

Abstract
Protease-activated receptor (PAR)-1 is a cellular receptor for thrombin that is activated after proteolytic cleavage. The contribution of PAR-1 to inflammatory cell–mediated renal injury was assessed in murine crescentic glomerulonephritis (GN). A pivotal role for thrombin in this model was demonstrated by the capacity of hirudin, a selective thrombin antagonist, to attenuate renal injury. Compared with control treatment, hirudin significantly reduced glomerular crescent formation, T cell and macrophage infiltration, fibrin deposition, and elevated serum creatinine, which are prominent features of GN. PAR-1–deficient (PAR-1−/−) mice, which have normal coagulation, also showed significant protection from crescentic GN compared with wild-type mice. The reductions in crescent formation, inflammatory cell infiltration, and serum creatinine were similar in PAR-1−/− and hirudin-treated mice, but hirudin afforded significantly greater protection from fibrin deposition. Treatment of wild-type mice with a selective PAR-1–activating peptide (TRAP) augmented histological and functional indices of GN, but TRAP treatment did not alter the severity of GN in PAR−/− mice. These results indicate that activation of PAR-1 by thrombin or TRAP amplifies crescentic GN. Thus, in addition to its procoagulant role, thrombin has proinflammatory, PAR-1–dependent effects that augment inflammatory renal injury.
Keywords: coagulation, kidney, cell-mediated immunity, hirudin, in vivo
Introduction
Coagulation is fundamental for hemostasis and is an integral part of inflammatory reactions. Inflammatory mediators promote coagulation by stimulating expression of procoagulant molecules on endothelial cells and macrophages. The capacity of the serine proteases of the coagulation pathway to promote inflammation by direct cellular effects is less well established. Potential roles for the cellular receptors for factor VIIa (tissue factor; reference 1), factor Xa (effector cell protease receptor; reference 2), and thrombin (protease-activated receptor [PAR]-1; reference 3) in cellular activation have been suggested by in vitro studies. In vivo, proinflammatory roles for coagulant proteins have been suggested by studies demonstrating that amelioration of Escherichia coli induced endotoxic shock after treatment with activated protein C 4 or anti–tissue factor antibodies 5.
Thrombin is a serine protease that cleaves fibrinogen to form fibrin monomers and uniquely cleaves cell surface receptors, known as PARs. Four members of this seven-transmembrane domain, G protein–coupled receptor family have recently been cloned and designated PAR-1 6 7, PAR-2 8, PAR-3 9, and PAR-4 10 11. Protease cleavage of these receptors creates a neo-NH2 terminus, which acts as a tethered ligand that binds to the seven-transmembrane segment of the PAR. PAR-1, -3, and -4 are cleaved by thrombin, whereas PAR-2 is cleaved by trypsin. The tethered neo-NH2 terminus activates the receptor, independent of thrombin or trypsin binding 6 12. Free peptides, as short as six amino acids, can mimic the neo-NH2 terminus and activate PARs.
PAR-1 13 and PAR-2 14 are expressed in the human kidney. PAR-1 is constitutively expressed on glomerular endothelium and mesangial and epithelial cells and on the endothelium of the interstitial renal vasculature 13. Activation of PAR-1 after thrombin cleavage can be mimicked by short peptides known as thrombin receptor–activating peptides (TRAPs) containing the initial amino acid sequence SFLLRN. TRAP has no protease activity and thus, in contrast to thrombin, TRAP is incapable of cleaving fibrinogen. For this reason, TRAP has been useful for probing fibrin-independent, receptor-mediated roles of thrombin. PAR-1 activation by thrombin or TRAP in vitro results in the production of proinflammatory mediators, including IL-8 15, E-selectin, and platelet-derived growth factor 16 by endothelial cells and monocyte chemoattractant protein 1 17 by mesangial cells.
Glomerulonephritis (GN) is the most common cause of end stage renal failure. Crescentic GN is a particularly severe and rapidly progressive form of GN, characterized by glomerular inflammatory cell infiltration, fibrin deposition, and local upregulation of procoagulant molecules. Studies of human 18 19 and experimental 20 crescentic GN have shown that local activation of the extrinsic coagulation pathway is important in the pathogenesis of glomerular injury. Glomerular deposition of fibrin is directly responsible for some of the injurious effects of extrinsic pathway activation 21 22. However, recent studies have indicated that fibrinogen-independent effects may also be involved 23 24. It is possible that direct cellular effects of serine proteases of the extrinsic coagulation pathway may promote inflammatory glomerular injury. Downregulation of thrombin receptor (PAR-1) antigen associated with upregulation of thrombin receptor (PAR-1) mRNA 13 in human crescentic GN suggests a possible role for thrombin signaling in this disease. This downregulation of the thrombin receptor protein in human GN is consistent with the activation and internalization of PAR-1 that occurs after thrombin cleavage in vitro 25.
Little is known about the direct proinflammatory effects of thrombin (or other coagulation serine proteases) in vivo. However, the recent generation of PAR-1 gene knockout (GKO) mice and the characterization of selective agonist peptides for this receptor have allowed the direct cellular effects of thrombin to be studied in vivo. PAR-1 GKO mouse embryos have a 50% incidence of mortality, but the survivors are phenotypically normal at birth 26 and have normal coagulation and grossly normal wound healing. These mice have absent PAR-1 expression, but their platelets respond normally to thrombin, predominantly through PAR-3 and PAR-4 11. These mice provide a unique opportunity to study the role of PAR-1–mediated thrombin effects in vivo, in the absence of coagulation disturbances.
The contribution of thrombin and PAR-1 to inflammatory renal injury was studied in vivo in a well characterized murine model of crescentic GN. Hirudin was used to selectively inhibit thrombin's serine protease activity, PAR-1 GKO mice were used to determine the effects of selective deficiencies of thrombin's cell receptor–mediated effects, and TRAP was administered to selectively activate PAR-1. The results show a major contribution of thrombin via PAR-1 to cell-mediated renal inflammation.
Materials and Methods
Animals
Studies were performed in male PAR-1–deficient (PAR-1− / −) mice between 8 and 10 wk of age. The generation and phenotypic description of these mice has been reported 26. These mice have been extensively backcrossed (>97%) into the C57BL/6 background. Male C57BL/6 mice were used as controls.
Induction of Crescentic GN
GN was induced in sensitized mice by a planted nephritogenic antigen. Mice were sensitized by subcutaneous injection of 2 mg of sheep globulin in 100 μl of Freund's complete adjuvant, given as divided doses in each flank. 10 d later, GN was initiated by intravenous administration of 3.6 mg of sheep anti–mouse glomerular basement membrane (GBM) globulin. This dose does not induce proteinuria in nonimmunized mice. All endpoints were assessed 10 d after administration of anti-GBM globulin.
Treatment Protocols
Hirudin.
Hirudin (Revasc; CIBA-Geigy Pharmaceuticals) was subcutaneously administered twice daily, at a dose of 2 mg/kg in 50 μl of normal saline, starting 4 h after administration of anti-GBM globulin. Normal saline (50 μl) was given according to the same protocol as control treatment.
TRAP.
TRAP, with the amino acid sequence SFLLRN, was synthesized by Prof. M. Hearn (Monash University) and was also subcutaneously administered twice daily, at a dose of 2 mg/kg in 50 μl of normal saline, starting 4 h after administration of anti-GBM globulin.
Histological Assessment
Crescents.
4-μm paraffin renal tissue sections were cut and stained with periodic acid-Schiff. Glomerular crescent formation was assessed in a blinded protocol. Glomeruli were considered to exhibit crescent formation when two or more layers of cells were observed in Bowman's space. A minimum of 50 glomeruli were assessed to determine the crescent score for each animal.
Glomerular T Cell and Macrophage Accumulation.
Spleen and kidney tissue was fixed in periodate lysine paraformaldehyde for 4 h, washed in 7% sucrose solution, and then frozen in liquid nitrogen–cooled isopentane. Tissue sections (6 μm) were stained to demonstrate macrophages and T cells using a three-layer immunoperoxidase technique as previously described 27. The primary mAbs were GK1.5 (anti–mouse CD4; American Type Culture Collection [ATCC]) and M1/70 (anti–mouse Mac-1; ATCC). Sections of spleen provided a positive control for each animal, and protein G–purified rat Ig was substituted for the primary mAb to provide a negative control. A minimum of 20 equatorially sectioned glomeruli were assessed per animal using a blinded protocol, and the results were expressed as cells per glomerular cross-section (c/gcs).
Fibrin Deposition.
Tissue sections (4 μm) were cut from snap-frozen kidney and stained by direct immunofluorescence with FITC-conjugated goat anti–mouse fibrinogen (Nordic). Glomerular fibrin deposition was assessed using a semiquantitative blinded protocol. Only glomeruli cut in or near the equatorial cross-section were included. A minimum of 30 glomeruli were scored (0 to +3) to determine a mean score for each animal. Glomeruli in which fluorescence was not different from background were scored 0, glomeruli with sparse fibrin deposition in the glomerular tuft were scored +1, glomeruli with prominent fibrin deposition in the glomerular tuft were scored +2, and glomeruli with prominent fibrin deposition in both the glomerular tuft and in Bowman's space were scored +3.
Functional Assessment of Renal Injury
Serum Creatinine.
Serum creatinine concentrations were measured by the alkaline picric acid method using an autoanalyzer calibrated to read low serum creatinine levels.
Proteinuria.
Mice were housed individually in cages to collect urine over the final 24 h of each experiment. Urinary protein concentrations were determined by a modified Bradford method 28 adapted to a microtiter plate assay. 24-h urinary protein excretion was calculated from the 24-h urine volume and the urinary protein concentration.
Assessment of Humoral Immune Responses to Sheep Globulin
Circulating Mouse Anti–Sheep Globulin Antibody.
Titers of circulating mouse anti–sheep globulin antibody levels were measured by ELISA. Microtiter plates were coated with normal sheep globulin (10 μg/ml) and incubated with mouse serum at a dilution of 1:100. Wells were then washed, and bound mouse anti–sheep globulin antibody was detected by incubation with horseradish peroxidase–conjugated sheep anti–mouse Ig (Amersham Corp.) at a dilution of 1:200 and then with 0.1 M 2,2′-azino-di-3-ethylbenzthiazoline sulfonate (ABTS; Boehringer Mannheim) in 0.02% H2O2 as a substrate. The absorbance was read at 405 nm on a microtiter plate reader (Dynatech Labs., Inc.), and the results were expressed as absorbance units (A405).
Glomerular Antibody Deposition.
Glomerular deposition of autologous antibody was assessed on frozen sections of renal tissue stained with FITC-conjugated sheep anti–mouse IgG (Silenus) using a semiquantitative blinded scoring protocol. Background fluorescence was given a score of 0. Faint linear staining of mouse Ig in glomeruli was scored as +1. Moderate linear staining was scored as +2, and the most intense staining was scored as +3. A minimum of 30 glomeruli were scored to provide a mean score for each animal.
Statistical Analysis
Results are expressed as the mean ± SEM. Group sizes were: normal (wild-type [WT]), nondiseased mice, n = 6; control (saline-treated) WT mice with GN, n = 10; hirudin-treated mice with GN, n = 6; PAR-1−/− mice with GN, n = 6; TRAP-treated WT mice with GN, n = 6; and TRAP-treated PAR-1−/− mice with GN, n = 6. The statistical significance of differences between groups was determined by ANOVA (analysis of variance) and Fisher's protected least significant differences.
Results
Thrombin Is an Important Mediator of Injury in Crescentic GN.
Administration of hirudin to block both the procoagulant and PAR-1–activating actions of thrombin afforded significant protection from the development of crescentic GN. Crescent formation and fibrin deposition are not observed in glomeruli of WT (Fig. 1 A) or PAR-1−/− mice (Fig. 1 B) before induction of GN. However, after sensitization to sheep globulin and administration of sheep anti–mouse GBM antibody, proliferative and crescentic GN was induced in WT mice (Fig. 1 C). Crescent formation occurred in 14 ± 3.1% of glomeruli. This was associated with prominent glomerular fibrin deposition (fibrin score 1.1 ± 0.1, normal = 0) and infiltration of CD4+ T cells (0.82 ± 0.07 c/gcs, normal = 0.01 ± 0.01 c/gcs) and macrophages (6.6 ± 0.6 c/gcs, normal = 0.02 ± 0.01 c/gcs) (Fig. 2), in keeping with the typical pathological features of a delayed-type hypersensitivity reaction. Previous studies have demonstrated that this model of GN results from a Th1-biased 29, MHC class II– 30 and CD4-dependent 31 immune response. The histological features of glomerular inflammation were associated with significant functional renal injury, indicated by increased serum creatinine (29 ± 2 μmol/liter; normal = 16 ± 2 μmol/liter; P < 0.001) and increased protein excretion in the urine (proteinuria 6.1 ± 0.5 mg/24 h; normal = 0.7 ± 0.1 mg/24 h; P < 0.001) (Fig. 3).
Figure 1.
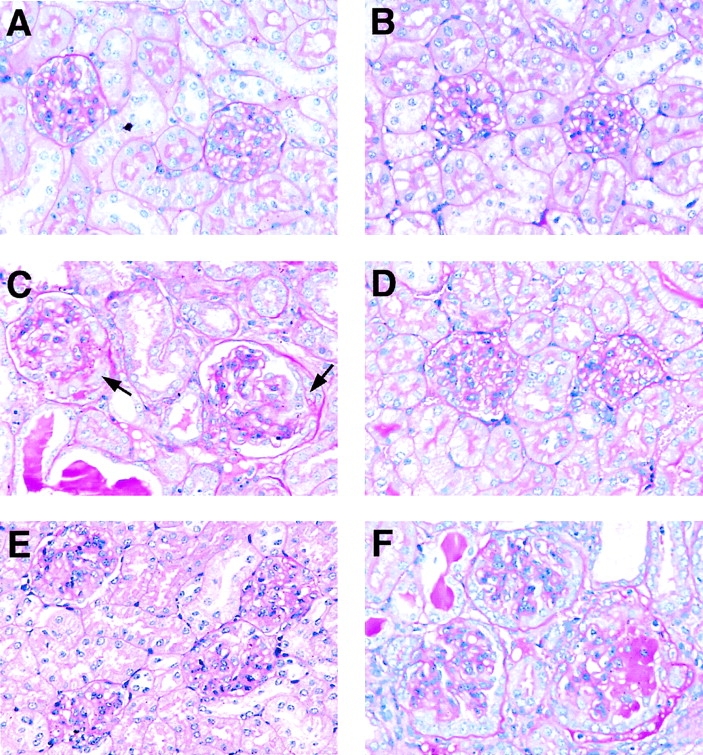
Histological features of normal and nephritic glomeruli from WT and PAR-1−/− mice (periodic acid-Schiff stain; magnification 200). (A) Glomeruli from a WT mouse before induction of GN. (B) Glomeruli from a PAR-1−/− mouse before the induction of GN. (C) Glomeruli from a WT mouse, 10 d after initiation of GN, showing proliferative changes in the glomerular tufts and crescents in Bowman's space (arrows). (D) Crescentic glomerular injury was markedly diminished by hirudin treatment in WT mice, although proliferative changes persisted in the glomerular tufts. (E) PAR-1−/− mice also showed marked attenuation of crescentic glomerular injury after the induction of GN. (F) Administration of TRAP markedly accentuated crescentic glomerular injury in WT mice developing GN.
Figure 2.
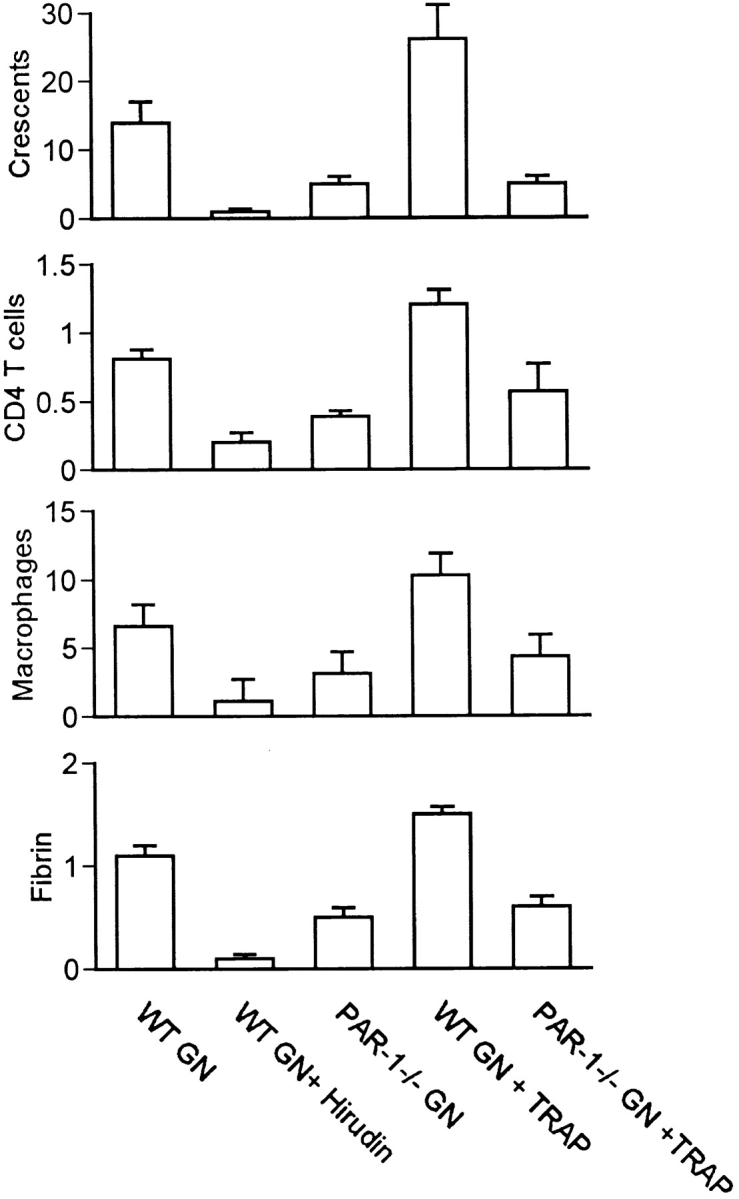
Histological parameters of injury in mice with GN. Panels show the incidence of glomerular crescent formation (percent of glomeruli), glomerular accumulation of CD4+ T cells and macrophages (c/gcs), and glomerular fibrin deposition (semiquantitative score, 0 to +3) in C57BL/6 mice treated with saline (WT GN, n = 10), hirudin (WT GN + Hirudin, n = 6), or TRAP (WT GN + TRAP, n = 6) and PAR-1−/− mice treated with saline (PAR-1−/− GN, n = 12) or TRAP (PAR-1−/− GN + TRAP, n = 6).
Figure 3.

Functional parameters of injury in normal mice and mice with GN. Panels show serum creatinine (μmol/liter) and proteinuria (mg/24 h) in normal, nondiseased C57BL/6 mice (Normal, n = 6) and in C57BL/6 mice with GN treated with saline (WT GN), hirudin (WT GN + Hirudin), or TRAP (WT GN + TRAP) and in PAR-1−/− mice with GN treated with saline (PAR-1−/− GN) or TRAP (PAR-1−/− GN + TRAP).
Mice treated with hirudin showed a marked reduction in the severity of crescentic GN, although mild proliferative changes persisted in the glomerular tuft (Fig. 1 D). The incidence of crescent formation (1.0 ± 0.4% of glomeruli, P = 0.0021), glomerular fibrin deposition (fibrin score 0.17 ± 0.04, P < 0.0001), and mononuclear inflammatory cell infiltration in glomeruli (CD4+ T cells 0.2 ± 0.03 c/gcs, P < 0.0001; macrophages 1.1 ± 0.1 c/gcs, P < 0.0001) were all markedly reduced compared with saline-treated mice (Fig. 2). Mice treated with hirudin also showed significant reduction in their functional indices of renal injury (serum creatinine 16 ± 2 μmol/liter, P < 0.0001; proteinuria 3.6 ± 0.6 mg/24 h, P < 0.0001) compared with saline-treated mice with GN (Fig. 3).
Hirudin treatment resulted in significant prolongation of the activated partial thromboplastin time (24 ± 4 s; control 18 ± 6 s; P < 0.004), an ex vivo measure of thrombin-dependent coagulation. Circulating platelet numbers were unaffected by hirudin (Table ). There was no evidence of abnormal bleeding from injection sites or spontaneous hemorrhage, suggesting that this dose of hirudin is therapeutically relevant. The circulating antibodies to the nephritogenic antigen (serum anti–sheep globulin antibody) and the glomerular deposition of autologous antibody in mice developing GN was unaffected by hirudin (Table ), indicating that attenuation of renal injury is not attributable to suppression of humoral immune responses.
Table 1.
Coagulation Parameters before and after Initiation of GN
| Group | Platelet count | APTT | ||
|---|---|---|---|---|
| Before GN | After GN | Before GN | After GN | |
| ×109 per liter | s | |||
| WT | 224 ± 12 | 234 ± 3 | 18 ± 1 | 18 ± 6 |
| WT + hirudin | 213 ± 10 | 252 ± 5 | 16 ± 4 | 24 ± 4 |
| PAR-1−/− | 253 ± 5 | 215 ± 8 | 18 ± 3 | 18 ± 4 |
| WT + TRAP | 257 ± 18 | 206 ± 13 | 17 ± 2 | 19 ± 4 |
| PAR-1−/− + TRAP | 286 ± 14 | 301 ± 11 | 20 ± 3 | 18 ± 6 |
APTT, activated partial thromboplastin time.
Table 2.
Serum Anti–Sheep Globulin Antibody and Glomerular Mouse Ig Deposition in Sensitized Mice 10 d after Administration of Sheep Anti-GBM Globulin
| Group | Serum anti–sheep globulin Ig (A405) | Glomerular mouse Ig |
|---|---|---|
| WT | 1.17 ± 0.15 | 2.7 ± 0.2 |
| WT + hirudin | 1.25 ± 0.17 | 2.5 ± 0.4 |
| PAR-1−/− | 1.25 ± 0.17 | 2.6 ± 0.1 |
| WT + TRAP | 1.21 ± 0.13 | 2.8 ± 0.2 |
| PAR-1−/− + TRAP | 1.18 ± 0.12 | 2.7 ± 0.3 |
PAR-1 Deficiency Protects against Development of Crescentic GN.
PAR-1−/− mice with absent PAR-1–mediated cellular functions but normal thrombin coagulant function and normal PAR-3– and -4–mediated platelet function showed marked protection from the development of GN (Fig. 1 E). The incidence of crescents was reduced (4.8 ± 1.3% of glomeruli, P = 0.0073) by 64% compared with saline-treated WT mice. Glomerular fibrin deposition (fibrin score 0.53 ± 0.09, P < 0.0001) and glomerular mononuclear inflammatory cell infiltration (macrophages 3.1 ± 0.3 c/gcs, P < 0.0001; CD4+ T cells 0.39 ± 0.04 c/gcs, P < 0.0001) was also significantly reduced (Fig. 2). Serum creatinine (15 ± 2 μmol/liter, P < 0.0001) was reduced to normal values. The reduction in proteinuria (4.8 ± 0.5 mg/24 h, P = 0.076) was not statistically significant. Apart from significantly greater reduction in glomerular fibrin deposition in hirudin-treated mice (P = 0.0048 compared with PAR-1−/−), hirudin and PAR-1 deficiency afforded similar protection from development of crescentic GN. Both hirudin−/− and PAR-1−/− mice show persistent proliferative changes in glomeruli. This suggests that the PAR-1–mediated effects of thrombin, rather than its procoagulant effects, dominate its contribution to glomerular inflammation and injury. PAR-1−/− mice had normal platelet numbers and normal coagulation (Table ) as previously reported 26. The circulating antibody titers to the sheep globulin and glomerular deposition of autologous antibody were the same in PAR-1−/−, WT, and hirudin-treated WT mice developing GN, indicating that PAR-1 deficiency did not affect the nephritogenic immune response (Table ).
Selective Activation of PAR-1 with TRAP Augments Renal Injury in Crescentic GN.
In contrast to the protective effects of PAR-1 deficiency, administration of TRAP to selectively activate PAR-1, independent of thrombin, resulted in marked exacerbation of renal injury in WT mice developing GN (Fig. 1 F). Treatment with TRAP increased the incidence of crescents by 86% (26 ± 5% of glomeruli, P = 0.0042) and markedly increased glomerular fibrin deposition (fibrin score 1.5 ± 0.07, P = 0.0042) compared with saline-treated WT mice. Glomerular macrophage (10.3 ± 0.7 c/gcs, P < 0.0001) and T cell recruitment (1.2 ± 0.11 c/gcs, P = 0.0002) was also significantly augmented (Fig. 2). TRAP treatment resulted in an elevation of serum creatinine (34 ± 1 μmol/liter, P = 0.03), but proteinuria (5.2 ± 0.7 mg/24 h, P = 0.28) was not significantly increased compared with saline-treated mice with GN (Fig. 3). The immune response to the nephritogenic antigen, as assessed by circulating anti–sheep globulin antibody levels and the glomerular deposition of autologous antibody, was unaffected by TRAP treatment (Table ).
The administration of TRAP to PAR-1−/− mice did not exacerbate GN injury, indicating that the effects of TRAP in this model are selectively related to PAR-1 activation. Crescent formation (5.0 ± 1.3%), glomerular fibrin deposition (score 0.63 ± 0.1), glomerular macrophage accumulation (4.3 ± 0.2 c/gcs), and T cell recruitment (0.57 ± 0.05 c/gcs) were all similar to the values in saline-treated PAR−/− mice with GN (Fig. 2). Functional indices of renal injury (creatinine 19 ± 2 μmol/liter; proteinuria 4.8 ± 0.7 mg/24 h; Fig. 3), circulating antibody titers, and glomerular deposition of autologous antibody (Table ) were similarly unaffected.
Discussion
The protective effects of hirudin in this murine model of crescentic GN demonstrate a pivotal proinflammatory role for thrombin in the development of inflammatory renal injury. Hirudin is a specific and selective inhibitor of the serine protease activity of thrombin 32. It has not been reported to block other serine proteases 33. Hirudin binds to the active center and fibrinogen-binding exosite of thrombin and blocks its capacity to cleave fibrinogen and PAR-1. Hirudin treatment markedly attenuated the histological features of glomerular inflammation: inflammatory cell recruitment, fibrin deposition, and crescent formation. This was associated with significant protection from the functional consequences of glomerular injury, increased serum creatinine and protein leakage into the urine. Although the reduction in proteinuria was not as profound as the reduction in crescent formation or serum creatinine, these effects still represent a substantial but incomplete abrogation of glomerular injury by hirudin. Proteinuria is well recognized as a more sensitive index of mild glomerular injury than either crescent formation or serum creatinine.
Glomerular hypercellularity observed in this disease was not markedly reduced by hirudin treatment, despite significant reductions in T cell and macrophage recruitment and functional renal injury. Neutrophil accumulation and proliferation of intrinsic cells in hirudin-treated (and PAR-1−/−) mice developing GN appeared to contribute to this persistent glomerular hypercellularity. This observation suggests that glomerular neutrophil recruitment and mesangial cell proliferation are not critically dependent on thrombin/PAR-1–mediated inflammatory events in this model. They are consistent with the view that mononuclear inflammatory cells are the major cellular effectors involved in crescent formation and that proliferative changes are an early stage in the development of crescentic GN.
Although the protection afforded by hirudin demonstrates an important proinflammatory role for thrombin in this model, it does not help to distinguish the procoagulant actions of thrombin from its PAR-1–mediated effects. The study of PAR-1−/− mice allowed the contribution of thrombin via cleavage of this cellular receptor to inflammatory renal injury to be distinguished from its contribution via cleavage of fibrinogen or other coagulant actions. A critical role for PAR-1 in the development of inflammatory renal injury associated with crescentic GN was demonstrated. In PAR-1 GKO mice, protection from renal injury was similar to that seen with hirudin treatment, as was the persistence of some glomerular hypercellularity. This suggests that receptor-mediated effects of thrombin (rather than its coagulant effects) are responsible for the majority of the thrombin contribution to renal injury in this model. Despite the normal systemic coagulation system and normal platelet numbers and function in PAR-1 GKO mice, these mice had significantly less fibrin deposition in their glomeruli during development of GN. This is consistent with attenuation of inflammatory injury in the glomerulus and demonstrates the positive feedback between inflammatory tissue injury and local fibrin deposition.
The administration of TRAP provided an alternative approach, which allowed augmentation of PAR-1 activation independent of the procoagulant effects of thrombin. This six–amino acid peptide selectively activates PAR-1. It has no enzymatic activity and cannot cleave fibrinogen. TRAP significantly augmented both histological and functional renal injury in WT mice developing crescentic GN. Both crescent formation and serum creatinine were significantly increased, demonstrating that selective augmentation of PAR-1 activation amplifies inflammatory renal injury. The absence of a significant increase in proteinuria in TRAP-treated mice is consistent with the observation that proteinuria may not increase despite profound renal injury, due to the severe reduction of glomerular filtration. The failure of TRAP to alter the manifestations of disease in PAR-1 GKO mice demonstrates the selectivity of TRAP as a PAR-1 agonist. PAR-1 antagonist peptides have recently been reported 34 and may provide a new therapeutic modality in inflammatory renal injury. In view of the problems associated with conventional anticoagulant therapy in human crescentic GN, these may provide a useful therapeutic advance.
In summary, an important role for thrombin, acting mainly through activation of its cellular receptor PAR-1, has been demonstrated in inflammatory renal injury. This is the first in vivo demonstration of a functional role for a member of the PAR family in inflammation. It provides a further demonstration of the important contribution of the direct cellular effects of procoagulant molecules to inflammatory tissue injury.
Acknowledgments
The technical assistance of Ms. J. Sharkey is gratefully acknowledged.
This work was supported by grants from the National Health and Medical Research Council of Australia, the Association Claude Bernard, and INSERM U489.
Footnotes
Abbreviations used in this paper: GBM, glomerular basement membrane; GKO, gene knockout; GN, glomerulonephritis; PAR, protease-activated receptor; TRAP, thrombin receptor–activating peptide; WT, wild-type.
References
- Mueller B.M., Ruf W. Requirement for binding of catalytically active factor VIIa in tissue factor–dependent experimental metastasis. J. Clin. Invest. 1998;101:1372–1378 . doi: 10.1172/JCI930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson A.C., Nachman R.L., Altieri D.C., Summers B.D., Ruf W.E., Hajjar D.P. Effector cell protease receptor-1 is a vascular receptor for coagulation factor Xa. J. Biol. Chem. 1996;271:28407–28413. doi: 10.1074/jbc.271.45.28407. [DOI] [PubMed] [Google Scholar]
- Mari B., Guerin S., Far D.F., Breitmayer J.P., Belhacene N., Peyron J.F., Rossi B., Auberger P. Thrombin and trypsin-induced Ca(2+) mobilization in human T cell lines through interaction with different protease-activated receptors. FASEB (Fed. Am. Soc. Exp. Biol.) J. 1996;10:309–316. doi: 10.1096/fasebj.10.2.8641564. [DOI] [PubMed] [Google Scholar]
- Taylor F.B., Jr., Chang A., Esmon C.T., D'Angelo A., Vigano-D'Angelo S., Blick K.E. Protein C prevents the coagulopathic and lethal effects of E. coli infusion in the baboon. J. Clin. Invest. 1987;97:918–925. doi: 10.1172/JCI112902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor F.B., Jr., Chang A., Ruf W., Morrissey J.H., Hinshaw L., Catlett R., Blick K., Edgington T.S. Lethal E. coli septic shock is prevented by blocking tissue factor with monoclonal antibody. Circ. Shock. 1991;33:127–134. [PubMed] [Google Scholar]
- Vu T.K., Hung D.T., Wheaton V.I., Coughlin S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Rasmussen U.B., Vouret-Craviari V., Jallat S., Schlesinger Y., Pages G., Pavirani A., Lecocq J.P., Pouyssegur J., Van Obberghan-Schilling E. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 1991;288:123–128. doi: 10.1016/0014-5793(91)81017-3. [DOI] [PubMed] [Google Scholar]
- Nystedt S., Emilsson K., Wahlestedt C., Sundelin J. Molecular cloning of a potential novel proteinase activated receptor. Proc. Natl. Acad. Sci. USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H., Connolly A.J., Zeng D., Kahn M.L., Zheng Y.W., Timmons C., Tram T., Coughlin S.R. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- Xu W.F., Andersen H., Whitmore T.E., Presnell S.R., Yee D.P., Ching A., Gilbert T., Davie E.W., Foster D.C. Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. USA. 1998;95:6642–6646. doi: 10.1073/pnas.95.12.6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn M.L., Zheng Y.W., Huang W., Bigornia V., Zeng D., Moff S., Farese R.V., Jr., Tam C., Coughlin S.R. A dual thrombin receptor system for platelet activation. Nature. 1998;394:690–694. doi: 10.1038/29325. [DOI] [PubMed] [Google Scholar]
- Vu T.K., Wheaton V.I., Hung D.T., Charo I., Coughlin S.R. Domains specifying thrombin-receptor interaction. Nature. 1991;353:674–677. doi: 10.1038/353674a0. [DOI] [PubMed] [Google Scholar]
- Xu Y., Zacharias U., Peraldi M.N., He C.J., Lu C., Sraer J.D., Brass L.F., Rondeau E. Constitutive expression and modulation of the functional thrombin receptor in the human kidney. Am. J. Pathol. 1995;146:101–110. [PMC free article] [PubMed] [Google Scholar]
- Bohm S.K., Kong W., Bromme D., Smeekens S.P., Anderson D.C., Connolly A., Kahn M., Nelken N.A., Coughlin S.R., Pagan D.G. Molecular cloning, expression and potential functions of the human proteinase-activated receptor-2. Biochem. J. 1996;314:1009–1016. doi: 10.1042/bj3141009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno A., Murakami K., Yamanouchi K., Watanabe M., Kondo T. Thrombin stimulates production of interleukin-8 in human umbilical vein endothelial cells. Immunology. 1996;88:76–81. doi: 10.1046/j.1365-2567.1996.d01-635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar R., de la Motte C.A., Poptic E.J., DiCorleto P.E. Thrombin receptor-activating peptides differentially stimulate platelet-derived growth factor production, monocytic cell adhesion, and E-selectin expression in human umbilical vein endothelial cells. J. Biol. Chem. 1994;269:13936–13941. [PubMed] [Google Scholar]
- Grandaliano G., Valente A.J., Abboud H.E. A novel biologic activity of thrombinstimulation of monocyte chemotactic protein production. J. Exp. Med. 1994;179:1737–1741. doi: 10.1084/jem.179.5.1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale T.J., Tipping P.G., Carson S.D., Holdsworth S.R. Participation of cell-mediated immunity in deposition of fibrin in glomerulonephritis. Lancet. 1988;2:421–424. doi: 10.1016/s0140-6736(88)90413-8. [DOI] [PubMed] [Google Scholar]
- Tipping P.G., Dowling J.P., Holdsworth S.R. Glomerular procoagulant activity in human proliferative glomerulonephritis. J. Clin. Invest. 1988;81:119–125. doi: 10.1172/JCI113282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holdsworth S.R., Tipping P.G. Macrophage induced glomerular fibrin deposition in experimental glomerulonephritis in the rabbit. J. Clin. Invest. 1985;76:1367–1374. doi: 10.1172/JCI112112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naish P.F., Evans D.J., Peters D.K. The effects of defibrination with ancrod in experimental allergic glomerular injury. Clin. Exp. Immunol. 1975;20:303–309. [PMC free article] [PubMed] [Google Scholar]
- Thomson N.M., Moran J., Simpson I.J., Peters D.K. Defibrination with ancrod in nephrotoxic nephritis in rabbits. Kidney Int. 1976;10:343–347. doi: 10.1038/ki.1976.120. [DOI] [PubMed] [Google Scholar]
- Erlich J.H., Holdsworth S.R., Tipping P.G. Tissue factor initiates glomerular fibrin deposition and promotes major histocompatibility complex class II expression in crescentic glomerulonephritis. Am. J. Pathol. 1997;150:873–880. [PMC free article] [PubMed] [Google Scholar]
- Cunningham M.A., Romas P., Hutchinson P., Holdsworth S.R., Tipping P.G. Tissue factor and factor VIIa receptor/ligand interactions induce pro-inflammatory effects in macrophages. Blood. 1999;94:3413–3420. [PubMed] [Google Scholar]
- Woolkalis M.J., DeMelfi T.M., Jr., Blanchard N., Hoxie J.A., Brass L.F. Regulation of thrombin receptors on human umbilical vein endothelial cells. J. Biol. Chem. 1995;270:9868–9875. doi: 10.1074/jbc.270.17.9868. [DOI] [PubMed] [Google Scholar]
- Connolly A.J., Ishihara H., Kahn M.L., Farese R.V., Jr., Coughlin S.R. Role of the thrombin receptor in development and evidence for a second receptor. Nature. 1996;381:516–519. doi: 10.1038/381516a0. [DOI] [PubMed] [Google Scholar]
- Tipping P.G., Huang X.R., Berndt M.C., Holdsworth S.R. A role for P selectin in complement independent, neutrophil mediated glomerular injury in mice. Kidney Int. 1994;46:79–88. doi: 10.1038/ki.1994.246. [DOI] [PubMed] [Google Scholar]
- Bradford M.M. A rapid and sensitive method for quantitation of microgram quantities of protein using the principle of protein dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Huang X.R., Holdsworth S.R., Tipping P.G. Evidence for delayed type hypersensitivity mechanisms in glomerular crescent formation. Kidney Int. 1994;46:69–78. doi: 10.1038/ki.1994.245. [DOI] [PubMed] [Google Scholar]
- Li S., Kurts C., Kontgen F., Holdsworth S.R., Tipping P.G. Major histocompatibility complex class II expression by intrinsic renal cells is required for crescentic glomerulonephritis. J. Exp. Med. 1998;188:597–602. doi: 10.1084/jem.188.3.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipping P.G., Huang X.R., Qi M., Van G.Y., Tang W.W. Crescentic glomerulonephritis in CD4- and CD8-deficient mice. Requirement for CD4 but not CD8 cells. Am. J. Pathol. 1998;152:1541–1548. [PMC free article] [PubMed] [Google Scholar]
- Stone S.R., Braun P.J., Hofsteenge J. Identification of regions of alpha-thrombin involved in its interaction with hirudin. Biochemistry. 1987;26:4617–4624. doi: 10.1021/bi00389a004. [DOI] [PubMed] [Google Scholar]
- Rydel T.J., Ravichandran K.G., Tulinsky A., Bode W., Huber R., Roitsch C., Fenton J.W. The structure of a complex of recombinant hirudin and human alpha-thrombin. Science. 1990;249:277–280. doi: 10.1126/science.2374926. [DOI] [PubMed] [Google Scholar]
- Bernatowicz M.S., Klimas C.E., Hartl K.S., Peluso M., Allegretto N.J., Seile S.M. Development of potent thrombin receptor antagonist peptides. J. Med. Chem. 1996;39:4879–4887. doi: 10.1021/jm960455s. [DOI] [PubMed] [Google Scholar]