

Abstract
Bone marrow (BM)-derived professional antigen-presenting cells (pAPCs) are required for the generation of cytotoxic T lymphocyte (CTL) responses to vaccinia virus and poliovirus. Furthermore, these BM-derived pAPCs require a functional transporter associated with antigen presentation (TAP). In this report we analyze the requirements for BM-derived pAPCs and TAP in the initiation of CTL responses to lymphocytic choriomeningitis virus (LCMV) and influenza virus (Flu). Our results indicate a requirement for BM-derived pAPCs for the CTL responses to these viruses. However, we found that the generation of CTLs to one LCMV epitope (LCMV nucleoprotein 396–404) was dependent on BM-derived pAPCs but, surprisingly, TAP independent. The study of the CTL response to Flu confirmed the existence of this BM-derived pAPC-dependent/TAP-independent CTL response and indicated that the TAP-independent pathway is ∼10–300-fold less efficient than the TAP-dependent pathway.
Keywords: mice; lymphocytic choriomeningitis virus; influenza virus; T lymphocytes, cytotoxic; immunity, cellular
Introduction
CTLs are a major arm of the immune response responsible for clearing viruses after they have infected cells. Fully differentiated CTLs (effector cells) can recognize infected cells and kill them, thereby eliminating sites of production and/or persistence of virus. CTLs are able to recognize these cells because they display fragments of viral proteins bound to MHC class I molecules on the cell surface 1. Most of these MHC class I–presented viral polypeptides are generated in the cytoplasm from the hydrolysis of viral proteins synthesized by the infected cell. The antigenic peptides are then transferred into the endoplasmic reticulum (ER) by the transporter associated with antigen presentation (TAP) transporter 2 3. Newly synthesized MHC class I molecules are assembled and retained in the ER. Upon binding a peptide, the class I molecule is released and is transported to the cell surface where it is displayed to the immune system. This pathway is known as the endogenous or classical MHC class I pathway 4.
We have recently examined how tissue-tropic viral infections (i.e., those that infect nonhematopoietic cells but not bone marrow [BM]-derived cells) stimulate precursor CTLs to become CTL effectors. We found that although nonhematopoietic cells infected with vaccinia or poliovirus displayed viral peptides on their surface MHC class I molecules, they were unable to initiate a CTL immune response. Instead, the generation of these responses required the presentation of the viral antigens by BM-derived professional APCs (pAPCs), presumably dendritic cells or possibly macrophages 5. These cells are present in all tissues wherein they acquire antigens and subsequently traffic to lymph nodes where they present these antigens to specific CD8+ T cells.
BM-derived pAPCs can acquire viral antigens in two different ways. One is if they are infected with the virus. In this case they present the viral antigens via the endogenous MHC class I pathway. The second way is by internalization of the viral antigens, which subsequently get presented on class I molecules. This latter mechanism is known as the exogenous MHC class I pathway or “cross priming” 6. The exogenous MHC class I pathway was shown to be the obligatory mechanism for generating CTL responses to poliovirus infections of nonhematopoietic cells 5.
The ability to take up and present exogenous antigens on MHC class I molecules is largely limited to dendritic cells and macrophages. Presentation of exogenous antigens on MHC class I molecules can occur by two distinct mechanisms. In one, the antigen is transferred from the endocytic compartment into the cytosol where it is degraded into oligopeptides. This mechanism requires the TAP transporter to ferry peptide to class I molecules in the ER 7. This pathway has been widely described in vitro, and is used for the generation of CTLs to poliovirus infections of nonhematopoietic cells in vivo 5. The second mechanism for presenting exogenous antigens on MHC class I molecules does not require the TAP transporter. In this situation, the peptides are generated in the endocytic compartment and somehow are acquired by class I molecule in this site or at the cell surface. This mechanism has only been demonstrated in vitro 8 9, and it is unknown whether it is operative in in vivo situations.
Here we report new experiments to address the role of BM-derived pAPCs in the CTL responses to lymphocytic choriomeningitis virus (LCMV) and influenza virus (Flu). Although our results confirm and generalize the notion that BM-derived pAPCs are required for the generation of antiviral CTL responses, the results were surprising in as much as TAP-independent antigen presentation by BM-derived cells could readily be demonstrated for natural epitopes of these two viruses in vivo. To our knowledge, the results reported here are the first to demonstrate that a TAP-independent pathway can be functional in vivo. However, our results also demonstrate that this TAP-independent pathway of antigen presentation is much less efficient than the TAP-dependent mechanism which, in vivo, seems to be the dominant pathway of MHC class I antigen presentation.
Materials and Methods
Cell Lines and Media.
EL4 (H-2b) and P815 (H-2d) cells were obtained from American Type Culture Collection. These cells were maintained in RPMI supplemented with 10% FCS, 2 mM l-glutamine, 50 μg/ml gentamicin, 0.01 M Hepes buffer, nonessential amino acids (all from Life Technologies), and 5 × 10−5 2-mercaptoethanol (Sigma-Aldrich).
Mice.
Mice were kept at the Animal Medicine Facility of the University of Massachusetts Medical Center. All experiments were performed under Institutional Animal Care and Use Committee–approved protocols.
Strains.
B6-Tap1tp 1Arp (TAP0/0), C57BL/6 (B6), DBA2 (D2), and (C57BL/6 × DBA2)F1 (B6D2) mice were obtained from The Jackson Laboratory at 6–8 wk of age.
Preparation of BM Chimeras.
To prepare chimeras, BM cells from 6–15-wk-old donor mice were treated with anti-Thy 1 antibody (M5/49.4.1; American Type Culture Collection) and complement to eliminate mature T cells, washed twice, and resuspended in PBS. Recipient mice were irradiated with 600 rads followed by a second irradiation with 500 rads 4 h later using a GammaCell 40 apparatus (Nordion). Supralethally irradiated chimeras received a single dose of 1,300 rads. Irradiated mice were reconstituted by intravenous inoculation of 4–6 × 106 BM cells from the different donors. To avoid rejection of donor MHC class I–negative TAP0/0 cells by host NK cells, chimeras also received an intraperitoneal injection of 10 μl rabbit anti–asialo GM1 gammaglobulin (Wako Chemicals) on the day of the transplant. This treatment was also employed in control chimeras receiving B6 BM. Chimeras were rested for 4–6 mo after reconstitution to allow for complete elimination of host-derived pAPCs. In some experiments double-irradiated chimeras were used. These were prepared by subjecting chimeras (4 mo after the first BM transplant) to a second irradiation and BM transplant.
Viruses and Peptides.
LCMV virus stocks (Armstrong strain, 4 × 105 PFU/ml) were a gift from Dr. Raymond Welsh (University of Massachusetts Medical School, Worcester, MA). Flu (A/PR8/34) was obtained as chicken alantoic fluid from infected, embryonated eggs. The virus stock contained 4,860 hemagglutination units (HAU) per ml, in which 1 HAU was considered as the dilution of virus stock (in 100 μl PBS) that agglutinated 50% of human red blood cells (group O, factor Rh+) resuspended at a concentration of 0.5% in 100 μl PBS. Both viruses were injected intraperitoneally at the indicated doses in 0.5 ml PBS and killed 8 (LCMV) or 15–20 d (Flu) after infection.
To analyze responses to the different viruses, we used synthetic versions of well-defined peptide epitopes. All of the peptides were custom synthesized by Research Genetics. The purity and identity of the peptides were determined by HPLC and mass spectrometry. The Flu epitopes were Flu nucleoprotein (np)366–374 (ASNENMETM 10) and Flu np147–155 (TYQRTRALV [10]), both derived from the np of A/PR/8/34. Flu np366–374 is a Db-binding peptide and the major CTL epitope for A/PR/8/34 in H-2b mice. Flu np147–155 binds to Kd and is the immunodominant epitope in H-2d mice. As LCMV epitopes, we used LCMV glycoprotein (gp)33–43 (KAVYNFATCGI 11), LCMV np396–404 (FQPQNGAFI [11]), LCMV gp276–278 (SGVENPGGYCL [11]), and LCMV np118–126 (RPQASGVYM 12). LCMV gp33–43 is both Db and Kb restricted, whereas LCMV np396–404 is Db restricted. These are the two immunodominant epitopes in H-2b mice. LCMV gp276–278 also binds to Db but is a subdominant epitope in H-2b mice. LCMV np118–126 is an Ld-binding peptide and the immunodominant epitope in H-2d mice.
CTL Assays.
Effector cells consisted of freshly collected spleen cells (LCMV) or spleen cells that had been restimulated in vitro for 5 d (Flu). For restimulation, 5 × 107 spleen cells were incubated in upright T25 tissue culture flasks (Falcon) with 1.5 × 106 EL4 or P815 cells that had been incubated with 1 μg/ml of the appropriate Flu peptide and 50 μg/ml mitomycin C (Sigma-Aldrich) for 1 h followed by extensive washing. CTL activity was measured using 51Cr-release assays. In these assays, effector cells were cultured with 6,000 Na51Cr-labeled target cells for 5 h at the indicated E/T ratios in 200 μl complete RPMI. The percentage of killing was calculated using the formula: (experimental release − spontaneous release)/(full release − spontaneous release) × 100, in which spontaneous release was the count obtained when targets cells were cultured in media in the absence of effector cells, and full release when target cells were lysed with 1% Triton X-100. Radioactivity was measured using a micro-beta tri-lux system (Wallak).
Results
Role of BM-derived pAPCs in the Response to LCMV
Mice with TAP-deficient BM-derived Cells Generate CTL Responses to LCMV.
To further investigate the role of BM-derived pAPCs in the generation of immune responses to viruses, we analyzed the presentation requirements to generate CTLs to LCMV. We chose this virus because in the mouse it induces extremely strong CTL responses. 8 d after infection, at the peak of the CTL response, ∼50–70% of the splenic CD8+ T cells are virus specific in B6 animals 13. Moreover, in contrast to viruses such as vaccinia and vesicular stomatitis virus, CTL responses to LCMV do not require B7-CD28 costimulation (data not shown, and reference 14). As B7 molecules are exclusively expressed on BM-derived pAPCs, this latter finding raised the possibility that other kinds of APCs might be capable of stimulating anti-LCMV responses.
To analyze the role of BM-derived pAPCs with a functional TAP in the response to viruses in vivo, we made TAP0/0→B6 chimeras (BM donor→BM recipient). TAP0/0 cells synthesize class I heavy and light chains normally, but express extremely low levels of cell surface MHC class I (both Db and Kb) because there are no peptides shuttled from the cytosol to the ER. When BM from TAP0/0 mice was transplanted into irradiated wild-type animals, the ability of the TAP0/0-derived cells to present antigen on MHC class I through the traditional pathway remained impaired, as evidenced by the marked reduction of cell surface MHC class I in spleen cells 5 15. However, CD8+ T cells developed normally and their numbers in the spleen of TAP0/0 →B6 chimeras were similar to those in control B6→B6 mice, because the TAP0/0 CD8+ T cells developed in the recipient's thymus where the radioresistant thymic epithelial cells expressed normal levels of MHC class I 5 15. Moreover, we have shown previously that the CD8+ T cells in the TAP0/0→B6 chimeras respond to immunization with wild-type dendritic cells pulsed with limiting amounts of peptide at least as well as in B6→B6 mice; therefore, in TAP0/0→B6 chimeras there is no defect in the repertoire of CD8+ T cells or in their responsiveness 5.
To analyze the role of BM-derived cells with a functional TAP in the anti-LCMV CTL response, TAP0/0→B6 and B6→B6 mice were infected with LCMV, and CTL activity against two dominant epitopes (LCMV np396–404 and LCMV gp33–43) and one subdominant epitope (LCMV gp276–286) was determined using peptide-pulsed EL4 cells as targets. The results were surprising and in sharp contrast with the results we reported previously for vaccinia virus. As seen in Fig. 1, freshly explanted spleen cells from TAP0/0→B6 or B6→B6 chimeras that had been infected 8 d before effectively killed targets that had been incubated with either LCMV np396–404 or LCMV gp33–43. To a lesser degree, these spleen cells also killed targets that had been pulsed with the subdominant epitope LCMV gp276–278. Although the response by TAP0/0→B6 mice was in general somewhat lower than in controls, they still generated strong CTL responses to LCMV. Similar results were obtained when the CD8+ T cell responses were measured by IFN-γ intracellular staining of in vitro–restimulated spleen cells (not shown).
Figure 1.

TAP0/0→B6 chimeras infected with LCMV generate CTL responses to dominant and subdominant LCMV epitopes. Chimeras (three/group) of the type TAP0/0→B6 (left panels) and B6→B6 (right panels) were infected with 4 × 103 PFU LCMV. 8 d later, mice were killed and their spleen cells were used in 51Cr-release assays against the targets indicated (right). Each line represents a single mouse. For each group and target, identical symbols indicate the same mouse.
Several hypotheses can explain these unexpected results: (a) the CTL response to LCMV did not require MHC class I presentation by BM-derived pAPCs; (b) the CTL response to LCMV was dependent on BM-derived pAPCs, but because LCMV is so efficient in inducing CTLs, the few residual pAPCs of host origin were enough to drive the CTL response; and (c) the CTL response to LCMV was dependent on BM-derived pAPCs but the LCMV peptides were presented in a TAP-independent manner. We designed experiments to address each of these hypotheses.
Double-irradiated TAP→B6 Chimeras Still Generate Responses to LCMV.
We attempted to test whether residual host (wild-type) BM-derived pAPCs were responsible for the anti-LCMV CTL response in the TAP→B6 chimeras by using more rigorous procedures to eliminate these cells.
It has been reported that a second irradiation and BM transplant into chimeras decreases the number of host- and BM-derived cells to almost undetectable levels. Similar results can be achieved by supralethally irradiating recipient mice (i.e., exposure to a single dose of 1,300 rads, instead of the usual split dosages we normally used of 500 + 600 rads 16 17 18). We made double-irradiated chimeras with TAP0/0→B6 recipients that were aged for 4 mo (to allow turnover of pAPCs), reirradiated (500 + 600 rads), and reconstituted again with BM similar to that of the original donor. Supralethally irradiated chimeras were made with B6D2 heterozygous recipients (homozygous mice do not survive these high levels of irradiation). Fig. 2 shows that TAP0/0→B6 double-irradiated chimeras infected with LCMV generated strong CTL responses to LCMV np396–404 and LCMV gp33–43 that were somewhat smaller than those of B6→B6 mice. Also, both groups of mice developed weak but detectable CTL responses to the subdominant epitope LCMV gp276–286. Similar results were obtained when supralethally irradiated chimeras were used (not shown). These experiments indicate that further depletion of host origin pAPCs does not inhibit the generation of CTLs in TAP0/0→B6 chimeric mice. However, these experiments cannot completely rule out that a few residual host- and BM-derived cells were driving the CD8+ T cell responses in these mice.
Figure 2.

Double irradiation does not abrogate the ability of TAP0/0 →B6 chimeras to generate CTL responses to LCMV epitopes. Double-irradiated chimeras (three/group) of the type TAP0/0→B6 (left panels) and B6→B6 (right panels) were infected with 4 × 103 PFU LCMV. 8 d later, mice were killed and their spleen cells were used in 51Cr-release assays against the targets indicated (right). Each line represents a single mouse. For each group and target, identical symbols indicate the same mouse.
BM-derived APCs Are Required in the CTL Response to an LCMV Epitope.
To test the hypothesis that the anti-LCMV CTL response in TAP0/0→B6 chimeric mice was due to TAP-independent antigen presentation, we generated chimeric mice with BM whose MHC molecules were unable to present the LCMV epitopes.
Specifically, we constructed parent→F1 chimeras of the type B6→B6D2, D2→B6D2, and control B6D2→B6D2. If BM-derived pAPCs were required for the generation of CTL responses, B6→B6D2 mice should only be responsive to H-2b–restricted epitopes, D2→B6D2 should only be responsive to H-2d–restricted epitopes, and B6D2→B6D2 mice should be responsive to both H-2b– and H-2d–restricted epitopes. We infected the parent→F1 chimeras with LCMV, and 8 d later the animals were killed and their spleen cells used in CTL assays against epitopes restricted to H-2b or H-2d. The results in Fig. 3 show that D2→B6D2 mice, which do not express Db on BM-derived cells of donor origin, failed to mount a CTL response to LCMV np396–404. This was despite Db expression in non–BM-derived cells. On the other hand, B6→B6D2 and B6D2→B6D2 controls (which express Db on both BM-derived and non–BM-derived cells) generated strong CTL responses to LCMV np396–404. The inability of D2→B6D2 mice to generate CTLs to LCMV np396–404 was not due to a generalized CD8+ T cell deficiency because they responded strongly to the Ld-restricted epitope LCMV np118–126. These results demonstrate that BM-derived pAPCs are required in the CTL response to LCMV np396–404. Importantly, these results also show that single-irradiated chimeras lack host (radioresistant) pAPCs that can support the response to LCMV np396–404. Therefore, the generation of CTLs to LCMV np396–404 in single, double, and supralethally irradiated TAP0/0→B6 chimeras as described above was not due to residual host- and BM-derived cells but to a TAP-independent mechanism of antigen presentation by donor-derived APCs.
Figure 3.
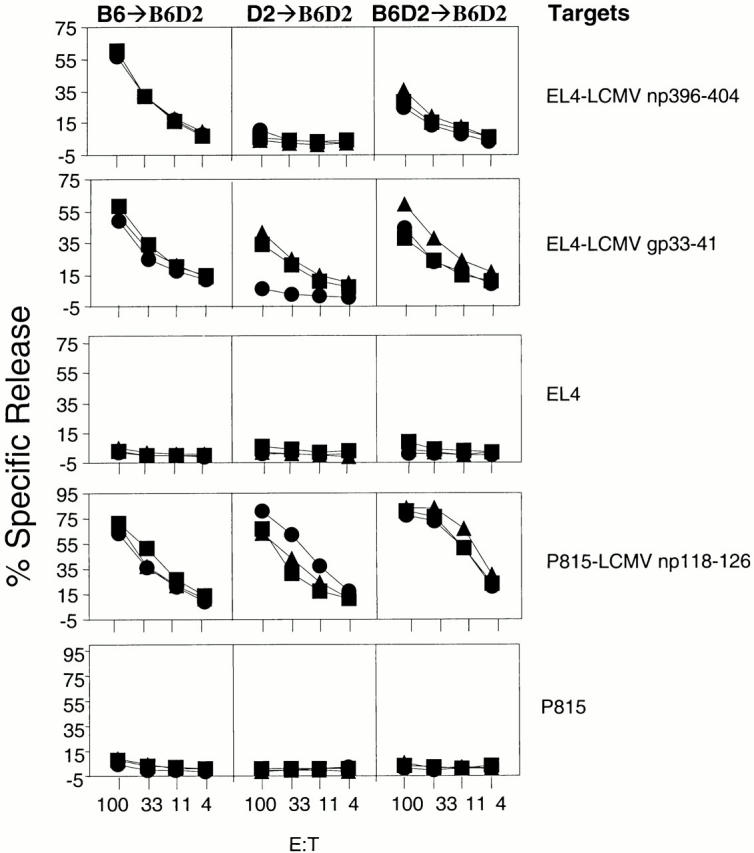
The CTL response to LCMV np396–404 is dependent on antigen presentation by BM-derived cells. Chimeras (three/group) of the type B6→B6D2 (left panels), D2→B6D2 (middle panels), and B6D2→B6 D2 (right panels) were infected with 4 × 103 PFU LCMV. 8 d later, mice were killed and their spleen cells were used in 51Cr-release assays against the targets indicated (right). Each line represents a single mouse. For each group and target, identical symbols indicate the same mouse.
Surprisingly, although the results for LCMV np396–404 were clear-cut, those for LCMV gp33–43 and LCMV np118–126 were not. We found that at the relatively high doses of LCMV we normally used (i.e., 4 × 103 PFU), D2→B6D2 not only generated responses to the Ld-restricted LCMV np118–126, but also to the Db-restricted LCMV gp33–43, albeit consistently lower than those generated by B6→B6D2 mice. Also, B6→B6D2 mice generated CTLs to the Ld-restricted LCMV np118–126 in single and double-irradiated chimeras (not shown). This could indicate that at high viral doses, residual pAPCs of host origin are initiating the response to these two epitopes, or that for these epitopes, nonhematopoietic cells are functioning as pAPCs. We favor the first hypothesis because in additional experiments with 200 PFU LCMV, the response to LCMV gp33–43 was abrogated in D2→B6D2 but not in B6→B6D2 chimeras (Fig. 4). In addition, experiments with supralethally irradiated mice seem to indicate that residual host pAPCs are responsible for the CTL responses to the high viral dose of LCMV. However, the health of the mice was not optimal at the time of these experiments, and we were unable to repeat the experiments a sufficient number of times to draw definitive conclusions on this point.
Figure 4.

At a low viral dose, the CTL response to LCMV gp33–41 is dependent on antigen presentation by BM-derived cells. Chimeras (three/group) of the type B6→B6D2 (left panels), D2→B6D2 (middle panels), and B6D2→B6 D2 (right panels) were infected with 200 PFU LCMV. 8 d later, mice were killed and their spleen cells were used in 51Cr-release assays against the targets indicated (right). Each line represents a single mouse. For each group and target, identical symbols indicate the same mouse.
Role of BM-derived pAPCs in the Response to Flu
The CTL Response to Flu Is Dependent on BM-derived pAPCs.
To determine the role of BM-derived pAPCs in the generation of CTLs to yet another virus, we decided to examine Flu for which the CTL response has been well characterized in the mouse 1. We inoculated B6→B6D2, D→B6D2, and B6D2→B6D2 mice with 73 HAU Flu. The results in the top panels of Fig. 5 clearly show that D2→B6D2 mice, which did not express the Db-restricting element on the BM-derived cells, did not generate CTLs to the Db-restricted epitope Flu np366–374, despite expression of Db on the recipient's nonhematopoietic cells. The lack of response was not due to the inability of D2→B6D2 mice to generate CTLs because they responded strongly to the Kd-restricted epitope Flu np147–155 (Fig. 5, bottom row, center panel). This clearly demonstrated the fundamental importance of BM-derived pAPCs in the generation of CTLs to Flu np366–374. The same groups of mice that were analyzed for the response to Flu np366–374 were also analyzed for the response to the Kd-restricted Flu np147–155 (Fig. 5, bottom panels). B6D2→B6D2 and D2→B6D2 mice generated strong responses to this epitope. This was expected because they expressed Kd in the vast majority of BM-derived cells, which were of donor origin. However, B6→B6D2 mice also generated CTL responses to Flu np147–155 despite the lack of expression of Kd in most BM-derived cells. To investigate whether this response was due to host- and BM-derived pAPCs that resisted irradiation, we generated supralethally irradiated chimeras (1,300 rads) of the types B6→B6D2 and B6D2→B6D2. The results in Fig. 6 show that supralethally irradiated B6→B6D2 mice did not respond to Flu np147–155 whereas B6D2→B6D2 mice did. This was not due to the inability of supralethally irradiated B6→B6D2 mice to mount CTL responses because they generated detectable responses to the Db-restricted Flu np366–374. These results demonstrate that BM-derived cells are required for the generation of CTLs to multiple epitopes of the Flu virus.
Figure 5.

The CTL response to Flu np366–374 is dependent on antigen presentation by BM-derived antigen-presenting cells. Chimeras (three/group) of the type B6→B6D2 (left panels), D2→B6D2 (middle panels), and B6D2→B6D2 (right panels) were infected with 73 HAU Flu A/PR8/34. CTL assays were performed with spleen cells that had been restimulated in vitro as described in Materials and Methods. Targets are as indicated. Each line corresponds to an individual mouse. Similar figures within a group of chimeras correspond to the same mouse (e.g., all squares in the B6→B6D2 group correspond to the same mouse recognizing different targets).
Figure 6.
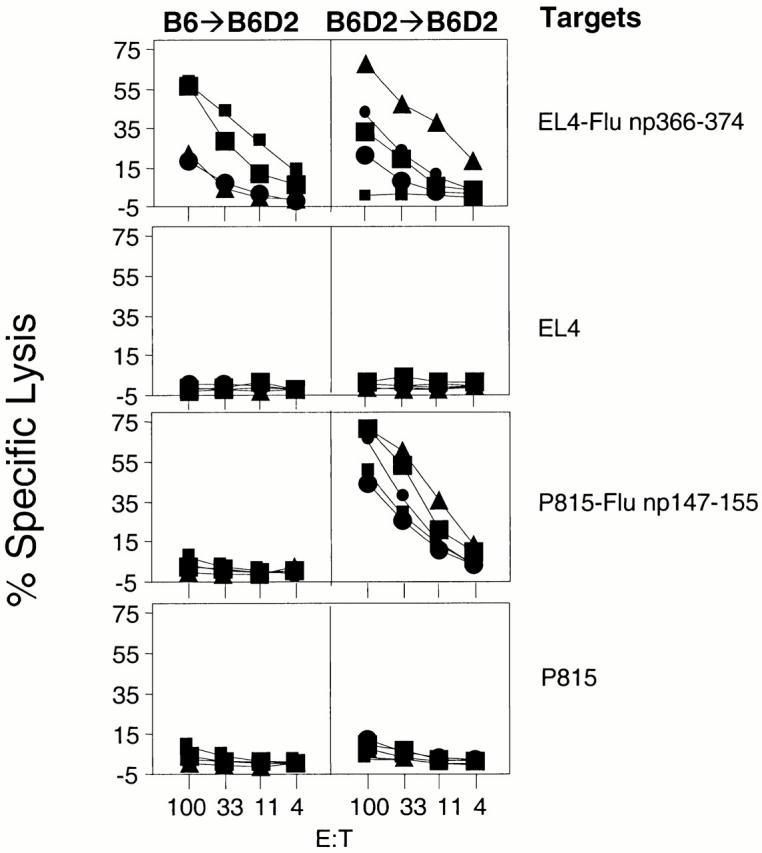
The CTL response to Flu np147–155 in B6→B6D2 is abrogated by supralethally irradiating recipient mice. Supralethally irradiated chimeras of the type B6→B6D2 (left panels; four animals) and B6D2→B6D2 (right panels; five animals) were infected with 73 HAU Flu A/PR8/34. CTL assays were performed with spleen cells that had been restimulated in vitro as described in Materials and Methods. Targets are as indicated. Each line corresponds to an individual mouse. Similar figures within a group of chimeras correspond to the same mouse (e.g., all squares in the B6→B6D2 group correspond to the same mouse recognizing different targets).
High Doses of Flu Generate CTLs That Are Dependent in a BM-derived APC but TAP Independent.
The results with LCMV np396–404 indicated the existence of a TAP-independent antigen presentation pathway that was operative in vivo. However, we had previously not found this pathway during vaccinia and poliovirus infection. Because LCMV is a mouse pathogen and replicates very well in the mouse, and vaccinia and poliovirus replicate in the adult mouse much less extensively, we hypothesized that the ability to generate CTLs in a TAP-independent fashion was at least partly dependent on a high antigenic load. To look at the relationship of antigenic load and the use of different pathways of MHC class I antigen presentation during viral infections in vivo, we investigated the role of TAP on BM-derived pAPCs in the CTL response to graded doses of Flu. This virus infects the mouse experimentally, but its ability to replicate is much more restricted than LCMV's. We inoculated TAP0/0→B6 chimeras or B6→B6 controls with either 2.4 (data not shown), 24, or 73 HAU Flu (Fig. 7). Mice were killed 15–20 d later and their spleen cells were restimulated in vitro for 5 d with mitomycin C–treated EL4 cells that had been incubated with Flu np366–374. Whereas control B6→B6 chimeras generated strong responses to high and low viral doses, TAP0/0→B6 chimeras failed to generate a CTL response to the Flu epitope at the lower doses. Conversely, TAP0/0→B6 chimeras still generated CTL responses to Flu when the viral dose was increased to 30 μl, albeit strongly diminished compared with their B6→B6 counterparts. The inability of TAP0/0→B6 mice to generate responses at the lower dose and diminished responses at the higher dose could not be attributed to an intrinsic defect of the CD8+ T cell repertoire of these mice because we and others have demonstrated previously that TAP0/0→B6 mice generate CTL responses at least as strong as B6→B6 mice when immunized with a virus construct that bypasses TAP 5 15 and when the number of antigen-bearing APCs or the concentration of peptide at the APC surface was a limiting factor 5. Therefore, the results with Flu confirm those with LCMV above indicating that in vivo, BM-derived pAPCs can present antigen independent of TAP function. However, this pathway required higher antigenic load than the classical TAP-dependent pathway.
Figure 7.

TAP0/0→B6 chimeras generate specific CTL responses to high but not low doses of Flu. Chimeras of the type TAP0/0→B6 (left panels) and B6→B6 (right panels) were infected with 24.3 (top panels; three mice per group) or 73 (bottom panels; five mice per group) HAU Flu A/PR8/34. CTL assays were performed with spleen cells that had been restimulated in vitro as described in Materials and Methods. Targets were Flu np366–374 pulsed EL4 cells (filled symbols) and EL4 targets (open symbols).
Discussion
In a previous report, we demonstrated that BM-derived pAPCs are required for the initiation of antivaccinia and antipolio CTL responses. In this report, we extend these findings to demonstrate that BM-derived pAPCs are also required in the CTL response to Flu and at least for two LCMV epitopes (LCMV np396–404 and low doses of LCMV gp33–43). Therefore, our results, together with the demonstration by other investigators that CTL responses to other antigens such as tumors, DNA vaccines, and bacteria are also dependent on BM-derived pAPCs 19 20 21 22 23 24 25 26, indicate a general requirement for BM-derived pAPCs in the initiation of most, if not all primary CTL responses.
In the case of LCMV np118–126, we have been unable to demonstrate a dependence on BM -derived APCs. Lenz et al. 26 in this issue had similar results with this epitope. From our combined data, it is possible to conclude that nonhematopoietic cells can initiate CTL responses to LCMV np118–126. However, it is more likely that a few residual host pAPCs (probably <1%) may be sufficient to stimulate these CTL responses. LCMV replicates extremely well in dendritic cells so that the few residual cells of host origin may become infected and efficiently stimulate CTL responses. Indeed, this is what we observe for CTL responses to Flu np147–155 in “single” versus supralethally irradiated chimeras. Therefore, it is likely that with high doses of LCMV, the few residual cells are still capable of initiating almost wild-type responses to LCMV gp33–43 and LCMV np118–126, but not to LCMV np396–404. The response to LCMV gp33–43 can be abrogated when the viral dose is reduced, but the CTL response to LCMV np118–126 remains. This difference between LCMV epitopes could be dependent on the efficiency of antigen processing, peptide affinity for MHC class I, and/or CD8+ T cell precursor frequency. Earlier studies with “parent→F1” chimeras 27 also support the concept that BM-derived pAPCs are required for the generation of anti-LCMV CTLs, even to the Ld-restricted epitope. Why the results of these authors differ from ours in the response to LCMV np118–126 is unclear. However, the disparity might be attributed to different strains of LCMV and/or different combinations of donor/recipient mouse strains, or it may be that they measured the responses using LCMV-infected targets rather than cells pulsed with the individual CTL epitopes. Nonetheless, their results strongly support our view that BM-derived pAPCs have a fundamental role in CTL responses.
In this report we also demonstrate anti-Flu and anti-LCMV CTL responses that are dependent on BM-derived pAPCs but independent of TAP function. As CTL responses are not generated to LCMV np396–404 and Flu np366–374 in chimeras reconstituted with H-2d BM, our irradiation protocol has been sufficient to eliminate the host pAPCs. Furthermore, we have found that peritoneal exudate macrophages obtained from TAP0/0→B6 mice are incapable of presenting a TAP-dependent epitope when infected with a recombinant vaccinia virus (not shown). Therefore, the generation of CTLs in TAP0/0→wild-type chimeras is likely to be due to TAP-independent presentation by BM-derived pAPCs rather than by radioresistant host (wild-type) pAPCs. Because LCMV np396–404 and Flu np366–374 are derived from proteins that do not translocate into the ER, these results are surprising and represent the first demonstration in vivo of TAP-independent presentation of such epitopes that we are aware of.
There are two possible explanations for the TAP-independent presentation of the LCMV and Flu epitopes. One is that these peptides are transported by an alternate mechanism from the cytosol into the ER of infected BM-derived pAPCs. Although such a mechanism has been suggested for an ER membrane protein 28, this seems to be exceptional, and in similar in vitro experiments TAP-independent presentation has not been observed for the LCMV and Flu epitopes 3 29. The other explanation is that these peptides do not require transport into the ER because they are binding to class I molecules in another location. In some cases, macrophages and dendritic cells can generate class I–presented peptides from exogenous antigens in endocytic compartments 8 9 30. Our data do not distinguish between these mechanisms. However, because there is abundant data that exogenous antigens do stimulate CTL responses in vivo 6 and can be the major source of antigen for generating CTL responses to a viral infection 5, we favor this second interpretation.
The TAP-independent mechanism of CTL generation appears to be substantially less efficient than the TAP-dependent route. Between 10–300-fold more Flu is needed to generate CTL responses in chimeras with TAP-deficient BM compared with controls. Moreover, for other viruses such as polio and vaccinia virus we fail to detect any TAP-independent response, including situations in which the CTL response is stimulated through the exogenous pathway of class I antigen presentation. This may be because the viral load is too low to stimulate CTLs through this pathway or because the antigenic epitopes fail to generate properly in the endocytic compartment. Although the TAP-independent pathway of antigen presentation is less efficient, it is likely to contribute to the generation of CTLs at least in some infections with high viral loads.
Acknowledgments
We are very grateful to Dr. Raymond Welsh for LCMV stocks and advice.
This work was supported by grants from the National Institutes of Health to K.L. Rock.
Footnotes
Abbreviations used in this paper: B6, C57BL/6; B6D2, (C57BL/6 × DBA2)F1; BM, bone marrow; D2, DBA2; ER, endoplasmic reticulum; Flu, influenza virus; gp, glycoprotein; HAU, hemagglutination unit(s); LCMV, lymphocytic choriomeningitis virus; np, nucleoprotein; pAPC, professional APC; TAP, transporter associated with antigen presentation; TAP0/0, B6-Tap1tp 1Arp.
References
- Townsend A., Bodmer H. Antigen recognition by class I-restricted T lymphocytes. Annu. Rev. Immunol. 1989;7:601–624. doi: 10.1146/annurev.iy.07.040189.003125. [DOI] [PubMed] [Google Scholar]
- Spies T., DeMars R. Restored expression of major histocompatibility class I molecules by gene transfer of a putative peptide transporter. Nature. 1991;351:323–324. doi: 10.1038/351323a0. [DOI] [PubMed] [Google Scholar]
- Powis S.J., Townsend A.R., Deverson E.V., Bastin J., Butcher G.W., Howard J.C. Restoration of antigen presentation to the mutant cell line RMA-S by an MHC-linked transporter. Nature. 1991;354:528–531. doi: 10.1038/354528a0. [DOI] [PubMed] [Google Scholar]
- York I.A., Rock K.L. Antigen processing and presentation by the class I major histocompatibility complex. Annu. Rev. Immunol. 1996;14:369–396. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]
- Sigal L.J., Crotty S., Andino R., Rock K.L. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398:77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- Rock K.L. A new foreign policyMHC class I molecules monitor the outside world. Immunol. Today. 1996;17:131–137. doi: 10.1016/0167-5699(96)80605-0. [DOI] [PubMed] [Google Scholar]
- Kovacsovics-Bankowski M., Rock K.L. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267:243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- Harding C.V., Song R. Phagocytic processing of exogenous particulate antigens by macrophages for presentation by class I MHC molecules. J. Immunol. 1994;153:4925–4933. [PubMed] [Google Scholar]
- Pfeifer J.D., Wick M.J., Roberts R.L., Findlay K., Normark S.J., Harding C.V. Phagocytic processing of bacterial antigens for class I MHC presentation to T cells. Nature. 1993;361:359–362. doi: 10.1038/361359a0. [DOI] [PubMed] [Google Scholar]
- Rotzschke O., Falk K., Deres K., Schild H., Norda M., Metzger J., Jung G., Rammensee H.G. Isolation and analysis of naturally processed viral peptides as recognized by cytotoxic T cells. Nature. 1990;348:252–254. doi: 10.1038/348252a0. [DOI] [PubMed] [Google Scholar]
- Gairin J.E., Mazarguil H., Hudrisier D., Oldstone M.B. Optimal lymphocytic choriomeningitis virus sequences restricted by H-2Db major histocompatibility complex class I molecules and presented to cytotoxic T lymphocytes. J. Virol. 1995;69:2297–2305. doi: 10.1128/jvi.69.4.2297-2305.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitton J.L., Tishon A., Lewicki H., Gebhard J., Cook T., Salvato M., Joly E., Oldstone M.B. Molecular analyses of a five-amino-acid cytotoxic T-lymphocyte (CTL) epitopean immunodominant region which induces nonreciprocal CTL cross-reactivity. J. Virol. 1989;63:4303–4310. doi: 10.1128/jvi.63.10.4303-4310.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murali-Krishna K., Altman J.D., Suresh M., Sourdive D.J., Zajac A.J., Miller J.D., Slansky J., Ahmed R. Counting antigen-specific CD8 T cellsa reevaluation of bystander activation during viral infection. Immunity. 1998;8:177–187. doi: 10.1016/s1074-7613(00)80470-7. [DOI] [PubMed] [Google Scholar]
- Kundig T.M., Shahinian A., Kawai K., Mittrucker H.W., Sebzda E., Bachmann M.F., Mak T.W., Ohashi P.S. Duration of TCR stimulation determines costimulatory requirement of T cells. Immunity. 1996;5:41–52. doi: 10.1016/s1074-7613(00)80308-8. [DOI] [PubMed] [Google Scholar]
- Huang A.Y., Bruce A.T., Pardoll D.M., Levitsky H.I. In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity. 1996;4:349–355. doi: 10.1016/s1074-7613(00)80248-4. [DOI] [PubMed] [Google Scholar]
- Kosaka H., Sprent J. Tolerance of CD8+ T cells developing in parent→F1 chimeras prepared with supralethal irradiationstep-wise induction of tolerance in the intrathymic and extrathymic environments. J. Exp. Med. 1993;177:367–378. doi: 10.1084/jem.177.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao E.K., Lo D., Sprent J. Strong T cell tolerance in parent→F1 bone marrow chimeras prepared with supralethal irradiation. Evidence for clonal deletion and anergy. J. Exp. Med. 1990;171:1101–1121. doi: 10.1084/jem.171.4.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron Y., Lo D., Sprent J. T cell specificity in twice-irradiated F1→parent bone marrow chimerasfailure to detect a role for immigrant marrow-derived cells in imprinting intrathymic H-2 restriction. J. Immunol. 1986;137:1764–1771. [PubMed] [Google Scholar]
- Huang A.Y., Golumbek P., Ahmadzadeh M., Jaffee E., Pardoll D., Levitsky H. Role of bone marrow-derived cells in presenting MHC class I-restricted tumor antigens. Science. 1994;264:961–965. doi: 10.1126/science.7513904. [DOI] [PubMed] [Google Scholar]
- Iwasaki A., Torres C.A., Ohashi P.S., Robinson H.L., Barber B.H. The dominant role of bone marrow-derived cells in CTL induction following plasmid DNA immunization at different sites. J. Immunol. 1997;159:11–14. [PubMed] [Google Scholar]
- Takashima A., Morita A. Dendritic cells in genetic immunization. J. Leukoc. Biol. 1999;66:350–356. doi: 10.1002/jlb.66.2.350. [DOI] [PubMed] [Google Scholar]
- Akbari O., Panjwani N., Garcia S., Tascon R., Lowrie D., Stockinger B. DNA vaccinationtransfection and activation of dendritic cells as key events for immunity. J. Exp. Med. 1999;189:169–178. doi: 10.1084/jem.189.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porgador A., Irvine K.R., Iwasaki A., Barber B.H., Restifo N.P., Germain R.N. Predominant role for directly transfected dendritic cells in antigen presentation to CD8+ T cells after gene gun immunization. J. Exp. Med. 1998;188:1075–1082. doi: 10.1084/jem.188.6.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattergoon M.A., Robinson T.M., Boyer J.D., Weiner D.B. Specific immune induction following DNA-based immunization through in vivo transfection and activation of macrophages/antigen-presenting cells. J. Immunol. 1998;160:5707–5718. [PubMed] [Google Scholar]
- Condon C., Watkins S.C., Celluzzi C.M., Thompson K., Falo L.D., Jr. DNA-based immunization by in vivo transfection of dendritic cells. Nat. Med. 1996;2:1122–1128. doi: 10.1038/nm1096-1122. [DOI] [PubMed] [Google Scholar]
- Lenz L.L., Butz E.A., Bevan M.J. Requirements for bone marrow–derived antigen presenting cells in priming cytotoxic T cell responses to intracellular pathogens. J. Exp. Med. 2000;192:1135–1142. doi: 10.1084/jem.192.8.1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkernagel R.M., Kreeb G., Althage A. Lymphohemopoietic origin of the immunogenic, virus-antigen-presenting cells triggering anti-viral T-cell responses. Clin. Immunol. Immunopathol. 1980;15:565–576. doi: 10.1016/0090-1229(80)90067-7. [DOI] [PubMed] [Google Scholar]
- Snyder H.L., Bacik I., Bennink J.R., Kearns G., Behrens T.W., Bachi T., Orlowski M., Yewdell J.W. Two novel routes of transporter associated with antigen processing (TAP)-independent major histocompatibility complex class I antigen processing. J. Exp. Med. 1997;186:1087–1098. doi: 10.1084/jem.186.7.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hombach J., Pircher H., Tonegawa S., Zinkernagel R.M. Strictly transporter of antigen presentation (TAP)-dependent presentation of an immunodominant cytotoxic T lymphocyte epitope in the signal sequence of a virus protein. J. Exp. Med. 1995;182:1615–1619. doi: 10.1084/jem.182.5.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R., Harding C.V. Roles of proteasomes, transporter for antigen presentation (TAP), and beta 2-microglobulin in the processing of bacterial or particulate antigens via an alternate class I MHC processing pathway. J. Immunol. 1996;156:4182–4190. [PubMed] [Google Scholar]