

Apoptotic cell suicide can be initiated by a plethora of stimuli that generally feed into one of two known cell death signaling pathways (Fig. 1; for review see references 1 and 2). Both pathways share many features, including molecular devices that spark caspase activation by proenzyme recruitment, oligomerization, and proximity-induced autocatalytic activation. Beyond this, however, their activities are relatively (but not absolutely) distinct. The ‘intrinsic’ pathway feeds cell death signals through the mitochondrion, which appears to act as a generic damage sensor and monitor of metabolic status. With the assistance of cytochrome c, cell death is initiated by the formation of a macromolecular complex (the apoptosome), which utilizes apoptotic protease activating factor (APAF)-1 to mediate the activation of caspase-9. The ‘extrinsic’ pathway transduces the signals of extracellular ‘death ligands’ belonging to the TNF superfamily (e.g., TNF-α, Fas ligand [FasL]/Apo1L/CD95L, Trail/Apo2L, Apo3L). The binding of these ligands to preassembled receptor complexes 3 4 triggers the activation of caspase-8 through the adapter molecule Fas-associated death domain (FADD)/Mort1 (plus others in some cases). Once the activation of initiator caspases occurs by either of these two routes, the pathways converge on the effector caspases (caspase-3 and caspase-7), which are the proteolytic engines of cell death. Each of the two pathways is modulated, as might be expected, by regulatory polypeptides such as FLICE (FADD-like IL-1ß–converting enzyme) inhibitory protein (cFLIP)/usurpin (‘extrinsic’ pathway) or Bcl-2 family members (‘intrinsic’ pathway), and these molecules have been used to examine the relationship between the two pathways in vitro, and more recently in vivo.
Figure 1.

Extrinsic and intrinsic cell death signaling pathways. The majority of proteolytic cleavage events that manifest the apoptotic phenotype are mediated by ‘effector’ caspases, such as caspase-3 and caspase-7, which become fully activated when the large and small subunits that are harbored within the dormant proenzyme are liberated after endoproteolysis by upstream ‘initiator’ caspases, such as caspase-8 and caspase-9. The initiator caspases themselves are activated by autoproteolytic activation after facilitated oligomerization. In the extrinsic pathway, this occurs as a consequence of ligand binding to ‘death receptor’ complexes, which leads to the recruitment of procaspase-8 via the adapter molecule FADD/Mort1. This pathway is modulated by the availability of the molecular components (putative type I and type II cells differ in this regard) and dominant-negative regulators such as decoy receptors and cFLIP/usurpin. For the majority of cell death stimuli, the intrinsic death signal is communicated through the mitochondrion by an unknown mechanism, which leads to several changes in the organelle, including the release of polypeptidic agents dangereux, such as cytochrome c and second mitochondria-derived activator of caspases (SMAC)/Diablo. This pathway is highly dependent on the stoichiometry of anti- versus proapoptotic Bcl-2 family members. When enabled, caspase-9 activation occurs at the hands of the oligomerization mediator APAF-1, which requires cytochrome c for the appropriate conformation. SMAC/Diablo helps to cross another apoptosis checkpoint by sequestering inhibitors of apoptosis protein (IAPs), which would otherwise block the actions of downstream effector caspases even in the presence of proteolytic maturation. Controversy surrounds the degree to which cross-talk occurs between the extrinsic and intrinsic pathways in vivo. At a molecular level, this appears to occur via the proteolysis of BID, which normally serves an antiapoptotic role within the intrinsic pathway until it is truncated (to tBID) by caspase-8 (derived from the extrinsic pathway), whereupon it promotes activation of the intrinsic pathway. In principle, signals from the extrinsic pathway may require the assistance of the intrinsic pathway, for example, when the signal strength is weak or when the IAP barrier is high and the actions of SMAC/Diablo become necessary.
With the identification of these two distinct apoptotic pathways, controversy raged as reports demonstrating that Bcl-2 did not inhibit CD95-mediated apoptosis were matched by an equal number of reports demonstrating that it did (CD95, a.k.a. Fas/Apo1, being the archetypal extrinsic death receptor). Then, in 1998, the group led by Peter Krammer and Marcus Peter (Scaffidi et al., reference 5) reported the existence of two distinct cell types that utilize distinct CD95 signaling pathways: type I cells undergo CD95-mediated apoptosis without the involvement of mitochondria, whereas type II cells require release of cytochrome c from mitochondria in order for CD95 to exert its apoptotic effects. At the molecular level, these two cell types differ principally in the amount of caspase-8 recruited to CD95 via the adapter molecule FADD/Mort1 to form the death-inducing signaling complex (DISC). Whereas type I cells contain large amounts of DISC in response to anti-CD95 antibodies, type II cells do not and thus are dependent on stimulation of the intrinsic apoptotic pathway to undergo cell death. These studies thus could explain why the extrinsic apoptotic pathway was insensitive to Bcl-2 overexpression in some cells (type I) but sensitive to Bcl-2 overexpression in other cells (type II) 6. The identification of Bid as the ‘go-between,’ transmitting signals from the extrinsic to the intrinsic pathway, provided a molecular basis for the cross-talk between the two pathways 7 8. Moreover, studies in vivo revealed that hepatocytes had all of the characteristics of the Krammer/Peter (reference 5) type II cell. Agonistic antibodies to CD95 cause massive liver apoptosis and lethality in mice due, at least in part, to liver failure. Overexpression of Bcl-2 in hepatocytes protects mice from anti-CD95–mediated liver failure 9 10. In addition, mice nullizygous for Bid, the key protein involved in the cross-talk between the intrinsic and extrinsic pathways, are also resistant to the liver failure that ensues after injection of anti-CD95 antibodies 11. The absence of Bid, however, does not alter the sensitivity of other cell types to anti-CD95 antibodies (e.g., thymocytes and fibroblasts). Taken together, these studies support the existence of two distinct responders to CD95 stimulation, one dependent solely on the extrinsic pathway and one dependent on both the extrinsic and intrinsic pathways.
The idea that cross-talk exists between extrinsic and intrinsic apoptotic pathways in certain cell types is now widely accepted; after all, cross-talk is a common theme in the world of signal transduction. However, a small number of groups remain cautious in accepting this notion. Andreas Strasser and colleagues point out that many studies use agonistic antibodies to CD95 to stimulate the extrinsic pathway. In their hands, these agonistic antibodies do not behave as the physiological form of the CD95L that, they claim, is multimeric in nature (either membrane-associated CD95L or cross-linked CD95L). While they do not observe significant hepatocyte apoptosis in the presence of anti-CD95 antibodies, these cells are sensitive to the CD95L when provided in a multimeric form 12. Moreover, the apoptotic cell death observed with the multimeric ligand is not inhibited by overexpression of Bcl-2. In response to this criticism, Schmitz et al. (Krammer group) demonstrated that the differential sensitivity to Bcl-2 in type I and type II cells is maintained when the extrinsic pathway is stimulated by the CD95L rather than agonistic antibodies to CD95 13. It is important to note, however, that the CD95L used in this study was trimeric rather than multimeric and thus differs from that used in the studies by Strasser and colleagues 14.
Is the nature of the ligand solely responsible for the discrepant results obtained by the groups of Krammer/Peter and colleagues and Strasser and colleagues in hepatocytes and other type II cells? Moreover, and importantly, is the multimeric CD95L actually a more physiological form of the ligand? Would mice defective in Bid retain resistance to hepatocellular injury in response to a more physiological stimulus of the CD95 pathway?
The nature of the stimulus of the CD95 pathway does not explain the fact that Strasser and colleagues do not observe significant cell death when type II cells are treated with anti-CD95 antibodies or trimeric CD95L, whereas Krammer and colleagues and several other groups do. These discrepant results might be due to differences in the ‘tone’ of the intrinsic pathway (i.e., the sensitivity of the intrinsic pathway to apoptotic stimuli) and the tone of the intrinsic pathway might be influenced by the array of growth factors and cytokines to which the cells are exposed. Villunger et al. elegantly illustrate this concept in the September 4 issue of this journal 15. They demonstrate that the proinflammatory cytokine G-CSF stimulates the extracellular signal–regulated kinase (ERK) pathway and inhibits spontaneous apoptosis of granulocytes as well as apoptosis induced by stimuli of the intrinsic pathway (etoposide and doxorubicin) but not apoptosis induced by stimulation of the extrinsic pathway with multimerized CD95L (granulocytes are resistant to weaker stimuli of the extrinsic pathway such as agonistic antibodies to CD95 or trimerized CD95L). Similarly, G-CSF prevents the death of granulocytes stimulated by inhibitors of the mitogen-activated protein kinase (MAPK) pathway. Given that G-CSF does not directly modulate the p38 MAPK pathway, Villunger et al. suggest that both signal transduction pathways converge to regulate the expression and/or activity of molecules that interfere specifically with the intrinsic but not the extrinsic pathway. They propose that the p38 MAPK and ERK pathways inhibit the intrinsic apoptotic pathway by shifting the delicate balance between pro- and antiapoptotic Bcl-2 family members, stimulating either the overexpression of antiapoptotic Bcl-2 family members or the downregulation of proapoptotic Bcl-2 family members (although Western blot analyses failed to detect significant differences in the expression of either pro- or antiapoptotic Bcl-2 family members in response to G-CSF). This hypothesis is supported by the observation that overexpression of Bcl-2 acts like G-CSF to protect granulocytes from spontaneous apoptosis as well as apoptosis stimulated by doxorubicin, cisplatin, and p38 MAPK inhibitors but not from multimeric CD95.
Although the mechanism by which the complex interplay between the signal transduction pathways and the intrinsic apoptotic pathway remains ill defined, this interplay is no less important and might be responsible for the differences in sensitivity of cells to anti-CD95 antibodies observed by Strasser and colleagues and Krammer and colleagues, as illustrated in Fig. 2. Alterations in the tone of the intrinsic apoptotic pathway may affect the ability of cells to respond to suboptimal stimulation of the extrinsic pathway in type II cells using anti-CD95 antibodies or trimeric CD95L (type I cells, for reasons that remain obscure, appear to support productive formation of the DISC (CD95, FADD/MORT1, caspase-8), irrespective of the strength of the stimulus). Type II cells, exposed to cytokines or growth factors that activate the signal transduction pathways and increase the resistance of mitochondria to the cytochrome c–releasing effects caused by cleavage of Bid, might behave as reported by Huang et al. (Strasser group; reference 12). These cells survive a weak CD95 stimulus (anti-CD95 antibodies or trimeric CD95L) that is dependent on the intrinsic pathway but die in the presence of a strong CD95 stimulus (e.g., multimeric CD95L) that is not dependent on the intrinsic pathway. In contrast, cells maintained in an environment impoverished in cytokines/growth factors might behave as reported by Scaffidi et al. (Krammer group; reference 5). These cells exhibit a proapoptotic response to a weak CD95 stimulus that is inhibitable by Bcl-2 overexpression or Bid deficiency.
Figure 2.
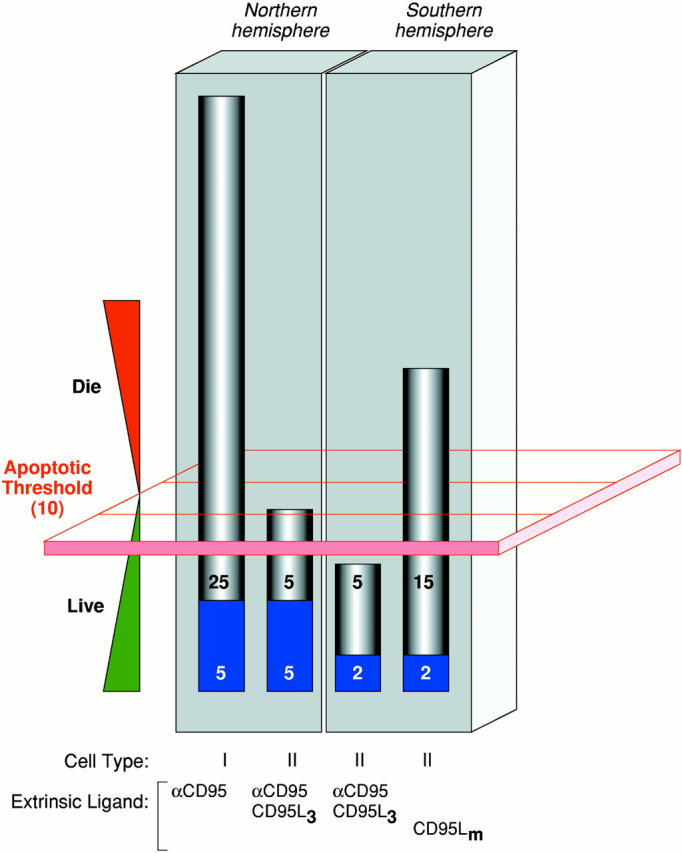
How apoptotic thresholding might influence experimental outcome. Many possible explanations may account for the differential sensitivity of cells from the ‘Northern hemisphere’ labs (e.g., Krammer/Peter) versus those from the ‘Southern hemisphere’ (e.g., Strasser) to CD95 stimulation (the Krammer/Peter labs are located in Germany and the Strasser lab is in Australia, hence the Northern and Southern hemisphere references.). Herein is illustrated one hypothesis. It assumes that most cell types have an apoptotic threshold that, when breached, results in apoptotic cell death. This apoptotic threshold (represented by the red plane) requires, say, 10 arbitrary apoptotic units (which could be, among other things, a critical mass of caspase catalytic activity). The ability of a cell to cross this threshold depends on the combined contributions of the extrinsic cell death pathway (gray) and the fraction of the intrinsic pathway (blue) that can be engaged through pathway cross-talk. Type I cells (first bar), in both the Northern and Southern hemispheres, harbor a very efficient DISC mechanism that alone can contribute the necessary apoptotic units to breach the threshold. Thus, regardless of the status of the intrinsic pathway, and even if it is completely blocked by the presence of Bcl-2, the type I cell can muster the necessary currency to engage the apoptotic machinery and die. Therefore, extrinsic and intrinsic pathways in type I cells appear independent (assuming intrinsic does not talk to extrinsic). In type II cells, DISC formation is weaker and the extrinsic pathway depends more on contributions from the intrinsic pathway. These cells behave differently in the Northern hemisphere versus the Southern hemisphere. In the Northern hemisphere, the combination of the extrinsic contribution and the fraction of the intrinsic pathway that it is able to engage is sufficient for apoptosis to proceed (second bar). Attenuation of the intrinsic component, by Bcl-2 overexpression for example, reduces the total number of apoptotic units to below the apoptotic threshold and the cells survive. The extrinsic pathway, therefore, appears to be modulated by Bcl-2. In the Southern hemisphere, the number of available apoptotic units that can be contributed by the intrinsic pathway may be lower. Thus, engagement of the extrinsic pathway with ligating CD95 (Fas/Apo1) antibody or trimerized ligand is not sufficient to mediate passage across the apoptotic threshold on its own (third bar). With a more efficient stimulus, mediated by multimerized CD95L, for example (fourth bar), a critical mass of the extrinsic pathway is enlisted to breach the apoptotic threshold, regardless of the contribution from the intrinsic pathway. Cell death in this scenario is therefore insensitive to Bcl-2 overexpression. The differences observed in the Northern and Southern hemispheres might therefore be explained by a combination of (a) the number of apoptotic units within the intrinsic pathway that can be recruited by cross-talk from the extrinsic pathway (lower in the South) and (b) the strength of the apoptotic stimulus (ligating antibodies and trimerized CD95L being equivalent but weaker than multimerized CD95L).
Do distinct apoptotic pathways exist when the tone of the intrinsic pathway is shifted toward an antiapoptotic state, whereas cross-talk exists between intrinsic and extrinsic pathways when the tone of the intrinsic pathway is shifted toward a proapoptotic state? This hypothesis would certainly reconcile the discrepancies observed between the groups of Krammer/Peter and colleagues and Strasser and colleagues. There are, however, other explanations as well that will no doubt emerge as the literature in this area evolves. But at least some things are clear. The existence of at least two distinguishable cell death–signaling pathways has been well established (extrinsic and intrinsic), and the identification of the major molecular components of these pathways provides a framework for predicting the circumstances under which each becomes engaged to initiate apoptosis. Similarly, and more recently, the discovery of molecules that betray the activities of one pathway to the other (such as Bid) account for the apparent cross-talk that many laboratories have observed, thus providing other obvious experiments to perform. Next comes context, and it is the in vivo circumstances that may largely dictate whether the two pathways remain isolated or cooperate in their tasks (e.g., cell type, signal type and strength, endogenous apoptotic tone, etc.). As with many biochemical pathways before it, ambiguity will reign supreme in apoptosis signaling before clarity emerges.
References
- Budihardjo I., Oliver H., Lutter M., Luo X., Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Green D.R. Apoptotic pathwayspaper wraps stone blunts scissors. Cell. 2000;102:1–4. doi: 10.1016/s0092-8674(00)00003-9. [DOI] [PubMed] [Google Scholar]
- Siegel R.M., Frederiksen J.K., Zacharias D.A., Chan F.K., Johnson M., Lynch D., Tsien R.Y., Lenardo M.J. Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science. 2000;288:2354–2357. doi: 10.1126/science.288.5475.2354. [DOI] [PubMed] [Google Scholar]
- Chan F.K., Chun H.J., Zheng L., Siegel R.M., Bui K.L., Lenardo M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–2354. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Schmitz I., Zha J., Korsmeyer S.J., Krammer P.H., Peter M.E. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem. 1999;274:22532–22538. doi: 10.1074/jbc.274.32.22532. [DOI] [PubMed] [Google Scholar]
- Luo X., Budihardjo I., Zou H., Slaughter C., Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- Li H., Zhu H., Xu C.J., Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- Lacronique V., Mignon A., Fabre M., Viollet B., Rouquet N., Molina T., Porteu A., Henrion A., Bouscary D., Varlet P. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat. Med. 1996;2:80–86. doi: 10.1038/nm0196-80. [DOI] [PubMed] [Google Scholar]
- Rodriguez I., Matsuura K., Khatib K., Reed J.C., Nagata S., Vassalli P. A bcl-2 transgene expressed in hepatocytes protects mice from fulminant liver destruction but not from rapid death induced by anti-Fas antibody injection. J. Exp. Med. 1996;183:1031–1036. doi: 10.1084/jem.183.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Roth K.A., Korsmeyer S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Huang D.C., Hahne M., Schroeter M., Frei K., Fontana A., Villunger A., Newton K., Tschopp J., Strasser A. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-x(L) Proc. Natl. Acad. Sci. USA. 1999;96:14871–14876. doi: 10.1073/pnas.96.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz I., Walczak H., Krammer P.H., Peter M.E. Differences between CD95 type I and II cells detected with the CD95 ligand. Cell Death Differ. 1999;6:821–822. doi: 10.1038/sj.cdd.4400569. [DOI] [PubMed] [Google Scholar]
- Huang D.C.S., Tschopp J., Strasser A. Bcl-2 does not inhibit cell death induced by the physiological Fas ligandimplications for the existence of type I and type II cells. Cell Death Differ. 2000;7:754–755. doi: 10.1038/sj.cdd.4400683. [DOI] [PubMed] [Google Scholar]
- Villunger A., O'Reilly L.A., Holler N., Adams J., Strasser A. Fas ligand, Bcl-2, granulocyte colony-stimulating factor, and p38 mitogen-activated protein kinaseregulators of distinct cell death and survival pathways in granulocytes. J. Exp. Med. 2000;192:647–658. doi: 10.1084/jem.192.5.647. [DOI] [PMC free article] [PubMed] [Google Scholar]