-1746.1991.tb01450.x. [DOI] [PubMed] [Google Scholar]
- Valkonen K.H., Wadstrom T., Moran A.P. Interaction of lipopolysaccharides of Helicobacter pylori with the basement membrane protein laminin. Infect. Immun. 1994;62:3640–3648 . doi: 10.1128/iai.62.9.3640-3648.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringer M., Valkonen K.H., Wadstrom T. Binding of vitronectin and plasminogen to Helicobacter pylori . FEMS Immunol. Med. Microbiol. 1994;9:29–34 . doi: 10.1111/j.1574-695X.1994.tb00470.x. [DOI] [PubMed] [Google Scholar]
- Wu K.C., Jackson L.M., Galvin A.M., Gray T., Hawkey C.J., Mahida Y.R. Phenotypic and functional characterization of the myofibroblasts, macrophages, and lymphocytes migrating out of the human gastric lamina propria following loss of epithelial cells. Gut. 1999;44:323–330 . doi: 10.1136/gut.44.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg S., Silverstein S.C. Phagocytosis. In: Paul W.E., editor. Fundamental Immunology. Raven Press; New York : 1993. pp. 941–964. [Google Scholar]
- Chmiela M., Czkwianianc E., Wadstrom T., Rudnicka W. Role of Helicobacter pylori surface structures in bacterial interaction with macrophages. Gut. 1997;40:20–24 . doi: 10.1136/gut.40.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Ooij C., Homola E., Kincaid E., Engel J. Fusion of Chlamydia trachomatis-containing inclusions is inhibited at low temperatures and requires bacterial protein synthesis. Infect. Immun. 1998;66:5364–5371 . doi: 10.1128/iai.66.11.5364-5371.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manor E., Sarov I. Fate of Chlamydia trachomatis in human monocytes and monocyte-derived macrophages. Infect. Immun. 1986;54:90–95 . doi: 10.1128/iai.54.1.90-95.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bernard M., Papini E., de Filippis V., Gottardi E., Telford J.L., Manetti R., Fontana A., Rappuoli R., Montecucco C. Low pH activates the vacuolating cytotoxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 1995;270:23937–23940 . doi: 10.1074/jbc.270.41.23937. [DOI] [PubMed] [Google Scholar]
- Molinari M., Galli C., Norais N., Telford J.L., Rappuoli R., Luzio J.P., Montecucco C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J. Biol. Chem. 1997;272:25339–25344 . doi: 10.1074/jbc.272.40.25339. [DOI] [PubMed] [Google Scholar]
- Papini E., de Bernard M., Milia E., Bugnoli M., Zerial M., Rappuoli R., Montecucco C. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc. Natl. Acad. Sci. USA. 1994;91:9720–9724 . doi: 10.1073/pnas.91.21.9720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg T.H., Swanson J.A. Measurement of phagosome-lysosome fusion and phagosomal pH. Methods Enzymol. 1994;236:147–160 . doi: 10.1016/0076-6879(94)36014-6. [DOI] [PubMed] [Google Scholar]
- Burns D.L. Biochemistry of type IV secretion. Curr. Opin. Microbiol. 1999;2:25–29 . doi: 10.1016/s1369-5274(99)80004-6. [DOI] [PubMed] [Google Scholar]
- Cornacchione P., Scaringi L., Fettucciari K., Rosati E., Sabatini R., Orefici G., Von Hunolstein C., Modesti A., Modica A., Minelli F. Group B streptococci persist inside macrophages. Immunology. 1998;93:86–95 . doi: 10.1046/j.1365-2567.1998.00402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolridge K.G., Ketley J.M. Campylobacter-host cell interactions. Trends Microbiol. 1997;5:96–102 . doi: 10.1016/S0966-842X(97)01004-4. [DOI] [PubMed] [Google Scholar]
- Karita M., Tummuru M.K., Wirth H.P., Blaser M.J. Effect of growth phase and acid shock on Helicobacter pylori cagA expression. Infect. Immun. 1996;64:4501–4507 . doi: 10.1128/iai.64.11.4501-4507.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukholm G., Tannaes T., Nedenskov P., Esbensen Y., Grav H.J., Hovig T., Ariansen S., Guldvog I. Colony variation of Helicobacter pyloripathogenic potential is correlated to cell wall lipid composition. Scand. J. Gastroenterol. 1997;32:445–454 . doi: 10.3109/00365529709025079. [DOI] [PubMed] [Google Scholar]
- Tompkins L.S., Falkow S. The new path to preventing ulcers. Science. 1995;267:1621–1622 . doi: 10.1126/science.7886448. [DOI] [PubMed] [Google Scholar]
- Blaser M.J. Helicobacter pylori phenotypes associated with peptic ulceration. Scand. J. Gastroenterol. Suppl. 1994;29:1–5 . [PubMed] [Google Scholar]
- Graham D.Y. Antibiotic resistance in Helicobacter pyloriimplications for therapy. Gastroenterology. 1998;115:1272–1277 . doi: 10.1016/s0016-5085(98)70100-3. [DOI] [PubMed] [Google Scholar]
- Neri M., Susi D., Bovani I., Laterza F., Porreca E., Cuccurullo F. Gastric mucosal infiltration by Helicobacter pylori favours bacterial survival after treatment. Aliment. Pharmacol. Ther. 1996;10:181–185. doi: 10.1046/j.1365-2036.1996.713893000.x. [DOI] [PubMed] [Google Scholar]