

Abstract
Mast cells are found in connective and mucosal tissues throughout the body. Their activation via immunoglobulin E (IgE)–antigen interactions is promoted by T helper cell type 2 (Th2) cytokines and leads to the sequelae of allergic disease. We now report a mechanism by which Th2 cytokines can regulate mast cell survival. Specifically, we find that interleukin (IL)-4 and IL-10 induce apoptosis in IL-3–dependent bone marrow–derived mast cells and peritoneal mast cells. This process required 6 d of costimulation with IL-3, IL-4, and IL-10, and expression of signal transducer and activator of transcription 6 (Stat6). Apoptosis was coupled with decreased expression of bcl-xL and bcl-2. While this process occurred independent of the Fas pathway, culture in IL-3+IL-4+IL-10 greatly sensitized mast cells to Fas-mediated death. Additionally, we found that IgE cross-linkage or stimulation with stem cell factor enhanced the apoptotic abilities of IL-4 and IL-10. Finally, IL-3–independent mastocytomas and mast cell lines were resistant to apoptosis induced by IL-3+IL-4+IL-10. These data offer evidence of Th2 cytokine–mediated homeostasis whereby these cytokines both elicit and limit allergic responses. Dysregulation of this pathway may play a role in allergic disease and mast cell tumor survival.
Keywords: interleukin 4, interleukin 10, mast cells, apoptosis, allergy
Introduction
Mast cells are inflammatory effector cells that are concentrated in connective and mucosal tissues. Their role in allergic diseases such as asthma, allergic rhinitis, food allergies, atopic dermatitis, and systemic anaphylaxis has been well-documented 1 2. When mast cells are activated through cross-linkage of their high-affinity FcεRI receptors, they release preformed and newly synthesized mediators including histamine, serine proteases, arachidonic acid metabolites, and cytokines. The result is an inflammatory response that includes vasodilation, bronchial and gastrointestinal smooth muscle contraction, and recruitment of leukocytes. There are three key components that contribute to mast cell–mediated allergic disease: the presence of environmental antigens, production of antigen-specific IgE, and mast cell activation and hyperplasia. The latter two of these components can be affected by Th2 cytokines, including IL-4 and IL-10. For example, IL-4 acts as a proliferative factor for mast cells and promotes Ig class switching in B cells from IgM to IgE, whereas IL-10 induces both mast cell proliferation and differentiation 3 4 5 6 7.
We have recently reported activities of IL-4 and IL-10 that argue for a homeostatic role of these cytokines in mast cell function. Using mouse bone marrow–derived mast cells (BMMCs), we demonstrated that IL-4 and IL-10 decrease expression and signaling of FcεRI and Kit 8 9. These data led us to postulate that Th2 cytokines may function in a homeostatic fashion. The proinflammatory roles of these Th2 cytokines (e.g., IgE production, short-term mast cell proliferation, and survival) may be balanced by negative regulatory effects (decreased expression and function of proinflammatory receptors). Supporting this homeostatic hypothesis is the observation that negative regulatory events occur only after several days of cytokine exposure, thus allowing for both pro- and antiinflammatory phases of the immune response.
This study reinforces the homeostatic role of IL-4 and IL-10. We show that combined stimulation with IL-4 and IL-10 induces apoptosis of IL-3–dependent BMMCs or of purified peritoneal mast cells. As with the regulation of IgE and Kit receptors, the apoptotic activity of IL-4 and IL-10 was not apparent until several days of culture. Our results demonstrate a role for IL-4 and IL-10 in the regulation of mast cell survival, and elucidate some of the mechanisms of this observation. Dysregulation of this homeostasis could underlie the pathophysiology of allergic disease.
Materials and Methods
Cells and Reagents.
BMMCs were derived from 6–12-wk-old C57BL/6, C57BL/6 × 129, C57BL/6 lpr, C57BL/6 gld (all from Taconic Farms), or BL/6 × 129 signal transducer and activator of transcription (Stat)6-deficient mice (donated by James Ihle, St. Jude Research Hospital, Memphis, TN) by culturing unseparated bone marrow cells at 5 × 105/ml in complete RPMI supplemented with 20% WEHI-3 conditioned medium (cRPMI/WEHI) for 3 wk. Mast cell phenotype was confirmed by flow cytometric analysis (FACScan™; Becton Dickinson, hereafter referred to as FACS®) using antibodies specific for Kit, CD13, IgE, or FcγRII/RIII, and by histochemical staining (data not shown). At the time of use, BMMC cultures were >99% mast cells. BMMCs were maintained in cRPMI/WEHI for up to 6 mo, but were generally used for experiments within 2 mo of reaching day 21 in culture. Peritoneal mast cells were isolated by peritoneal lavage, using ice-cold Hank's balanced salt solution lacking Mg2+ or Ca2+ and phenol red (Biofluids). Peritoneal lavage exudates were centrifuged at 150 g for 5 min. Pellets were gently drained, and resuspended in 72.5% Percoll (Amersham Pharmacia Biotech). Samples were centrifuged for 7 min at 300 g. Pellets were collected and cultured in complete RPMI at 37°C for 1 h to remove any residual macrophages. Nonadherent cells were removed and plated in the indicated cytokines. Mast cells comprised 85–90% of the resultant cell population, as determined by Wright-Giemsa staining.
Cytokines and Antibodies.
Murine IL-3, IL-4, and IL-10 were purchased from BioSource International. PE-coupled streptavidin and rat anti–mouse TNF-α, FITC-labeled annexin V, unlabeled mouse IgE, and biotinylated antibodies specific for Fas, FasL, and hamster IgG were purchased from BD PharMingen. Rat anti–mouse IgE and goat F(ab′)2 anti–rat IgG (H+L) were purchased from Southern Biotechnology Associates, Inc. Antibodies to bcl-2 and bcl-xL were obtained from Santa Cruz Biotechnology, Inc., and Transduction Laboratories, respectively.
Analysis of Apoptosis.
For analysis of mast cell apoptosis, cells were cultured at ∼3.0 × 105 cells/ml in IL-3 (5 ng/ml), or in IL-3 with IL-4 (10 ng/ml) and/or IL-10 (10 ng/ml) for 6 d. On day 4, cells were fed by replacing half of the medium and cytokines. On day 6, cells were fixed overnight in a solution of 150 μl 70% ethanol, 25 μl 1× PBS, and 25 μl fetal bovine serum and then stained in a solution of 94% 1× PBS, 50 μg/ml propidium iodide (PI), 100 μg/ml RNase A, and 10−4 mM EDTA. Fluorescent intensities were determined by FACS® analysis. Annexin V staining was performed with the TACS Annexin V-FITC kit from Trevigen, according to the manufacturer's specifications. For cell counts, BMMCs were prepared as described above and then analyzed by FACS® for 20-s periods of time over which total numbers of viable cells were recorded.
Fas and FasL Expression and Function.
At day 3 of culture in IL-3 alone or in IL-3 plus IL-4 and/or IL-10, BMMCs were incubated with 0.3 μl 2.4G2 rat anti–mouse FcγRII/RIII ascites per 100 μl for 10 min at 4°C, followed by 10 μg/ml of biotinylated anti-Fas or anti-FasL in 1× PBS, 3% fetal bovine serum, and 0.1% sodium azide (FACS buffer) for 30 min. After being washed in FACS buffer, cells were incubated for 30 min with 10 μg/ml of PE-coupled streptavidin and analyzed by FACScan™. To investigate the role of Fas signaling on BMMCs, cells were cultured and analyzed as described above for PI staining, except that 20 μg/ml of anti-Fas Ab was added on day 3 of culture.
bcl-2 and bcl-xL mRNA Analysis.
RNA was harvested from BMMCs at day 6 with Trizol (GIBCO BRL), and an RNase Protection assay (RPA) was performed using a kit, probe, and protocol from the RiboQuant System (BD PharMingen). Quantitation of mRNA expression was determined using a Phosphorimaging 445si System (Molecular Dynamics).
bcl-2 and bcl-xL Protein Analysis.
Whole cell lysates were prepared in 20 mM Tris-HCl, pH 7.5, 135 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM sodium vanadate, 0.05 mM NaF, 0.5% NP-40, and 1% Triton X-100. 20 μg of cleared lysates was resolved on a 12% SDS-PAGE acrylamide gel and transferred to nitrocellulose using semidry blot transfer. The blot was blocked for 1 h in TBST buffer (25 mM Tris, pH 7.8, 125 mM NaCl, and 0.25% Tween 20) containing 5% nonfat milk and then incubated in mouse anti–bcl-xL antibody (1:500 dilution; Upstate Biotechnology) overnight at 4°C in TBST/5% nonfat milk. The blot was washed in TBST buffer, and specific reactive proteins were detected using enhanced chemiluminescence (Amersham Pharmacia Biotech). The blot was then stripped of antibody by incubation of the blot in 2% SDS and 62.5 mM Tris buffer, pH 6.7, at 50°C for 30 min, rinsed in TBST buffer, reblocked in TBST/5% nonfat milk, and probed for anti–bcl-2 antibody (1:1,000; Santa Cruz Biotechnology, Inc.) by the same procedure.
IgE Cross-Linkage Studies.
BMMCs were cultured in IL-3 (5 ng/ml) for 3 d before activation. We have found this necessary for maximal cytokine production (our unpublished observations). Cells were then incubated with IgE (10 μg/ml) for 45 min at 4°C in cRPMI+IL-3 (5 ng/ml), washed, and incubated with rat anti–mouse IgE (5 μg/ml) for 30 min at 37°C, followed by goat F(ab′)2 anti–rat IgG (H+L, 5 μg/ml) for 5 h at 37°C. A subset of cells was stained for expression of TNF-α production to confirm cytokine production. After 5 h of activation, IL-4 and IL-10 were added to cultures, and BMMCs were further incubated for 6 d, with feeding on day 4, and assessed for apoptosis by PI staining.
Statistical Analysis.
Analyses of variance were performed at α = 0.05 using Statmost software (DataMost).
Results
IL-4 and IL-10 Induce Apoptosis in BMMCs.
To investigate the role of IL-4 and IL-10 in mast cell apoptosis, BMMCs were cultured in IL-3 alone or in IL-3 with IL-4 and/or IL-10. The length of culture depended on the technique for assessing apoptosis. For detecting the early stages of apoptosis, cells were incubated for 3 d, followed by staining with FITC-coupled annexin V. Annexin V binds phosphatidylserine found on the outer leaflet of cells undergoing “membrane flipping,” an event characteristic of early apoptosis 10. As shown in Fig. 1 A, BMMCs cultured in IL-3+IL-4+IL-10 demonstrated increased annexin V staining compared with cells cultured in IL-3 alone. BMMCs cultured in IL-3+IL-4 or in IL-3+IL-10 showed no change in annexin V binding (data not shown), indicating that dual stimulation with IL-4 and IL-10 was required to induce apoptosis.
Figure 1.
IL-4 and IL-10 induce apoptosis in BMMCs. (A) BMMCs were cultured in IL-3 alone or in IL-3+IL-4+IL-10 for 3 d, stained with FITC–annexin V, and analyzed by flow cytometry. Cells cultured in IL-3+IL-4+IL-10 had a relative mean fluorescent intensity of 49.0 compared with 20.3 for cells incubated in IL-3 alone. (B) BMMCs were cultured in either IL-3 alone or in IL-3+IL-4+IL-10 for 6 d, stained with PI, and analyzed by flow cytometry. Cells with less than G1/G0 DNA content fall within the M1 region and are considered apoptotic. (C) BMMC apoptosis expressed in percentages as determined by PI staining. Results are means and standard errors from 38 experiments using 9 individual BMMC populations. BMMCs were cultured for 6 d with the combinations of cytokines shown in the bar graph. *P < 0.05. (D) Time course of apoptosis as determined by PI staining. *P < 0.05. (E) Counts of viable BMMCs cultured in IL-3 alone, IL-3+SCF, or IL-3+IL-4+IL-10 over a 6-d period. Cell numbers were determined by running samples through FACS® over 20-s intervals. Data shown are for one BMMC population and are representative of three experiments using five different BMMC populations.


A later stage of apoptosis is characterized by the systematic fragmentation of the cell's DNA by its own nucleases. Decreases in DNA content can be determined by PI staining. For this procedure, cells were cultured for 6 d in IL-3 alone or in IL-3 with IL-4 and/or IL-10 before analysis. BMMCs cultured in IL-3 alone showed very little subdiploid DNA content, whereas those cultured in IL-3+IL-4+IL-10 showed an increase in subdiploid DNA (Fig. 1 B). Fig. 1 C shows a summary of PI staining results obtained from multiple experiments. Neither IL-4 nor IL-10 alone was able to induce apoptosis in IL-3–treated BMMCs after 6 d of culture. Percentages of apoptotic BMMCs were 11.7 (± 0.6%) for IL-3+IL-4–treated cells and 9.8 (± 0.8%) for IL-3+IL-10–treated cells compared with 7.6 (± 0.4%) for cells treated in IL-3 alone. Only culture of IL-3–treated BMMCs with IL-4 and IL-10 was able to induce significant apoptosis, seen in 30.1% (± 2.4%) of cells after 6 d. Apoptosis appeared to continue with time in culture, with an increased percentage of apoptotic cells over a 14-d time course (Fig. 1 D). We also found this effect to be dose responsive, with little or no apoptosis observed when the concentration of IL-4 or IL-10 was decreased <5 ng/ml (data not shown).
In Fig. 1 E, the number of viable BMMCs is shown for cells cultured in IL-3 alone or in IL-3+IL-4+IL-10 over 6 d. The number of viable BMMCs cultured in IL-3+IL-4+IL10 increased sharply and peaked at day 4 before diminishing on day 5. By day 6, the number of cells treated with IL-3+IL-4+IL-10 was slightly less than the baseline number of IL-3–cultured BMMCs. These data argue that while costimulation with IL-4 and IL-10 induces mast cell proliferation, this mitogenic activity declines with time in culture, resulting in no net increase in cell numbers compared with cells cultured in IL-3 alone.
Importantly, similar cultures containing IL-3 plus stem cell factor (SCF) consistently demonstrated equal or greater rates of proliferation than cultures receiving IL-3+IL-4+IL-10, yet demonstrated very low rates of apoptosis (Fig. 2, and data not shown). These data, coupled with the fact that our cultures were maintained at a low cell density and fed every 4 d, argue against exhaustion of medium components as an explanation for the observed apoptosis.
Figure 2.

IL-4 and IL-10 induce apoptosis in BMMCs cultured in IL-3 and SCF. Cells were stimulated for 6 d and assessed by PI staining. Resulting means and standard errors are from three experiments using four different BMMC populations. *P < 0.05.
IL-4 and IL-10 Induce Apoptosis in BMMCs Cultured in IL-3 with SCF.
Since SCF is likely to be present under conditions of a Th2 response and is a potent mast cell survival factor, we considered its effect on IL-4– and IL-10–induced BMMC apoptosis. As shown in Fig. 2, BMMCs cultured in IL-3+SCF were 4.9% apoptotic. In contrast, cells cultured in IL-3+SCF+IL-4+IL-10 were 63.3% apoptotic. These data were similar to those obtained using IL-3 alone: in both cases, costimulation with IL-4 and IL-10 induces apoptosis of cultured mast cells even when survival factors such as IL-3 or SCF are present.
IL-4 and IL-10 Induce Apoptosis in Freshly Isolated Peritoneal Mast Cells.
Although BMMCs are a well-studied assay system, we wished to determine if cytokine-induced apoptosis could also be observed in mature mast cells stimulated ex vivo. Peritoneal mast cells were purified from eight C57BL/6 × 129 mice by Percoll gradient centrifugation. These mature mast cells can be maintained in culture if stimulated with IL-3. To assess apoptosis induced by IL-4 and IL-10, peritoneal mast cells were cultured in a manner identical to the BMMC assay system, and stained with PI.
As shown in Fig. 3, peritoneal mast cells cultured in IL-3+IL-4 or in IL-3+IL-4+IL-10 demonstrated a significant increase in apoptosis compared with cells cultured in IL-3 alone. The total number of cells (viable plus apoptotic, obtained by timed flow cytometer assessment) in each of these culture conditions was remarkably similar, with a minimum of 3, 694 (± 154) observed with cells cultured in IL-3+IL-4+IL-10, and a maximum of 4,719 (± 22) observed with cells cultured in IL-3 alone. These data corroborate our results presented in Fig. 1 and Fig. 2, and further support our theory that costimulation with IL-4+IL-10 leads to apoptosis that limits mast cell expansion.
Figure 3.

Stimulation with IL-4+IL-10 induces apoptosis in freshly isolated peritoneal mast cells. Peritoneal mast cells were cultured for 6 d in the indicated cytokines, and assessed for apoptosis by PI staining. Data shown represent the mean percent apoptotic cells of triplicate samples from one of two representative experiments. *P < 0.05.
Fas and FasL Expression in BMMCs Treated with IL-4 and IL-10.
Since mast cells have been reported to express the apoptotic signaling protein Fas 11, we investigated its role in IL-4– and IL-10–induced apoptosis. Fas surface expression on BMMCs cultured for 3 d with IL-3+IL-4 or with IL-3+IL-10 was not significantly different than cells treated with IL-3 alone (data not shown). However, when BMMCs were cultured in IL-3+IL-4+IL-10, Fas expression increased by >80% (Fig. 4 A). RPA analysis reinforced these data, showing increased mRNA encoding Fas after 3 d of culture with IL-3+IL-4+IL-10 compared with IL-3–treated cells (data not shown).
Figure 4.
IL-4 and IL-10 upregulate expression of Fas, but not FasL. (A) BMMCs were cultured in IL-3 alone or in IL-3+IL-4 and/or IL-10 for 3 d, then stained for Fas expression. Cells treated with IL-3 and stained with hamster IgG served as control. For clarity, histograms for BMMCs treated with IL-3 and IL-4 or with IL-3 and IL-10 are omitted; no significant change in Fas expression occurred in these conditions. Data shown are representative of three independent experiments using four BMMC populations. (B) BMMCs were cultured as described above in A, and stained for FasL expression. Histograms for BMMCs treated with IL-3 and IL-4 or with IL-3 and IL-10 are omitted for clarity; FasL expression for BMMCs cultured in these cytokines are no different than for cells treated with IL-3 alone. Data shown are representative of three independent experiments using four BMMC populations. (C) BMMCs from lpr and gld mice were cultured in IL-3 or in IL-3 with IL-4 and/or IL-10 for 6 d, and assessed for apoptosis by PI staining. Data are means and standard errors from four experiments. *P < 0.05, comparing BMMCs cultured in IL-3+IL-4+IL-10 to the same BMMC population cultured in IL-3 alone. WT, wild-type.


Induction of apoptosis through the Fas receptor requires cross-linking by FasL 12, which can be made by BMMCs 13. We assessed the effect of IL-4+IL-10 stimulation on FasL surface expression on BMMCs. Unlike Fas expression, BMMCs cultured in IL-3+IL-4+IL-10 did not exhibit detectable FasL staining (Fig. 4 B).
The lack of FasL staining indicated that cell contact–mediated Fas killing was an unlikely explanation for the observed BMMC apoptosis. However, FasL is also produced as a soluble protein 12. To further investigate the role of Fas/FasL in IL-4+IL-10–induced apoptosis, we cultured BMMCs from lpr (Fas-deficient) and gld (FasL-deficient) mice and treated them in IL-3 alone or in IL-3 with IL-4 and/or IL-10. As illustrated in Fig. 4 C, culture in IL-3+IL-4+IL-10 induced apoptosis in BMMCs derived from lpr (25.6 ± 3.7%), gld (37.3 ± 4.0%), and wild-type (27.0 ± 1.9%) mice at comparable levels (P > 0.05). These data indicated that neither Fas nor FasL expression was required for apoptosis induction.
IL-4 and IL-10 Sensitize BMMCs to Fas-mediated Death.
Although Fas expression was not required for the observed cytokine-induced apoptosis, the upregulation of Fas expression by culture in IL-3+IL-4+IL-10 led us to question if Fas signaling is altered by the culture conditions. To assess Fas-mediated BMMC apoptosis, BMMCs were cultured in IL-3 alone or in IL-3 plus IL-4 and/or IL-10 for 3 d before the addition of anti-Fas antibody. Cells were then cultured for an additional 3 d and assessed for apoptosis by PI staining. As shown in Fig. 5, Fas cross-linkage had no effect on survival of BMMCs cultured in IL-3 alone. However, culture in IL-3+IL-4, or in IL-3+IL-4+IL-10 greatly increased the sensitivity of BMMCs to Fas-mediated apoptosis. Thus, functional activation of Fas was altered by the presence of IL-4 or IL-4+IL-10. The ability of IL-4 to increase Fas-mediated death without altering Fas expression levels indicated that upregulation of Fas expression was not strictly required for this effect.
Figure 5.

IL-4 and IL-10 sensitize BMMCs to Fas-mediated apoptosis. BMMCs were cultured in the indicated cytokines for 3 d, after which anti-Fas antibody was added to a final concentration of 20 μg/ml. Apoptosis was assessed by PI staining on day 6. Data shown are representative of three independent experiments using five BMMC populations. *P < 0.05, comparing BMMCs cultured with anti-Fas to cells cultured without anti-Fas.
Surface IgE Cross-Linkage Enhances Apoptosis Induced by IL-4 and IL-10.
Mast cell activation via IgE receptor signaling is a hallmark of allergic disease. Since this activation results in endogenous cytokine production that might alter our observed signaling by IL-4 and IL-10, we assessed BMMC apoptosis after cross-linkage of surface-bound IgE. BMMCs were allowed to bind IgE, then were washed, and bound IgE was cross-linked with rat anti–mouse IgE, followed by goat anti–rat IgG. After 5 h of stimulation, BMMCs were cultured in IL-3 alone or in IL-3 plus IL-4 and/or IL-10 for 6 d, followed by PI staining. Control cultures received no IgE cross-linkage. A subset of activated cells was fixed and stained for intracellular expression of TNF-α to confirm IgE receptor signaling (data not shown).
Surprisingly, IgE cross-linkage alone was sufficient to induce apoptosis in ∼25% of cells (Fig. 6), a result not previously reported. The extent of apoptosis was increased when BMMCs were activated by IgE cross-linkage and cultured in IL-3+IL-10 or in IL-3+IL-4+IL-10. On average, 87% apoptosis was noted in cultures containing IL-3+IL-4+IL-10. Similar data were obtained when BMMCs were activated with plate-bound IgE (data not shown). These data indicate that FcεRI-mediated mast cell activation, commonplace in allergic disease, may induce apoptosis and greatly enhances apoptotic signaling by IL-4+IL-10.
Figure 6.

IgE cross-linkage induces BMMC apoptosis and enhances Th2 cytokine–mediated apoptosis. BMMCs were unstimulated or activated by IgE cross-linkage as described in Materials and Methods, followed by culture in the indicated cytokines for 6 d. Apoptosis was assessed by PI staining. Data shown are means and standard errors of three samples from one of three representative experiments. *P < 0.05, comparing BMMCs cultured with IgE cross-linkage to cells cultured without IgE cross-linkage.
IL-4 and IL-10 Decrease mRNA and Protein Levels of bcl-2 and bcl-xL.
bcl-2 and bcl-xL have been shown to inhibit apoptosis 14. Therefore, changes in expression of either of these proteins might contribute to our observed apoptosis. We investigated the effects of IL-4 and IL-10 on mRNA and protein levels of these proteins, using RPA and Western blot analysis. As shown in Fig. 7 A, levels of bcl-2 and bcl-xL mRNA expression were lower in BMMCs cultured in IL-3+IL-4+IL-10 than cells treated with IL-3 alone. Costimulation with IL-4 and IL-10 was not required to decrease bcl-2 and bcl-xLexpression, as BMMCs cultured in IL-3+IL-4 or IL-3+IL-10 also showed diminished levels of mRNA for these proteins. Western blot analysis corroborated these results (Fig. 7 B): bcl-2 and bcl-xL protein levels were decreased in BMMCs after culture in IL-3 with IL-4 and/or IL-10, with an almost total absence of detectable protein in cells cultured in IL-3+IL-4+IL-10.
Figure 7.

IL-4 and IL-10 decrease mRNA and protein levels of bcl-2 and bcl-xL. (A) For RPA analysis, BMMCs were cultured for 6 d in IL-3 alone or IL-3 plus IL-4, IL-10, or both IL-4 and IL-10. mRNA for the housekeeping genes L32 and glyceraldehyde 3-phosphate dehydrogenase (GADPH) serve as loading controls. For BMMCs treated in IL-3+IL-4+IL-10, mRNA levels decreased by 76% for bcl-2 and 71% for bcl-xL compared with mRNA levels for BMMCs cultured in IL-3 alone. (B) Western blot analysis of bcl-2 and bcl-xL protein expression in total cell lysates from BMMCs cultured in IL-3 alone or IL-3 plus IL-4 and/or IL-10 for 6 d. Molecular masses are shown in parentheses. Data shown in both A and B are from one of two independent experiments using three BMMC populations.
IL-4 and IL-10 Do Not Induce Apoptosis in BMMCs Derived from Stat6 Knockout Mice.
The IL-4 receptor signals through multiple pathways (for a review, see reference 15), of which Stat6 has been argued to be one of the most critical. To assess a role for Stat6 in our observed apoptosis system, BMMCs derived from Stat6-deficient mice were cultured in IL-3 alone or in IL-3 with IL-4 and/or IL-10, and assessed for apoptosis by PI staining. Stat6-deficient BMMCs were completely resistant to apoptosis induced by IL-3+IL-4+IL-10, as shown in Fig. 8. These data indicate that expression of Stat6 is absolutely required for the induction of apoptosis by IL-3+IL-4+IL-10. The role of Stat6 in this process remains to be determined and is a central part of our current investigations.
Figure 8.

IL-4 and IL-10 do not induce apoptosis in Stat6-deficient mast cells. BMMCs from Stat6-deficient mice were cultured in the indicated cytokines, and assessed for apoptosis after 6 d of culture. Apoptosis in BMMCs from Stat6-deficient mice treated with IL-3+IL-4+IL-10 was not significantly greater than for Stat6-deficient cells cultured in IL-3 alone (P > 0.05). Results shown are means and standard errors from 14 experiments using 7 Stat6-deficient BMMC populations. WT, wild-type.
IL-4 and IL-10 Fail to Induce Apoptosis in Factor-independent Mast Cell Lines.
Both BMMCs and peritoneal mast cells are factor-dependent, primary cell populations. However, many mast cell studies employ factor-independent mast cell lines, some of which are mastocytoma tumors. To study the effect of IL-4 and IL-10 on these cell lines, we stimulated five different factor-independent mast cell lines with IL-3+IL-4+IL-10 for 6 d. Fig. 9 is a sample histogram of PI staining data for the P815 factor-independent mast cell line. P815 cells treated with IL-3+IL-4+IL-10, with IL-3 alone, or with no cytokines had similar fluorescent staining patterns. Mean percentage levels of apoptosis for P815 cells as well as for the MMC 14, MMC 34, BMDMC-B, and CFTL-12 factor-independent mast cell lines treated with IL-4 and IL-10 are shown in Table . These data were very similar to those observed with P815 cells: IL-3+IL-4+IL-10 failed to induce apoptosis after 6 d in any of these cell lines. It appears that Th2 cytokine-induced apoptosis is lost in these transformed, factor-independent cells.
Figure 9.
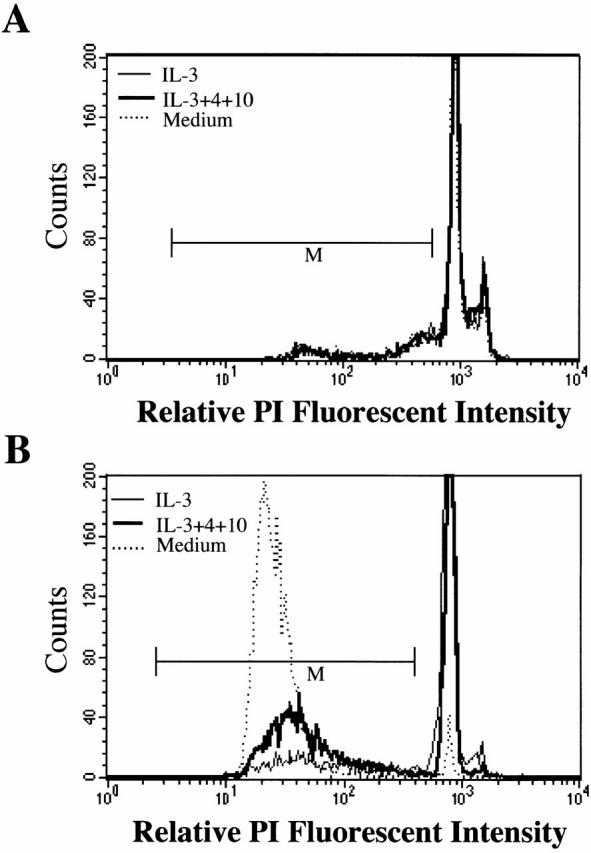
Factor-independent mast cells fail to undergo apoptosis when cultured in IL-3+IL-4+IL-10. P815 mastocytoma cells (A) or wild-type BMMCs (B) were cultured in the absence of cytokines, with IL-3 alone, or with IL-3+IL-4+IL-10 for 6 d. Apoptosis was determined by PI staining. Data shown are representative of three independent experiments that gave similar results.
Table 1.
IL-4+IL-1–induced Apoptosis in Mastocytoma Cell Lines: Data Shown Are Percentage of Apoptotic Cells
| Culture conditions | |||||
|---|---|---|---|---|---|
| Cell line | None | IL-3 | IL-3+IL-4 | IL-3+IL-10 | IL-3+IL-4+IL-10 |
| MMC 14 | 15.9 ± 1.7 | 19.4 ± 4.8 | 18.6 ± 3.3 | 16.9 ± 3.2 | 22.9 ± 4.2 |
| MMC 34 | 20.4 ± 3.0 | 18.7 ± 2.9 | 21.4 ± 3.7 | 21.2 ± 4.6 | 19.1 ± 3.3 |
| P815 | 21.6 ± 2.8 | 18.5 ± 2.3 | 17.1 ± 3.4 | 19.4 ± 2.2 | 19.8 ± 3.4 |
| BMDMC-B | 17.1 ± 5.3 | 17.1 ± 1.4 | 14.8 ± 2.0 | 17.3 ± 3.0 | 16.7 ± 4.0 |
| CFTL-12 | 25.2 ± 6.3 | 20.7 ± 4.9 | 21.3 ± 5.0 | 22.2 ± 6.0 | 22.2 ± 4.7 |
Cells were cultured in the indicated cytokines for 6 d, followed by assessment of apoptosis by PI staining, as detailed in Materials and Methods. Data shown are mean ± SE from three independent experiments.
Discussion
Although IL-4 and IL-10 have each been reported to influence apoptosis of myeloid cells 16 17 18 19 20 21 22, conflicting data exist in the mast cell field. IL-4 and IL-10 have been demonstrated to promote mast cell survival, development, and proliferation 3 4 5 6 7. Accordingly, we observed an increase in BMMC numbers after culture in IL-3+IL-4+IL-10. However, by day 3 of these cultures we detected surface phosphatidylserine expression, an event characteristic of early apoptosis 10 23. Increased numbers of pyknotic cells were observed by day 5, and DNA fragmentation was detected by day 6.
Induction of apoptosis by IL-4 and IL-10 was not observed solely with BMMCs, since freshly isolated peritoneal mast cells, cultured ex vivo as pure populations, yielded similar data. The ability of IL-4 to induce peritoneal mast cell apoptosis without IL-10 costimulation was an unexpected finding that may be specific to the phenotype of mast cells used. Peritoneal mast cells are a fully mature mast cell, whereas BMMC cultures have been argued to be incompletely differentiated. The importance of this finding remains to be determined, but the role of Th2 cytokines in mast cell apoptosis appears to be consistent regardless of these phenotypic nuances. Further, Oskeritzian et al. 24 recently reported that IL-4 can directly induce apoptosis of purified human mast cells. Thus, IL-4–mediated regulation of mast cell survival does not appear to be isolated to our assay system.
It should be stated clearly that BMMC apoptosis induced by IL-3+IL-4+IL-10 was not due to exhaustion of medium components. BMMCs were maintained at low concentrations, medium was replenished every 4 d, and cytokines were present in concentrations sufficient to support proliferation. Furthermore, BMMCs stimulated with IL-3 and SCF for 6 d proliferated to an equal or greater extent than BMMCs cultured in IL-3+IL-4+IL-10, but showed less apoptosis than cells treated with IL-3 alone.
Despite the lack of a Fas requirement for apoptosis induced by IL-3+IL-4+IL-10, Fas function was greatly enhanced in these cultures. Although Fas cross-linkage had no effect on the viability of BMMCs cultured in IL-3 alone, there was a dramatic increase in Fas-mediated apoptosis (>90% apoptotic cells in several experiments) after culture with IL-3+IL-4 or with IL-3+IL-4+IL-10. As mast cells would likely be exposed to a FasL-bearing population in vivo, our data offer a mechanism by which Th2 cytokines may “prime” mast cells for apoptosis. Given that BMMCs cultured in IL-3+IL-4 showed significant Fas-induced apoptosis exclusive of changes in Fas expression, changes in Fas signaling rather than expression may explain our observations. In support of this, initial experiments have demonstrated an increase in Fas-associated death domain (FADD) mRNA after IL-4 stimulation of BMMCs (data not shown). Thus, it is possible that IL-4 may alter the “quality” of the Fas signal, sensitizing mast cells to Fas-mediated apoptosis.
Mast cells are frequently activated by cross-linkage of IgE bound to FcεRI. This results in the activation of numerous signaling pathways, which might alter apoptotic signals conveyed by IL-4+IL-10. Thus, our theory that Th2 cytokines such as IL-4 and IL-10 serve as feedback factors limiting mast cell growth and activation must account for cell activation status. To this end, we show that mast cells activated by surface IgE cross-linkage undergo limited (25%) apoptosis in a 6-d culture. This is the first report of which we are aware that implicates FcεRI signaling in mast cell apoptosis. More impressive was the finding that nearly 90% of BMMCs activated by IgE cross-linkage before stimulation with IL-4+IL-10 undergo apoptosis. These findings are analogous to our Fas data, where one signal “primes” mast cells for apoptotic signaling by a second stimulus. Importantly, both Fas and FcεRI signaling are likely to occur in mast cells in vivo, especially during an inflammatory response. As such, the death of nearly all mast cells exposed to dual signaling by Th2 cytokines with either Fas or FcεRI may be a clinically relevant means of limiting mast cell hyperplasia.
Stimulation with IL-4 and/or IL-10 led to decreased mRNA and protein levels of bcl-2 and bcl-xL after 6 d in culture. Similar to our observations of increased Fas-mediated cell killing after IL-4 signaling, combined stimulation with IL-4 and IL-10 was not necessary to reduce bcl-2 and bcl-xL expression. These proteins have been shown to block the effects of the Fas-induced apoptotic pathway 25 26; thus, their absence may contribute not only to the observed cytokine-induced apoptosis in BMMCs but also to the ability of these cytokines to prime BMMCs for Fas-mediated death. This question is the focus of current study.
The mechanism of apoptosis induced by culture in IL-3+IL-4+IL-10 remains to be clarified. However, use of Stat6-deficient BMMCs indicates that expression of this transcription factor is strictly required. This observation fits well with our existing theory of IL-4–mediated regulation of mast cell function. Although IL-4 is known to promote mast cell survival and proliferation in short-term cultures 4 5, we find that longer stimulation (>3 d) leads to diminished expression and function of the key signal transduction proteins FcεRI and Kit 8 9. As with apoptosis, IL-4–mediated regulation of FcεRI and Kit requires Stat6 expression. Thus, after 3 d of stimulation with Th2 cytokines, mast cells appear to receive several downregulatory signals leading to their inactivation and death. All of these signals require Stat6 expression.
Under normal conditions, mast cell hyperplasia during a Th2 response is time limited. For example, Nippostrongylus brasiliensis infection of rats induces a strong Th2 response with profound intestinal mastocytosis. This mast cell expansion peaks at day 16 after infection, and resolves after ∼5 wk 27. Th2-mediated diseases such as allergic asthma are frequently associated with eosinophilia, but true mastocytosis is not usually noted (for example, see references 28, 29). Thus, Th2 cytokines may be initial stimulators of mast cell proliferation, but long-term exposure does not appear to lead to chronic in vivo mastocytosis. It is interesting to note that Stat6-deficient mice have a greatly exacerbated mast cell hyperplasia during intestinal nematode infection or after IL-4 treatment 30. Also, recent reports indicate that IL-4–deficient mice have a twofold increase in peritoneal mast cell numbers 31. It is tempting to speculate that the defect in both strains of mice is related to a failure of normal apoptotic controls that require IL-4 receptor–mediated Stat6 activation.
Mast cell hyperplasia leading to neoplastic growth is noted in a collection of diseases that begin with limited clonal mast cell expansion and progress to systemic mastocytosis. This progression from a focal mastocytoma to systemic mastocytosis is associated with aberrant Kit signaling, thought to lead to unlimited mast cell proliferation 32 33. In an attempt to mimic this scenario, we investigated the effect of IL-4 and IL-10 on factor-independent mast cell lines, some of which are known to bear c-kit mutations homologous to those found in human mastocytosis 34. Our studies found that no combination of IL-3, IL-4, or IL-10 had any effect on apoptosis of these mast cell lines.
If the lack of apoptosis in factor-independent mast cell lines could be attributed to constitutive Kit signaling, these data would appear to directly contradict the SCF-augmented apoptosis shown in Fig. 2. Some key points may explain the different data obtained with primary versus transformed cell populations. First, primary mast cells respond to Kit in a ligand-dependent manner that is limited by downregulation of Kit after SCF binding. Constitutively active forms of Kit in mastocytomas have neither constraint placed on them. The result may yield both a quantitative and qualitative difference in signaling, with chronic Kit activation circumventing the very death that is enhanced by limited Kit signaling. In support of this theory, Piao and colleagues demonstrated that a constitutively active form of Kit underwent autophosphorylation at different sites than wild-type Kit, and led to altered substrate activation 35. Second, other mutations might explain the lack of apoptosis observed with factor-independent mast cells. At present, we find that these lines have reduced expression of IL-4Rα and constitutive activation of several DNA binding proteins (our unpublished observations). It appears that analyzing the role of mutant Kit receptors may require a cell transfection system to limit the inherent variability encountered with spontaneously transformed cell lines. How SCF–Kit interactions interplay with Th2 cytokines is an important issue worthy of further study.
Although not addressed by our findings, other Th2 cytokines may function in positively or negatively regulating mast cell apoptosis. IL-5 and IL-6 have been reported to support human mast cell survival 29. While IL-13 bears many similarities to IL-4 and is produced by mast cells 36, we have been unable to show IL-13–mediated regulation of FcεRI or Kit (Ryan, J.J., unpublished findings). Thus, the role of Th2 cytokines other than IL-4 and IL-10 in eliciting mast cell apoptosis is unknown. Clarifying this issue may offer insight into the interplay of cytokine networks and their regulation of allergic disease.
The current data, coupled with those we have recently reported 8 9, offer a view of Th2 cytokines that includes not only their initiating allergic inflammation, but also controlling it. This homeostatic role of Th2 cytokines is summarized in Fig. 10. Initially eliciting mast cell proliferation and IgE synthesis, Th2 cytokine signaling is largely proinflammatory. However, continued stimulation for 3 d or longer inhibits expression and function of the proinflammatory/prosurvival mast cell receptors FcεRI and Kit. Subsequently, IL-4 and IL-10 directly induce mast cell apoptosis and facilitate Fas-mediated killing. We have recently found these cytokines to also be potent inhibitors of mast cell development from bone marrow progenitors (our unpublished findings), a result augmenting the downregulatory role of Th2 cytokines. The resulting effect of these Th2 cytokines is control of mast cell function throughout the course of an inflammatory response. The actions of IL-4 and IL-10 on mast cells are characterized by an initial increase in proliferation and mediator production followed by a self-limiting resolution of inflammatory activity. This homeostatic mechanism may explain the absence of chronic mast cell hyperplasia in some allergic diseases. The loss of such controls could contribute to neoplastic transformation.
Figure 10.

Model of Th2 cytokine–mediated homeostasis in mast cell inflammatory responses. Initial production of IL-4 and IL-10 elicits IgE synthesis and mast cell proliferation. IgE–antigen interactions activate mast cells, causing production of inflammatory mediators, with the resulting sequelae noted in allergic disease. This process may be reversed after 3 d, when IL-4 and IL-10 inhibit FcεRI and Kit function, and ultimately induce apoptosis of both mature mast cells and mast cell precursors. The result is a balance of pro- and antiinflammatory reactions, yielding time-limited mast cell hyperplasia and inflammatory reactions coordinated by the Th2 cytokines IL-4 and IL-10.
Acknowledgments
The authors would like to thank Hey Jin Chong and Marlena Cain for their help with experiments, Don Purkall and Dr. Shaun Ruddy for the generous use of their flow cytometry facilities, and John Myers for his technical assistance. We would also like to thank Dr. Thomas Huff for many useful conversations, and Dr. William Paul for his assistance with this manuscript.
This work was supported in part by generous grants from the American Cancer Society (IN-105, to J.J. Ryan), the Thomas F. Jeffress and Kate Miller Jeffress Memorial Trust (J-457, to J.J. Ryan), and National Institutes of Health grant 1R01-AI43433 (to J.J. Ryan).
Footnotes
Abbreviations used in this paper: BMMC, bone marrow–derived mast cell; PI, propidium iodide; RPA, RNase protection assay; SCF, stem cell factor; Stat, signal transducer and activator of transcription.
References
- Schwartz L.B., Huff T.F. Biology of mast cells and basophils. In: Middleton E., Reed C.E., Ellis E.F., Adkinson N.F., Yunginger J.W., Busse W.W., editors. AllergyPrinciples and Practice. Mosby Co; St. Louis: 1993. pp. 135–168. [Google Scholar]
- Sutton B.J., Gould H.J. The human IgE network. Nature. 1993;366:421–428. doi: 10.1038/366421a0. [DOI] [PubMed] [Google Scholar]
- Paul W.E. Interleukin-4a prototypic immunoregulatory lymphokine. Blood. 1991;77:1859–1870. [PubMed] [Google Scholar]
- Mosmann T.R., Bond M.W., Coffman R.L., Ohara J., Paul W.E. T-cell and mast cell lines respond to B-cell stimulatory factor 1. Proc. Natl. Acad. Sci. USA. 1986;15:5654–5658. doi: 10.1073/pnas.83.15.5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi Y., Kanakura Y., Fujita J., Takeda S.I., Nakano T., Tarui S., Honjo T., Kitamura Y. Interleukin-4 is an essential factor for in vitro clonal growth of murine connective tissue mast cells. J. Exp. Med. 1987;165:268–273. doi: 10.1084/jem.165.1.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson-Snipes L., Dhar V., Bond M.W., Mosmann T.R., Moore K.W., Rennick D.M. Interleukin 10a novel stimulatory factor for mast cells and their progenitors. J. Exp. Med. 1991;173:507–510. doi: 10.1084/jem.173.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rennick D.M., Hunte B., Holland G., Thompson-Snipes L. Cofactors are essential for stem cell factor-dependent growth and maturation of mast cell progenitorscomparative effects of interleukin-3 (IL-3), IL-4, IL-10, and fibroblasts. Blood. 1995;85:57–65. [PubMed] [Google Scholar]
- Ryan J.J., Desimone S., Klisch G., McReynolds L., Han K., Kovacs R., Mirmonsef P., Huff T.F. IL-4 inhibits mouse mast cell FcεRI expression through a STAT6-dependent mechanism. J. Immunol. 1998;161:6915–6923. [PubMed] [Google Scholar]
- Mirmonsef P., Shelburne C.P., Yeatman C.F., II, Chong H.J., Ryan J.J. Inhibition of Kit expression by IL-4 and IL-10 in murine mast cellsrole of STAT6 and phosphatidylinositol 3′-kinase. J. Immunol. 1999;163:2530–2539. [PubMed] [Google Scholar]
- Koopman G., Reutelingsperger C.P.M., Kuijten G.A.M., Keehen R.M.J., Pals S.T., van Oers M.H.J. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- Hartmann K., Wagelie-Steffen A.L., von Stebut E., Metcalfe D.D. Fas (CD95, APO-1) antigen expression and function in murine mast cells. J. Immunol. 1997;159:4006–4014. [PubMed] [Google Scholar]
- Peter M.E., Krammer P.H. Mechanisms of CD95 (APO-1/Fas)-mediated apoptosis. Curr. Opin. Immunol. 1998;10:545–551. doi: 10.1016/s0952-7915(98)80222-7. [DOI] [PubMed] [Google Scholar]
- Wagelie-Steffen A.L., Hartmann K., Vliagoftis H., Metcalfe D.D. Fas ligand (FasL, CD95L, APO-1L) expression in murine mast cells. Immunology. 1998;94:569–574. doi: 10.1046/j.1365-2567.1998.00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams J.M., Cory S. The Bcl-2 protein familyarbiters of cell survival. Science. 1998;281:1322–1326. doi: 10.1126/science.281.5381.1322. [DOI] [PubMed] [Google Scholar]
- Nelms K., Keegan A.D., Zamorano J., Ryan J.J., Paul W.E. The IL-4 receptorsignaling mechanisms and biological functions. Annu. Rev. Immunol. 1999;17:701–738. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- Mangan D.F., Robertson B., Wahl S.M. IL-4 enhances programmed cell death in stimulated human monocytes. J. Immunol. 1992;148:1812–1816. [PubMed] [Google Scholar]
- Manabe A., Coustan-Smith E., Kumagi M., Behm F.G., Raimondi S.C., Pui C.-H., Campana D. Interleukin-4 induces programmed cell death (apoptosis) in cases of high-risk acute lymphoblastic leukemia. Blood. 1994;83:1731–1737. [PubMed] [Google Scholar]
- Yamamoto M., Kawabata K., Fujihashi K., McGhee J.R., van Dyke T.E., Bamberg T.V., Hiroi T., Kiyono H. Absence of exogenous interleukin-4-induced apoptosis of gingival macrophages may contribute to chronic inflammation in periodontal diseases. Am. J. Pathol. 1996;148:331–339. [PMC free article] [PubMed] [Google Scholar]
- Gooch J.L., Lee A.V., Yee D. Interleukin-4 inhibits growth and induces apoptosis in human breast cancer cells. Cancer Res. 1998;58:4199–4205. [PubMed] [Google Scholar]
- Wedi B., Raap U., Lewrick H., Kapp A. IL-4-induced apoptosis in peripheral blood eosinophils. J. Allergy Clin. Immunol. 1998;102:1013–1020. doi: 10.1016/s0091-6749(98)70340-9. [DOI] [PubMed] [Google Scholar]
- Cox G. IL-10 enhances resolution of pulmonary inflammation in vivo by promoting apoptosis of neutrophils. Am. J. Pathol. 1996;271:566–571. doi: 10.1152/ajplung.1996.271.4.L566. [DOI] [PubMed] [Google Scholar]
- Itoh K., Hirohata S. The role of IL-10 in human B cell activation, proliferation, and differentiation. J. Immunol. 1995;154:4341–4350. [PubMed] [Google Scholar]
- Darzynkiewicz Z., Juan G., Li X., Gorczyca W., Murakami T., Traganos F. Cytometry in cell necrobiologyanalysis of apoptosis and accidental cell death (necrosis) Cytometry. 1997;27:1–20. [PubMed] [Google Scholar]
- Oskeritzian C.A., Wang Z., Kochan J.P., Grimes M., Du Z., Chang H.-W., Grant S., Schwartz L.B. Recombinant human (rh)IL-4-mediated apoptosis and rhIL-6-mediated protection of rh-stem cell factor-dependent human mast cells derived from cord blood mononuclear cell progenitors. J. Immunol. 1999;163:5105–5115. [PubMed] [Google Scholar]
- Schneider T.J., Grillot D., Foote L.C., Nunez G.E., Rothstein T.L. Bcl-x protects primary B cells against Fas-mediated apoptosis. J. Immunol. 1997;159:4834–4839. [PubMed] [Google Scholar]
- Itoh N., Tsujimoto Y., Nagata S. Effect of bcl-2 on Fas antigen-mediated cell death. J. Immunol. 1993;151:621–627. [PubMed] [Google Scholar]
- Befus A.D., Bienenstock J. Immunologically-mediated intestinal mastocytosis in Nippopstrongylus brasiliensis-infected rats. Immunology. 1979;38:95–101. [PMC free article] [PubMed] [Google Scholar]
- Stampfli M.R., Jordana M. Eosinophilia in antigen-induced airways inflammation Can. Respir. J. 5Suppl. A1998. 31A 35A [PubMed] [Google Scholar]
- Yanagida M., Fukamachi H., Ohgami K., Kuwaki T., Ishii H., Uzumaki H., Amano K., Tokiwa T., Mitsui H., Saito H. Effects of the T-helper 2-type cytokines, interleukin-3 (IL-3), IL-4, IL-5, and IL-6 on the survival of cultured human mast cells. Blood. 1995;86:3705–3714. [PubMed] [Google Scholar]
- Urban J.E., Noben-Trauth N., Donaldson D.D., Madden K.B., Morris S.C., Collins M., Finkelman F.D. IL-13, IL-4Rα, and STAT6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis . Immunity. 1998;8:255–264. doi: 10.1016/s1074-7613(00)80477-x. [DOI] [PubMed] [Google Scholar]
- Chen H.J., Lycke N., Enerback L. Surface and gene expression of immunoglobulin E receptors on mast cells and mast cell numbers in interleukin-4-gene knockout mice. Immunology. 1999;96:544–550. doi: 10.1046/j.1365-2567.1999.00727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garriga M.M., Friedman M.M., Metcalfe D.D. A survey of the number and distribution of mast cells in the skin of patients with mast cell disorders. J. Allergy Clin. Immunol. 1988;82:425–432. doi: 10.1016/0091-6749(88)90015-2. [DOI] [PubMed] [Google Scholar]
- Nagata H., Worobec A.S., Oh C., Chowdhury B.A., Tannebaum S., Suzuki Y., Metcalfe D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with associated hematologic disorder. Proc. Natl. Acad. Sci. USA. 1995;92:10560–10564. doi: 10.1073/pnas.92.23.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimura T., Furitsu T., Morimoto M., Isozaki K., Nomura S., Matsuzawa Y., Kitamura Y., Kanakura Y. Ligand-independent activation of c-kit receptor tyrosine kinase in a murine mastocytoma cell line P815 generated by a point mutation. Blood. 1994;83:2619–2626. [PubMed] [Google Scholar]
- Piao X., Paulson R., van der Geer P., Pawson T., Bernstein A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc. Natl. Acad. Sci. USA. 1996;93:14665–14669. doi: 10.1073/pnas.93.25.14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burd P., Thompson W., Max E., Mills F. Activated mast cells produce interleukin 13. J. Exp. Med. 1995;181:1373–1380. doi: 10.1084/jem.181.4.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]