

Abstract
The combination of hemorrhagic pneumonitis and rapidly progressive glomerulonephritis is a characteristic feature of Goodpasture's syndrome (GPS), an autoimmune disease resulting from the interaction of pathogenic anti–collagen type IV (C-IV) antibodies with alveolar and glomerular basement membranes. Lack of a suitable animal model for this fatal disease has hampered both a basic understanding of its etiology and the development of therapeutic strategies. We now report a novel model for GPS using mice deficient in a central regulatory receptor for immunoglobulin (Ig)G antibody expression and function, the type IIB Fc receptor for IgG (FcγRIIB). Mutant mice immunized with bovine C-IV reproducibly develop massive pulmonary hemorrhage with neutrophil and macrophage infiltration and crescentic glomerulonephritis. The distinctive linear, ribbon-like deposition of IgG immune complex seen in GPS was observed along the glomerular and tubulointerstitial membranes of diseased animals. These results highlight the role of FcγRIIB in maintaining tolerance and suggest that it may play a role in the pathogenesis of human GPS.
Keywords: Goodpasture's syndrome, type IV collagen, Fc receptor, autoimmunity, alveolar/glomerular basement membrane
Introduction
Patients with Goodpasture's syndrome (GPS) display a characteristic rapid and progressive glomerulonephritis and hemorrhagic pneumonitis with often fatal results 1 2 3. The presence of anti–collagen type IV (C-IV) antibodies and the characteristic ribbon pattern of immune complex deposition along the basement membranes of both lung and glomeruli has lead to a proposed pathogenic model for this disease 4 5 6. Autoantibodies to common components of the basement membranes of these two organs develop, notably to the α3 domain of C-IV, are deposited 7, and trigger effector cell activation and inflammatory sequelae. The mechanism(s) that result in the loss of tolerance and the development of autoantibodies to C-IV are unknown. Investigations into the proposed pathogenesis of this disease and development of therapeutic strategies have been hampered by the lack of a suitable animal model. Attempts to develop such a model for GPS by immunizing sheep 8 or rats 9 10 with homologous or heterologous glomerular basement membrane preparations have resulted in only partial success. For example, Abate et al. 10 induced glomerulonephritis and hemorrhagic pneumonitis in Wistar-Kyoto rats by immunization with the α3 chain of C-IV, but the observed lesions were only marginal. Similarly, injecting anti–glomerular basement membrane antibody into mice or their immunization with recombinant protein for the NC1 domain of the α3 chain of C-IV resulted in little or only marginal alveolar hemorrhage, in spite of the development of glomerular disease 11. These studies support the conclusion that immunization with C-IV alone is insufficient to trigger the full spectrum of GPS, thus pointing to the contribution of additional susceptibility factors necessary to the development of full-blown disease.
We have previously shown that FcγRIIB-deficient (FcγRIIB−/−) mice of the H-2b haplotype are susceptible to type II collagen–induced arthritis 12, a model for rheumatoid arthritis in humans, which previously could only be induced in susceptible rodents of particular MHC haplotypes, specifically H-2q or H-2r in mice. The etiology of the arthritis seen in these FcγRIIB−/− animals could be attributed to the enhanced humoral immune response to type II collagen and augmented proinflammatory mediator release by effector cells such as macrophages 12. The inhibitory Fc receptor for IgG immune complexes, FcγRIIB, plays a central role in both the afferent and efferent limbs of the immune response, shaping the antibody repertoire, maintaining peripheral tolerance, and setting thresholds for effector cell activation. Animals deficient in this receptor have generalized enhanced antibody responses 13 and heightened inflammation in all antibody-mediated classes of hypersensitivity reactions 12 14. These observations prompted us to test whether FcγRIIB may regulate responses to C-IV and be a critical factor in GPS development. Our data indicate that immunization of FcγRIIB−/− animals with C-IV results in autoimmunity and culminates in GPS-like disease, suggesting a role for the FcγRIIB regulatory pathway in the etiology of this autoimmune disease.
Materials and Methods
Animals.
FcγRIIB−/− mice were generated in the 129/SvJ (H-2b) and C57BL/6 (B6, H-2b) hybrid background as described previously 13. FcRγ−/− mice 15 were backcrossed to B6 mice over six generations. These mice and their wild-type counterparts (129/B6 hybrids and B6 mice for FcγRIIB−/− and FcRγ−/−, respectively) were kept and bred in the Animal Unit of The Institute of Development, Aging and Cancer (Tohoku University, Japan), an environmentally controlled and specific pathogen–free facility. B6 mice were obtained from Charles River Labs. Japan, Inc. All experiments were performed on 8-wk-old male and female mice.
C-IV Immunization.
Bovine C-IV (Cellmatrix IV®) was obtained from Nitta Gellatin, Inc. The C-IV solution (3.0 mg/ml in 1 mM HCl, pH 3.0) was neutralized by adding 1 mM NaOH (final concentration) before emulsifying with Freund's adjuvant. Mice were immunized at the tail base with 150 μg of C-IV emulsified in CFA containing Mycobacterium tuberculosis strain H37Rv (Wako Pure Chemical Industries, Ltd.) and boosted at the same location with 150 μg of C-IV plus IFA (Wako Pure Chemical Industries, Ltd.) 2, 4, and 6 wk later. The mice were killed 56 d later and processed for histopathological examinations.
Assay for Detection of Serum Anti–C-IV Antibodies.
Blood was collected from the subocular plexus of mice into microcentrifuge tubes containing EDTA on ice, and plasma was prepared. Serum antibody titers were measured by ELISA. Antibodies to bovine C-IV were detected in a 96-well microplate assay (Falcon; Becton Dickinson Labware) in which wells were coated with 50 μl/well of a 20 μg/ml solution of bovine C-IV in PBS at 4°C for overnight, washed three times with PBS containing 0.05% Tween 20 and 0.1% BSA, and then blocked with 250 μl/well of PBS containing 0.2% BSA at 4°C for overnight. Antibodies to mouse C-IV were detected by the use of the BIOCOAT® cellware mouse C-IV 96-well plate assay (Becton Dickinson Labware). The diluted serum (1:2,500–5,000) was added at 50 μl/well and allowed to react overnight at 4°C. The wells were washed three times with PBS containing 0.05% Tween 20, incubated with 50 μl of a 1:200 dilution of goat anti–mouse IgG1, IgG2a, IgG2b, IgM, or IgA coupled to horseradish peroxidase (Sigma Chemical Co.) at 4°C for 2 h, washed three times with PBS containing 0.05% Tween 20, and developed at room temperature for 30 min with 0.1 ml of TrueBlue Peroxidase Substrate (Kirkegaard & Perry Labs.). The OD at 450 nm was read using a Biolumin960 Microplate Reader (Molecular Dynamics Japan, Inc.).
Evaluation of Renal Functions.
Serum samples at 56 d were inspected for blood urea nitrogen (BUN) and serum creatinine (Cr) levels by the urease GLDH-UV method and ELISA using a TOSHIBA TBA-80FR. Proteinuria was monitored by the tetrabromophenol blue reaction assay using the Micro AUTION MA-4260 (Kyoto Daiichi Kagaku Co.).
Histological Study and Immunohistochemistry.
Mice were killed by cervical dislocation at day 56. Their lungs and kidneys were removed and fixed in 10% (vol/vol) neutral buffered formalin, followed by embedding in paraffin. The lung specimens were sectioned at 5 μm and stained with hematoxylin and eosin or periodic acid-Shiff (PAS). The kidney sections were stained with PAS. Alternatively, formalin-fixed and paraffin-embedded lung and kidney section (5 μm) were deparaffinized in xylen and rehydrated through graded ethanols. After washing with distilled water, sections were incubated in PBS containing STUF (Serotec Target Unmasking Fluid; Serotec Ltd.) for 10 min at 90°C and washed again three times with PBS. Sections were then incubated for 30 min at room temperature with affinity-purified, fluorescein-conjugated goat F(ab′)2 fragments (H + L chain) anti–mouse IgG, IgM, or C3 (Zymed Labs., Inc.). After washing three times in PBS, slides were mounted and examined with an Olympus BX50 microscope equipped with epifluorescence using an Olympus BH2-RFL-T3 mercury lamp and appropriate optics. Crescentic glomerulonephritis were counted in at least 50 glomeruli randomly selected in a histologic section from each mouse.
Cell Separation and Transfer Experiments.
FcγRIIB−/− mice were immunized with C-IV as described above. Splenocyte suspensions from the diseased FcγRIIB−/− mice were prepared at day 56, treated with 0.144 M NH4Cl for 1 min for depletion of erythrocytes, and then transferred intravenously (2 × 107 cells per mouse) to either FcγRIIB−/− or wild-type naive mice. Alternatively, splenocytes were separated to B220+ and B220− cells by magnetic sorting (B220 MACS® microbeads; Miltenyi Biotec) before cell transfer. These recipient mice were then boosted 7 d later with 150 μg of C-IV in IFA and killed 21 d later.
Results and Discussion
FcγRIIB−/− mice, immunized with bovine C-IV in CFA and boosted at 2, 4, and 6 wk with antigen in IFA, developed a GPS-like disease with pulmonary hemorrhage (Fig. 1) and glomerulonephritis (Fig. 2). Nearly all immunized FcγRIIB−/− mice developed pulmonary hemorrhage, whereas none of the wild-type controls displayed evidence of disease (Table ; 22 of 24 [92%] versus 0 of 14 [0%], respectively; P < 0.001). The appearance of the diseased lungs (Fig. 1 A) revealed extensive hemorrhage diffusely scattered throughout the organ, in sharp contrast to the results reported in previous attempts to induce GPS in rats and mice 9 10 11, in which only regional hemorrhagic patches were observed. Histopathological examinations of a diseased lung indicate massive hemorrhage with infiltrating neutrophils and macrophages (Fig. 1B–D), whereas lung samples from wild-type mice were normal (Fig. 1F and Fig. G). The diseased animals displayed bilateral alveolar hemorrhage (data not shown) as well as alveolar capillaritis with extensive infiltration of neutrophils and macrophages (Fig. 1 D). During this experiment, five of the immunized FcγRIIB−/− mice died of tracheal obstruction caused by hemoptysis (Table ).
Figure 1.
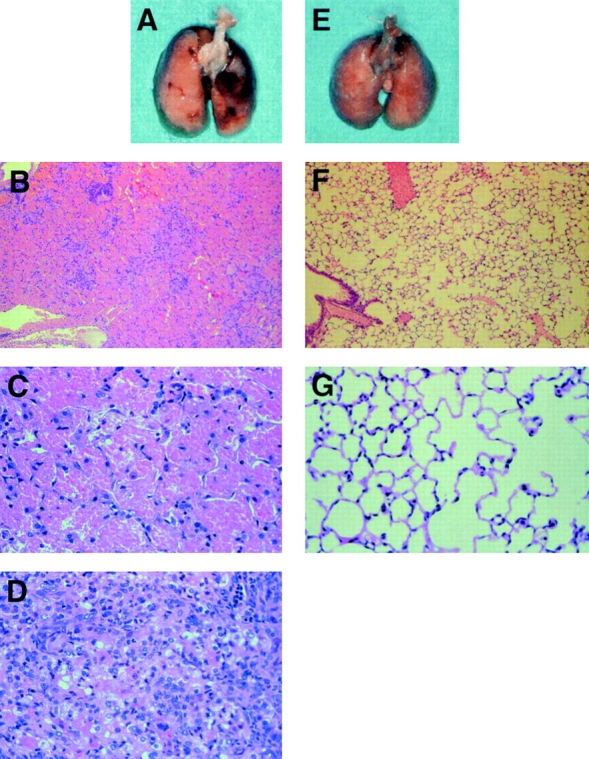
Development of pulmonary hemorrhage in mice with disruption of the FcγRIIB gene immunized with bovine C-IV. A–D are from FcγRIIB−/− mice, and E–G are from wild-type mice. B–D, F, and G are representative pictures stained with hematoxylin and eosin. Original magnifications: B and F, ×25; C, D, and G, ×100.
Figure 2.

Development of glomerular changes in mice with disruption of FcγRIIB. Histological findings of the kidney stained with PAS after immunization with C-IV in FcγRIIB−/− (A and B) and wild-type mice (C and D). Original magnifications: A and C, ×25; B and D, ×100. Wild-type mice yielded no distinct findings, but FcγRIIB−/− mice showed crescentic glomerulonephritis.
Table 1.
Summary of the Mice Immunized with C-IV
| Genotype | Infiltration of inflammatory cells in lung | Lung hemorrhage | Tubulo- interstitial nephritis | Glomerulo- nephritis | Crescentic glomeruli | Mortality rate | BUN | Cr |
|---|---|---|---|---|---|---|---|---|
| (%) | % | (%) | mg/dl | mg/dl | ||||
| FcγRIIB−/− | 22/24 (92) | 19/24 (79) | 22/24 (92) | 22/24 (92) | 33.8 ± 25.0 | 5/24 (21) | 45.7 ± 16.0 | 0.15 ± 0.06 |
| Wild type | 0/16 (0) | 0/16 (0) | 0/16 (0) | 0/16 (0) | 0 | 0/16 (0) | 23.3 ± 8.6 | 0.06 ± 0.02 |
| FcRγ2/− | 0/14 (0) | 0/14 (0) | 0/14 (0) | 0/14 (0) | 0 | 0/14 (0) | 23.4 ± 8.6 | 0.07 ± 0.03 |
| C57BL/6 | 0/7 (0) | 0/7 (0) | 0/7 (0) | 0/7 (0) | 0 | 0/7 (0) | 29.2 ± 5.7 | 0.03 ± 0.03 |
Histopathological examination of the kidneys in immunized FcγRIIB−/− animals revealed typical crescentic glomerulonephritis and tubulointerstitial nephritis, with infiltration of neutrophils and multinuclear giant cells and hyaline droplet degeneration of the tubules in nearly all mice tested (22 of 24 mice tested [92%]; Fig. 2A and Fig. B), whereas kidneys from wild-type mice did not show any pathological changes (Fig. 2C and Fig. D). Immunized FcγRIIB−/− mice showed higher BUN and serum Cr levels than controls (Table ), further confirming the compromised renal function. In addition, all diseased animals showed proteinuria, with mean protein levels of 533 mg/dl in the urine (data not shown). Immunohistochemical staining of glomeruli from immunized FcγRIIB−/− animals with FITC-labeled anti–mouse IgG demonstrated linear depositions of IgG immune complexes along the glomerular membrane (Fig. 3 A), in a characteristic ribbon-like pattern, and tubulointerstitial membrane (Fig. 3 B). In contrast, the deposition of IgG was hardly detectable in immunized wild-type mice (Fig. 3 C). Neither IgM nor C3 deposition was detected along the glomerular basement membranes in the diseased mice (data not shown).
Figure 3.

Deposition of IgG along the glomerular basement membrane in FcγRIIB−/− mice. Immunofluorescence staining of the kidney in FcγRIIB−/− (A and B) and wild-type mice (C). FcγRIIB−/− mice show linear deposition of mouse IgG along the glomerular (A) and tubulointerstitial basement membranes (B), whereas the deposition of IgG was undetectable in immunized wild-type mice (C).
Histological examination of eye, heart, and liver samples taken from immunized FcγRIIB−/− animals at necropsy did not reveal any evidence of inflammatory disease (data not shown), confirming that the disease process evident in the susceptible animals was not the result of a diffuse systemic inflammatory process. However, the possibility that subclinical autoimmune infiltration of other organs (e.g., gut, central nervous system) occurs has not been ruled out. In the absence of bovine C-IV immunization, FcγRIIB−/− mice do not develop any abnormalities in pulmonary and renal function (data not shown), indicating the necessity of an inductive stimulus to trigger disease in this susceptible background. The identification of FcγRIIB as a susceptibility factor in GPS and the ability to induce disease upon immunization with bovine C-IV offers the opportunity to investigate other presumptive triggers in the RIIB−/− mouse, such as toxic oxygen, hydrocarbons, or viruses, all of which have been suggested as possible inducers of human GPS 16.
The pulmonary and renal lesions of GPS are attributed to anti–glomerular basement membrane antibodies, which bind to common antigenic sites in the lung and kidney and activate inflammatory effector responses. Consistent with this pathogenic model, the antibody response of FcγRIIB−/− mice to bovine C-IV immunization is enhanced relative to wild-type controls, with elevated titers of anti–bovine C-IV IgG1, IgG2a, IgG2b, and IgM antibodies observed (Fig. 4A–D). Anti–C-IV IgA was not detected during the 8-wk experimental period of this study (Fig. 4 E), despite the recent description of IgAκ GPS in humans 17. Importantly, FcγRIIB−/− mice, but not wild-type or FcRγ−/− mice, showed elevated autoantibody responses to mouse C-IV (Fig. 4F–H), suggesting that murine GPS depends on the autoantibody production in this susceptible background. Although this enhanced antibody response may account for the GPS disease that develops in these animals, it is likely that contributions from enhanced effector cell responses to deposited anti–C-IV autoantibodies are a significant factor in the development of disease, as described in the enhanced macrophage responses in immune complex–mediated alveolitis 18 and in collagen-induced arthritis 12. In support of this notion, the alveolar and glomerular diseases could be transferred only to FcγRIIB−/− animals (five of nine mice tested) but not to wild-type mice (zero of eight mice tested) by injecting C-IV–sensitized splenocytes from FcγRIIB−/− mice into nonsensitized animals. Disease could not be transferred by injecting either B220+ or B220− splenocytes from C-IV–sensitized FcγRIIB−/− mice into nonsensitized RIIB−/− mice (zero of five mice tested), suggesting the additional requirement of sensitized T and B cell cooperation in disease development (data not shown).
Figure 4.

Concentration of anti–bovine C-IV and anti–mouse C-IV antibodies in sera from mice immunized with bovine C-IV. Anti–bovine C-IV antibody levels of IgG1, IgG2a, IgG2b, IgM, and IgA subclasses (A–E) and anti–mouse C-IV levels of total IgG, IgG1, IgG2a, and IgG2b (F–I) in wild-type (•), FcγRIIB−/− (□), and FcRγ−/− (▵) mice. Wild-type mice were used as controls for FcγRIIB−/− mice as described in Materials and Methods. Each point represents the mean ± SD. Statistical analysis was performed using Student's t test between FcγRIIB−/− and wild-type mice: *P < 0.05 and **P < 0.01.
In this study, several differences have been observed between this animal model and human GPS. First, we found the lack of C3 deposition in the kidneys (data not shown) and the absence of anti–C-IV antibodies of the IgA isotype (Fig. 4 E) as well as the low mortality rate (Table ) in this model. In human GPS, the deposition of IgM or C3 was detected along the glomerular basement membranes in some patients 19, and the deposition of IgA was reported in one patient 17. It remains to be investigated which factor(s) can induce IgM, C3, or IgA deposition in the murine kidneys in the present model. Second, untreated GPS in humans carries a much higher mortality rate; persons with the full-blown syndrome die within a few months 19, compared with the mortality rate of 21% at day 56 in the present model (Table ), although the clinical severity of lung hemorrhage is almost the same for diseased animals and human GPS (Fig. 1). The reason for the differing lifespans of diseased mice and humans might partially depend on the difference of the resistance to hypoxia caused by alveolar hemorrhage. In our estimation, the surviving animals at day 56 could also be alive at day 70. Additional immunizations of these surviving animals with C-IV would increase their mortality rate.
The observation that FcγRIIB deficiency creates a permissive immunological environment for the development of anti–C-IV autoantibodies offers further support for a role for this receptor in the mechanism of maintaining tolerance. FcγRIIB inhibits activation responses triggered by cross-linking of the B cell antigen receptor (BCR), establishing cellular thresholds for lymphocyte stimulation 20 21. In addition, FcγRIIB is capable of triggering apoptosis when it is cross-linked in the absence of the BCR 22. The activities associated with FcγRIIB are manifested in the peripheral lymphoid compartment, during the process of affinity maturation and memory generation in the germinal center (reference 22 and Tew, J., T. Manser, and J.V. Ravetch, manuscript submitted for publication). FcγRIIB expression on B cells is proposed to prevent the activation, proliferation, and expansion of low-affinity autoreactive cells in the periphery. Deficiency in FcγRIIB function removes this checkpoint and thereby contributes to the development of autoimmunity. The data presented here on the role of FcγRIIB in GPS suggest that alterations in the function or expression of the FcγRIIB gene could be a susceptibility factor in the pathogenesis of the human disease. This novel model for GPS offers the opportunity to dissect the etiological mechanisms of disease development and the identification of agents that trigger autoantibody production in a susceptible background, thereby offering new insights into the development of new therapeutic approaches for the treatment of this disease.
Acknowledgments
We are grateful to Dr. Katsuhiko Nakata, Muneo Hamada, and Miwa Yamamoto (Nara Research and Development Center, Santen Pharmaceutical Co., Nara, Japan) for their pathological examinations of eyes, Dr. Masahito Ebina (Department of Respiratory Oncology and Molecular Medicine, Institute of Development, Aging and Cancer, Tohoku University, Japan) for his helpful discussions, and Naomi Takagi for secretarial support.
This work is supported by research grants from the Ministry of Education, Science, Sports and Culture of Japan (to T. Takai and T. Nukiwa), the Nakatomi Health Science Foundation, the Novartis Foundation, the Yamanouchi Foundation for Research on Metabolic Disorders, CREST, JST (to T. Takai), and from the National Institutes of Health and the Juvenile Diabetes Foundation (to J.V. Ravetch).
References
- Goodpasture E.W. The significance of certain pulmonary lesions in relation at the etiology of influenza. Am. J. Med. Sci. 1919;158:863–870. doi: 10.1097/MAJ.0b013e31818fff94. [DOI] [PubMed] [Google Scholar]
- Kalluri R. Goodpasture syndrome. Kidney Int. 1999;55:1120–1122. doi: 10.1046/j.1523-1755.1999.0550031120.x. [DOI] [PubMed] [Google Scholar]
- Stantom M.C., Tange J.D. Goodpasture's syndrome (pulmonary haemorrhage associated with glomerulonephritis) Aust. Ann. Med. 1958;7:132–144. doi: 10.1111/imj.1958.7.2.132. [DOI] [PubMed] [Google Scholar]
- Lener R.A. The role of antiglomerular basement membrane antibody in the pathogenesis of human glomerulonephritis. J. Exp. Med. 1987;126:989–1004. doi: 10.1084/jem.126.6.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunwar S. Alveolar basement membranemolecular properties of the noncollagenous domain (hexamer) of collagen IV and its reactivity with Goodpasture autoantibodies. Am. J. Respir. Cell Mol. Biol. 1991;5:107–112. doi: 10.1165/ajrcmb/5.2.107. [DOI] [PubMed] [Google Scholar]
- Savage C.O., Pusey C.D., Bowman C., Rees A.J., Lockwood C.M. Antiglomerular basement membrane antibody mediated disease in the British Isles. Br. Med. J. 1986;292:1980–1984. doi: 10.1136/bmj.292.6516.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saus J., Wieslander J., Langeveld J.P.M., Quinones S., Hudson B.G. Identification of the Goodpasture antigen as the α3 (IV) chain of collagen IV. J. Biol. Chem. 1988;263:13374–13380. [PubMed] [Google Scholar]
- Steblay R.W. Anti-glomerular basement membrane glomerulonephritis. Am. J. Pathol. 1979;96:875–878. [PMC free article] [PubMed] [Google Scholar]
- Couser W.G., Darby C., Salant D.J., Adler S., Stilmant M.M., Lowenstein L.M. Anti-GBM antibody-induced proteinuria in isolated perfused rat kidney. Am. J. Physiol. 1985;249:F241–F250. doi: 10.1152/ajprenal.1985.249.2.F241. [DOI] [PubMed] [Google Scholar]
- Abate M., Kalluri R., Corona D., Yamaguchi N., McCluskey R.T., Hudson B.G., Andres G., Zoja C., Remuzzi G. Experimental Goodpasture's syndrome in Wistar-Kyoto rats immunized with α3 (IV) chain of the type IV collagen. Kidney Int. 1998;54:1550–1561. doi: 10.1046/j.1523-1755.1998.00153.x. [DOI] [PubMed] [Google Scholar]
- Kalluri R., Danoff T.M., Okada H., Neilson E.G. Susceptibility to anti-glomerular basement membrane disease and Goodpasture syndrome is linked to MHC class II genes and the emergence of T-cell-mediated immunity in mice. J. Clin. Invest. 1997;100:2263–2275. doi: 10.1172/JCI119764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa T., Kubo S., Yoshino T., Ujike A., Matsumura K., Ono M., Ravetch J.V., Takai T. Deletion of Fcγ receptor IIB renders H-2b mice susceptible to collagen-induced arthritis. J. Exp. Med. 1999;189:187–194. doi: 10.1084/jem.189.1.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai T., Ono M., Hikida M., Ohmori H., Ravetch J.V. Augmented humoral and anaphylactic responses in FcγRII-deficient mice. Nature. 1996;379:346–349. doi: 10.1038/379346a0. [DOI] [PubMed] [Google Scholar]
- Clynes R., Ravetch J.V. Cytotoxic antibodies trigger inflammation through Fc receptors. Immunity. 1995;3:21–26. doi: 10.1016/1074-7613(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Takai T., Li M., Sylvestre D., Clynes R., Ravetch J.V. FcR γ chain deletion results in pleiotrophic effector cell defects. Cell. 1994;76:519–529. doi: 10.1016/0092-8674(94)90115-5. [DOI] [PubMed] [Google Scholar]
- Kelly P.T., Haponik E.F. Goodpasture syndromemolecular and clinical advances. Medicine. 1994;73:171–185. doi: 10.1097/00005792-199407000-00001. [DOI] [PubMed] [Google Scholar]
- Fervenza F.C., Terreros D., Boutaud A., Hudson B.G., Williams R.A., Jr., Donadio J.V., Jr., Schwab T.R. Recurrent Goodpasture's disease due to a monoclonal IgA-kappa circulating antibody. Am. J. Kidney Dis. 1999;34:549–555. doi: 10.1016/s0272-6386(99)70084-3. [DOI] [PubMed] [Google Scholar]
- Clynes R., Maizes J.S., Guinamard R., Ono M., Takai T., Ravetch J.V. Modulation of immune complex–induced inflammation in vivo by the coordinate expression of activation and inhibitory Fc receptors. J. Exp. Med. 1999;189:179–186. doi: 10.1084/jem.189.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser R.S., Müller N.L., Colman N., Paré P.D. Goodpasture's syndrome and idiopathic pulmonary hemorrhage. In: Fraser R.S., Müller N.L., Colman N., Paré P.D., editors. Diagnosis of Diseases of the Chest, 4th Ed. Vol. 3. W.B. Saunders Co; Philadelphia: 1999. pp. 1757–1769. [Google Scholar]
- Amigorena S., Bonnerot C., Drake J.R., Choquet D., Hunziker W., Guillet J.G., Webster P., Sautes C., Mellman I., Fridman W.H. Cytoplasmic domain heterogeneity and functions of IgG Fc receptors in B lymphocytes. Science. 1992;256:1808–1812. doi: 10.1126/science.1535455. [DOI] [PubMed] [Google Scholar]
- Ono M., Bolland S., Tempst P., Ravetch J.V. Role of the inositol phosphatase SHIP in negative regulation of the immune system by the receptor FcγRIIB. Nature. 1996;383:263–266. doi: 10.1038/383263a0. [DOI] [PubMed] [Google Scholar]
- Pearse R.N., Kawabe T., Bolland S., Guinamard R., Kurosaki T., Ravetch J.V. SHIP recruitment attenuates FcγRIIB-induced B cell apoptosis. Immunity. 1999;6:753–760. doi: 10.1016/s1074-7613(00)80074-6. [DOI] [PubMed] [Google Scholar]