

Abstract
Mature B cells can alter their antibody repertoires by several mechanisms, including immunoglobulin heavy chain variable region (VH) replacement. This process changes the antigen combining site by replacing a portion of the original VH/diversity/heavy chain joining region (VHDJH) rearrangement with a corresponding portion of a new VH segment. This exchange can involve cryptic heptamer-like sequences embedded in the coding regions of VH genes.
While studying the B lymphocytes that expand in the synovial tissues of patients with rheumatoid arthritis (RA), clones with VHDJH variants that were apparently generated by VH replacement were identified with surprising frequency (∼8%). Examples of multiple independent VH replacement events occurring in distinct progeny clones were also identified. These secondary VH rearrangements were documented at both the cDNA and genomic DNA levels and involved several heptamer-like sequences at four distinct locations within VH (three sites in framework region 3 and one in complementarity determining region 2). The identification of blunt-ended double-stranded DNA breaks at the embedded heptamers and the demonstration of recombinase activating gene (RAG) expression suggested that these rearrangements could occur in the synovial tissues, presumably in pseudo-germinal centers, and that they could be mediated by RAG in a recognition signal sequence–specific manner. The presence of VH mutations in the clones that had undergone replacement indicated that these B cells were immunocompetent and could receive and respond to diversification signals. A relationship between these secondary VH gene rearrangements and the autoimmunity characteristic of RA should be considered.
Keywords: antibody diversity, recombinase activating genes, immune tolerance, autoimmunity, rheumatoid factor
Introduction
Antibody diversity is initially established in developing B lymphocytes in the bone marrow by the ordered rearrangement of V, (D), and J elements 1 2. These rearrangements occur via site-specific recombination events that classically involve recognition signal sequences (RSS) comprised of conserved heptamers and less conserved nonamers 3 4 5. As this process is largely random, receptors can be assembled that possess sufficient affinity for autoantigens to necessitate their elimination. However, such cells may be afforded another opportunity to express a tolerant receptor by undergoing secondary V region gene rearrangements that can involve either the H or L chain loci 6 7 8 9 10 11 12 13. These secondary rearrangements change a major portion of the antigen-combining site so the new receptor is significantly different from the original. The new receptor can consist of a completely new VLJL rearrangement or a VHDJH rearrangement that is modified by the replacement of a portion of the originally rearranged V segment (“VH replacement” [9]). If these secondary rearrangements provide the B cell with an acceptable antigenic specificity, it proceeds to the periphery as a functional B lymphocyte. This process of secondary rearrangement, presumably initiated by autoreactivity, has been termed receptor editing 6 7 8.
In the periphery, mature B cells can develop additional receptor/antibody diversity during the germinal center (GC) reaction. Although this process was long considered to be limited to the accumulation of point mutations 14, recent studies suggest that more extensive changes in the primary repertoire can occur by a variety of mechanisms. These mechanisms include the insertion and/or deletion of DNA segments of variable lengths 12 15 16, and the opportunity to again undergo V gene rearrangement. As these secondary rearrangements are thought to be initiated in those B cells undergoing the GC reaction that lack adequate affinity for the selecting antigen, this process appears to be different from receptor editing and therefore has been termed receptor revision 11 12 17 18.
Furthermore, receptor revision could potentially lead to autoreactivity if the drive to clonal diversification created self-reactive receptors that were ineffectively selected against, because of either genetic or acquired defects in immunoregulation. Therefore, this process could play an important role in the development of certain autoimmune disorders.
Rheumatoid arthritis (RA) is a relatively common, chronic, destructive arthropathy that results from an inflammatory synovitis of peripheral joints 19. These inflammatory processes appear to be mediated by cells of the myeloid, macrophage, and lymphoid lineages 20. Although there has been considerable controversy regarding the relative importance of the various cell types and their products in the inflammatory reactions of RA, it seems likely that all cell lineages participate in disease pathogenesis. B lymphocytes may not be necessary to initiate the inflammatory reactions characteristic of RA 21, but B cells and their products can perpetuate and potentiate these responses 22 23 24 25. This perpetuation most likely involves at least two functions of B cells: their abilities to present antigen to T cells and to elaborate antibodies. Central to each of these functions is the (auto)antigenic specificity encoded in the V regions of the B cell's Ig molecules. These antigenic specificities include the classical anti-IgG/rheumatoid factor (RF) reactivity 26 27 as well as other less well-defined reactivities that presumably are directed at tissue autoantigens 28 29 30 and possibly etiologic environmental antigens 31 32 33. Furthermore, a recent murine model that has some features similar to the human disease 34 suggests that B cells and their products could play an essential role in the disease process 35 36.
Histologically, the synovial tissues of some RA patients contain collections of B and T lymphocytes and follicular dendritic cells that resemble GCs 37 38. These pseudo-GCs possess at least some of the functions of typical GCs, i.e., they appear to be able to support clonal amplification and Ig V gene diversification as measured by the accumulation of new mutations. In the course of studying the clonally amplified B lymphocytes that develop V gene point mutations in the synovial tissues of RA patients, we identified a series of clones that exhibited VHDJH variants that were apparently generated by VH replacement. However, unlike previous studies of VH replacement in murine B cell lines 39 40 41 42 and transgenic mice 6 7 8 43 and in transformed 44 45 46 and normal human B lymphocytes 18 45, these secondary VH rearrangements occurred relatively frequently and involved several heptamer-like sequences at four distinct locations within VH. Our data suggest that these rearrangements can occur in situ and could be mediated by products of the recombinase activating genes (RAGs) in an RSS-specific manner. A preliminary report of these findings was presented previously 47.
Materials and Methods
Patient Samples.
Synovial tissue was obtained from three patients who fulfilled the American College of Rheumatology criteria for the diagnosis of RA 48. For patient 1 (a 29-yr-old Hispanic male), synovial tissue was collected from the right and left hips at the time of bilateral joint replacements; these samples are labeled ST1R or ST1L, respectively. For patient 2 (a 25-yr-old Black female) and 3 (a 56-yr-old White female), synovial tissue was collected from individual knee joints; these samples are labeled ST2 and ST3. Each sample was digested with collagenase type IV, DNase I (both from Worthington Biochemical Corporation), and hyaluronidase (Sigma-Aldrich) to obtain single cell suspensions. Mononuclear cells were isolated from these cell suspensions by density gradient centrifugation using Ficoll-Paque (Amersham Pharmacia Biotech) and then cryopreserved as viable cells in 20% fetal bovine serum with 10% dimethyl sulfoxide using a programmable cell freezing machine (CryoMed).
Isolation of DNA and RNA and Preparation of cDNA.
Genomic DNA was isolated from mononuclear cells using the Puregene DNA isolation kit (Gentra Systems) and total RNA was isolated using Ultraspec RNA (Biotech Laboratories). 1 μg of RNA was reverse transcribed to cDNA using 200 U Moloney murine leukemia virus reverse transcriptase (GIBCO BRL/Life Technologies), 1 U RNase inhibitor (5 Prime 3 Prime), and 20 pmol oligo dT primer in a total volume of 20 μl. These reagents were incubated at 42°C for 1 h, heated to 65°C for 10 min to stop the reactions, and then diluted to a final volume of 100 μl.
PCR Amplification of Ig VH Gene DNA and cDNA.
The sequences of the primers used in these reactions were published previously 49. Genomic DNA (100 ng) was amplified using a sense VH1 family–specific framework region (FR)1 primer in conjunction with an antisense JH consensus primer. These reactions were carried out in 50 μl using 5 pmol of each primer and were cycled with a 9600 GeneAmp System (PerkinElmer) as follows: denaturation at 94°C for 40 s, annealing at 65°C for 45 s, and extension at 72°C for 40 s. After 35 cycles, extension was continued at 72°C for an additional 10 min. cDNA was amplified using the same sense primer paired with either a Cμ, Cγ, or Cα antisense primer 50. However, the reactions were cycled as follows: denaturation at 94°C for 45 s, annealing at 65°C for 45 s, and extension at 72°C for 45 s. After 35 cycles, extension was continued at 72°C for an additional 10 min.
Creation of DNA and cDNA Libraries and Sequencing of Selected Clones.
VH1-specific DNA and cDNA libraries were created from the PCR products described above. 10 ng of each PCR product was ligated into pCR2.1 vector and transformed into INVαF′ competent cells using the TA cloning kit (Invitrogen). Transformed INVαF′ cell colonies were identified and isolated by color selection after overnight growth on Luria-Burtani (LB) agar plates containing X-Gal and ampicillin. Plasmids were isolated from each selected colony using Wizard minipreps (Promega) after overnight growth in LB broth containing kanamycin. The DNA sequences of individual molecular clones were determined using Big Dye DNA sequencing kit (PerkinElmer) with M13 forward and reverse primers and an automated genetic analyzer ABI PRISM 377 (Applied Biosystems). The error rate calculated for these sequence analyses was 8.36 × 10−4, based on the detection of 13 mutations in 15,554 nucleotides of CH genes of these and other human cDNA VHDJH-CH transcripts from RA synovial B cells. All VH gene sequences discussed in this study have been entered in EMBL/GenBank/DDBJ under accession nos. AF308542–AF308567.
PCR Conditions for Ig VH Gene Fingerprinting Assay.
The original Ig VH gene fingerprinting assay 51 was modified 52 into two stages, starting with either genomic DNA or cDNA as templates. The sequences of the primers used in these reactions and the details of the two PCR stages have been published previously 52.
Analyses of Ig DNA Breaks by Ligation-mediated PCR.
To avoid adventitious breaks, genomic DNA was prepared by the agarose plug method 53 from 2–3 × 105 CD19+ cells isolated using magnetic beads as described 54. To identify blunt-ended double-stranded (ds) DNA breaks, ligation-mediated (LM)-PCR was employed 55. In brief, linker ligation was performed in 25–50-μl reactions using T4 DNA ligase and the linker-ligated DNA was diluted with ligation buffer to adjust the amounts of DNA used in the subsequent seminested PCR 54. 2 μl of linker-ligated DNA was amplified using a sense VH1 family–specific leader primer in conjunction with an antisense linker primer (GCGGTGACCCGGGAGATCTGAATTCAC). These reactions were performed in 25 μl using 5 pmol of each primer and were cycled as follows: denaturation at 94°C for 30 s, annealing at 63°C for 45 s, and extension at 72°C for 45 s. After 35 cycles, extension was continued at 72°C for an additional 10 min. Next, 2 μl of each PCR product was reamplified with 5 pmol of VH1 family–specific FR1 primer and the same antisense linker primer. The reactions were carried out in 25 μl and cycled as described above. After 30 cycles, extension was continued at 72°C for an additional 10 min. The VH LM-PCR products were visualized with ethidium bromide staining.
Analyses of RAG-1 Expression by Nested Reverse Transcription PCR.
RAG-1 was amplified from cDNA by nested PCR using specific primer pairs that flank a 5.2-kb intron in the germline gene: sense, TGCAGACATCTCAACACTTTGGCCAG; antisense 1, TTTCAAAGGATCTCACCCGGAACAGC; antisense 2, AGCTTAAATTTCCATTCTGAATT. This PCR strategy has been used successfully to analyze RAG-1 expression in tonsilar GC B cells 56.
Results
Clonal Amplification among Synovial Tissue B Lymphocytes
HCDR3 length is a useful estimator of clonal diversity in polyclonal B cells 51. Comparisons of HCDR3 lengths generated from both the genomic DNA and cDNA of the same sample indicate the relative frequencies and activation states of individual B cell clones in polyclonal populations 52. Using this combined approach, we analyzed the B lymphocytes infiltrating the synovial tissues from four different joints of three RA patients. Widespread oligoclonality was found among the IgM-, IgG-, and IgA-expressing B cells in the four synovial tissue samples (52; see Fig. 3 and Fig. 4, and data not shown). Based on comparisons of the genomic DNA and cDNA fingerprinting assays, these clonal expansions involved both activated and memory B cells. Such clonal expansions are a common and characteristic feature of the B cell repertoires of RA patients 38 52 57 58 59, and they suggest in situ activation and growth of B lymphocytes with restricted B cell receptors (BCRs) that may be reactive with local (auto)antigens.
Figure 3.

cDNA clones exhibiting VH replacement at the 5′ FR3 embedded heptamer. Two VH1-IgG+ clones from ST1L (G11 and G19) are aligned as in Fig. 1. The original VHDJH rearrangement is listed above the new VHDJH rearrangement.
Figure 4.

cDNA clones exhibiting VH replacement at a heptamer embedded between the 5′ and 3′ FR3 heptamers (mid-position FR3 heptamer). See the legend to Fig. 1.
cDNA Sequence Analyses of VH1-expressing B Cell Clones Reveal Evidence Consistent with VH Replacement at Three Distinct Sites in FR3
To determine if diversification had occurred among the progeny of these expanded B cell clones, we prepared VH1-IgM– and VH1-IgG–expressing libraries from cDNA of the ST1R and ST1L synovial samples and determined the cDNA sequences of 95 random clones. The VH1 family was chosen for analysis based on the results of the VH fingerprinting analyses. As expected, several expanded B cell clones were found in each sample as indicated by the presence of identical HCDR3 sequences. In many instances, these clones contained distinct point mutations in their VH genes, indicating that intraclonal diversification had occurred (data not shown).
Five sets of cDNA clones (Table ) were especially interesting because they contained clonal progeny that exhibited a significant discordance in the sequence between the 3′ and the 5′ regions of their rearranged VHDJH genes (see Fig. 2 Fig. 3 Fig. 4 Fig. 5). Among the members of these clones, there was an average of >99.9% similarity (usually 100% identity) from the distal portions of FR3 through HCDR3 to the end of the CH sequence. In contrast, there was an average of <81.6% sequence similarity between the clonal members 5′ of these portions of FR3. This discordance is consistent with VH replacement (represented schematically in Fig. 1). However, unlike previous examples of this phenomenon that involved rearrangements to a single site in FR3 6 7 8 18 39 40 41 42 43 44 45 46, the rearrangements detected in the synovial tissue cDNA clones occurred at three distinct sites spanning FR3 (see Fig. 2 Fig. 3 Fig. 4 Fig. 5).
Table 1.
Molecular Characterization of the RA Synovial Tissue B Cell Clones That Underwent VH Replacement
| Type of diversification | No. of mutations | |||||||
|---|---|---|---|---|---|---|---|---|
| Clone | Source | No. of identicalsequences | VH gene(s) used | Ig isotype | VH mutation | VH replacement | FRR/S | CDRR/S |
| ST1R M5 | cDNA | 1 | VH 1–58 | IgM | − | − | 0/0 | 0/0 |
| ST1R M1 | cDNA | 1 | VH 1–58 | IgM | + | − | 0/1 | 0/0 |
| ST1R M8 | cDNA | 1 | VH 1–58 | IgM | + | − | 0/1 | 0/0 |
| ST1R M26 | cDNA | 1 | VH 1–58 | IgM | + | − | 1/0 | 0/0 |
| ST1R M6 | cDNA | 1 | VH 1–58/VH 1–18 | IgM | + | + | 5/2 | 1/0 |
| ST1R M31 | cDNA | 1 | VH 1–58/VH 1–24 | IgM | + | + | 1/2 | 0/0 |
| ST1R M10 | cDNA | 1 | VH 1–58/VH 1–69 | IgM | + | + | 10/5 | 4/1 |
| ST1R M17 | cDNA | 1 | VH 1–08 | IgM | + | − | 4/4 | 4/1 |
| ST1R M9 | cDNA | 1 | VH 1–08/VH 1–69 | IgM | + | + | 0/1 | 0/0 |
| ST1R G1 | cDNA | 1 | VH 3–09/VH 1–02 | IgG | + | + | 17/3 | 18/1 |
| ST1R G2 | cDNA | 1 | VH 3–09/VH 1–69 | IgG | + | + | 11/6 | 3/2 |
| ST1L G12 | cDNA | 1 | VH 1–69 | IgG | + | − | 8/3 | 2/1 |
| ST1L G2 | cDNA | 8 | VH 1–69 | IgG | + | − | 7/4 | 2/1 |
| ST1L G32 | cDNA | 1 | VH 1–69 | IgG | + | − | 7/4 | 2/2 |
| ST1L G22 | cDNA | 1 | VH 1–69 | IgG | + | − | 9/4 | 2/1 |
| ST1L G11 | cDNA | 1 | VH 1–69 | IgG | + | − | 9/5 | 5/1 |
| ST1L G19 | cDNA | 1 | VH 1–69/VH 1–08 | IgG | + | + | 10/6 | 4/1 |
| ST1L G8 | cDNA | 1 | VH 1–08 | IgG | + | − | 10/5 | 4/1 |
| ST1L G27 | cDNA | 1 | VH 1–08 | IgG | + | − | 13/4 | 4/1 |
| ST1L G29 | cDNA | 1 | VH 1–08/VH 1–69 | IgG | + | + | 7/2 | 3/1 |
| ST2 14 | DNA | 5 | VH 1–46 | Indeterminate | − | − | 0/0 | 0/0 |
| ST2 10 | DNA | 1 | VH 1–46/VH 1–02 | Indeterminate | + | + | 5/3 | 6/2 |
| ST2 6 | DNA | 2 | VH 1–58 | Indeterminate | + | − | 0/0 | 1/0 |
| ST2 31 | DNA | 1 | VH 1–18/VH 1–46 | Indeterminate | + | + | 3/1 | 0/0 |
| ST2 2 | DNA | 2 | VH 1–46 | Indeterminate | − | − | 0/0 | 0/0 |
| ST2 26 | DNA | 1 | VH 1–46/VH 1–69 | Indeterminate | + | + | 6/2 | 0/0 |
The nucleotide sequences of 95 cDNA clones and 36 genomic DNA clones were determined. Only those clones that contained progeny exhibiting VH replacement are listed. R, replacement; S, silent.
Figure 2.

cDNA clones exhibiting VH replacement at the 3′ FR3 embedded heptamer. Two VH1-IgG+ clones from ST1L (G27 and G29) are aligned centrally with their most similar germline gene counterparts listed above and below. The gaps in the sequences represent the boundaries between the various FR and CDR. The 3′ FR3 embedded heptamers are displayed with black backgrounds and the HCDR3 are boxed. Nucleotide differences between the two clones are indicated by dots and identities are indicated by asterisks. Nucleotide differences between the individual clones and their germline counterparts are indicated by letters and the identities are indicated by dashes. Note that the HCDR3 of the two clones are identical, whereas the VH segments upstream of the heptamer are very different between the two clones and resemble different germline genes. Based on either genealogical trees or point mutations, the VHDJH rearrangement of the original B cell clone is listed above the new VHDJH rearrangement.
Figure 5.
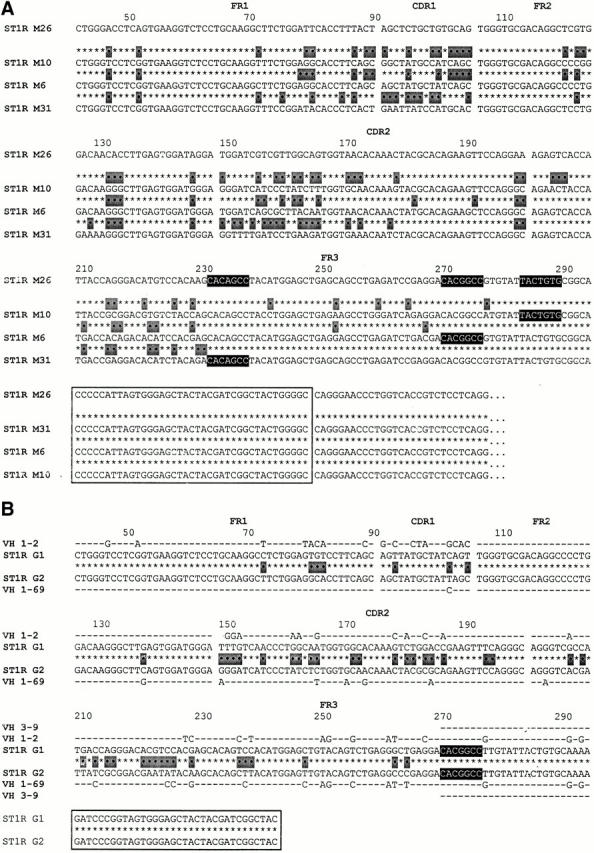
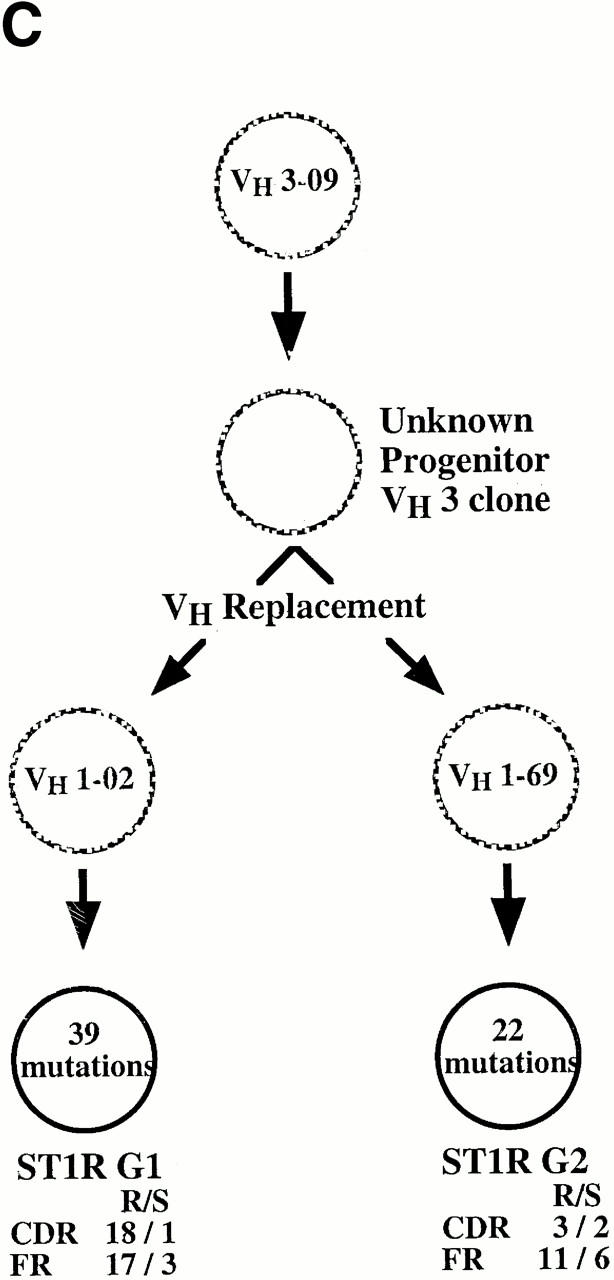
cDNA clones exhibiting multiple distinct secondary rearrangements using different heptamers. (A) Three distinct replacement events that occurred in the progeny of the parental ST1R M26 clone are listed below the original clone. Note that each clone used a different embedded heptamer to accomplish the replacement event. The most similar germline genes for M10 (1–69), M6 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18, and M31 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 are not listed. (B) Two distinct replacement events that occurred in the progeny (ST1R G1 and G2) of a VH3–09–expressing B cell clone. It is clear that the original clone expressed a VH3 gene because the nucleotides downstream from the embedded heptamer are identical to VH 3-09 and are not found in any VH1 family genes.
Figure 1.

Schematic representation of VH replacement occurring at the four heptameric sites defined in this study. The top panel illustrates a typical VH to DJH rearrangement. The lower panels illustrate the four types of VH replacement documented in this study. VH segment replacement occurred at four discrete heptamer-like sequences, three located in FR3 (3′ FR3 heptamer, mid-position FR3 heptamer, and 5′ FR3 heptamer) and one located in CDR2 (CDR2 heptamer).
Rearrangements Involving the 3′ FR3 Heptamer.
Fig. 2 lists an example of VH replacement occurring at the 3′ end of FR3 of two IgG-expressing clones from ST1L (clones G27 and G29). In this and the following figures, the sequence of the original B cell clone is listed above the new “replaced” B cell clone, with the most similar germline gene counterparts listed above and below the original and new rearrangements, respectively. The replacement in Fig. 2 occurs at an embedded heptamer of reverse orientation (TACTGTG; displayed with black background in the figure) that has been identified at the end of murine and human VH genes 39 40 41 60. These clones display 100% identity from the embedded heptamer to the end of the IgG CH sequence determined. In contrast, the 2 clones differ at 48 positions 5′ of the embedded heptamer (80.2% similarity).
When a homology search was made for the most likely germline counterpart of the gene segments 5′ of the heptamer, G27 was 90.6% similar to the VH 1–8 germline gene, whereas clone G29 was only 84.3% similar to the 1–8 germline gene. However, the 5′ segment of clone G29 was 94.6% similar to the VH 1–69 germline gene. These comparisons suggest that the two clones diverged by interchanging major portions of the VH 1–8 and 1–69 genes up to the position of the embedded heptamer.
Rearrangements Involving the 5′ FR3 Heptamer.
The IgG-expressing clones listed in Fig. 3 demonstrate VH replacement occurring at the 5′ end of FR3. These rearrangements utilize a heptamer (CACAGCC) embedded in an orientation like that found at the 3′ end of unrearranged VH genes. This heptamer lies 52 nucleotides upstream of the 3′ heptamer mentioned above.
As in the previous examples, there is 100% identity between the 2 clones from the embedded heptamer to the end of the CH sequence, but only 82.6% similarity between these 2 clones upstream of the heptamer (33 differences). The upstream portion of clone G11 was most similar to the VH 1–69 germline gene (93.7%), whereas the same portion of clone G19 was most similar to the VH 1–08 germline gene (92.6%).
Rearrangements Involving a Heptamer Embedded between the 5′ and 3′ FR3 Sites.
The clones listed in Fig. 4 illustrate VH replacement occurring at a heptamer (CACGGCC) located between the two sites mentioned above. This “mid-position FR3 heptamer” is oriented like the 5′ FR3 heptamer. The examples listed in the figure (M17 and M9) are members of an IgM-expressing clone from a different anatomic site of patient no. 1. In this case, there is only a one-nucleotide difference between the two clones from the heptamer to the end of the IgM CH sequence; this difference occurs outside of the HCDR3 in FR4. However, there are 43 nucleotide differences upstream of the heptamer (81.2% similarity). The 5′ portion of M9 derives from the VH 1–69 germline gene (99.6% similar), whereas the same portion of clone M17 is from the VH 1–8 germline gene (94.2% similar to 1–8 and only 81.9% similar to 1–69).
Multiple Distinct Rearrangements Using Different Heptamers within the Same Original B Cell Clones.
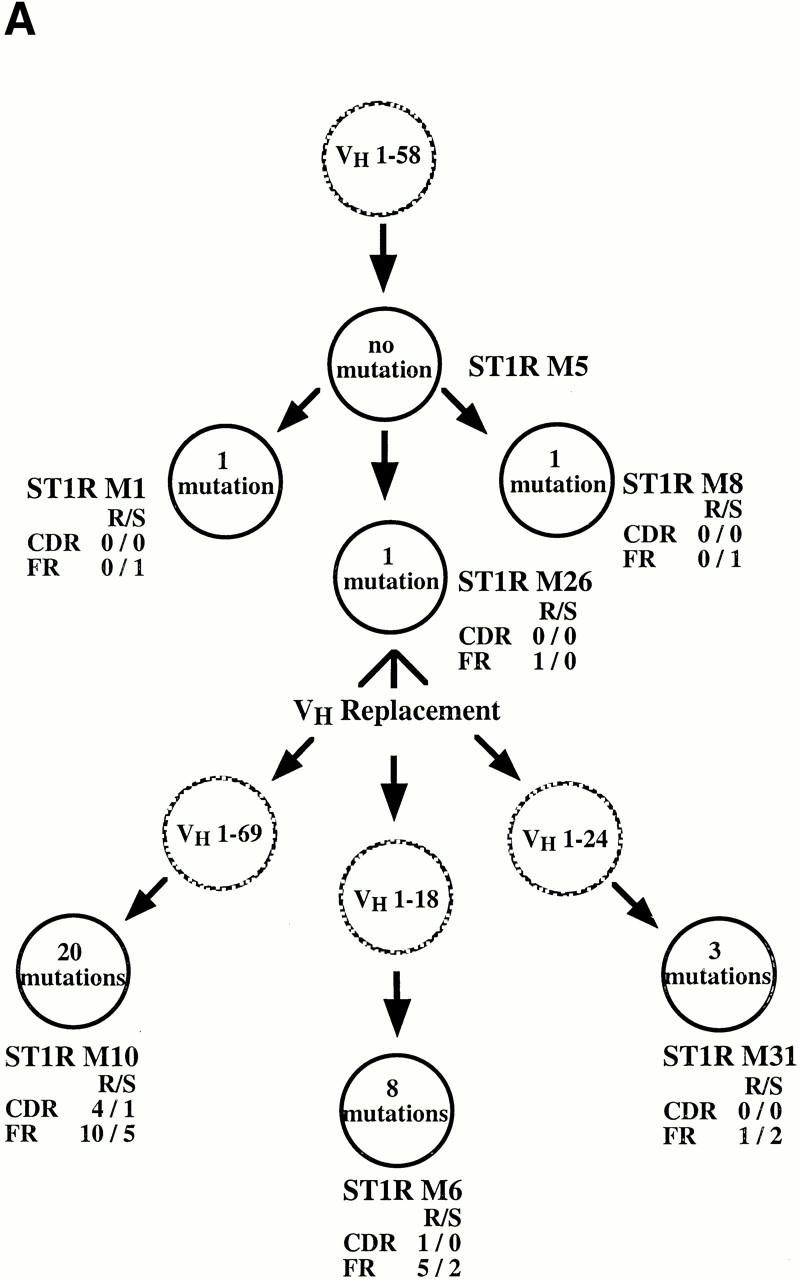
We detected two examples of multiple independent VH gene replacement events occurring in distinct progeny of single progenitor B cell clones. Fig. 5 A illustrates three replacements in the progeny of the ST1R M26 clone, each occurring at a different heptameric site. Thus, the rearranged M26 VHDJH was replaced at the 3′ FR3 heptamer by a new VH 1–69 germline gene to become clone M10. In addition, a replacement occurred in a different subclone involving a new VH 1–18 germline gene at the mid-position FR3 heptamer, yielding clone M6. Finally, a replacement using the VH 1–24 germline gene occurred at the 5′ FR3 heptamer, giving rise to clone M31.
The examples in Fig. 5 B represent two distinct VH1 gene replacement events (VH 1–2 and 1–69 segments) that occurred in a clone that originally used a VH3 gene (VH 3–09). Both of these rearrangements used the same mid-position FR3 heptamer. The identity of the original clone as a member of the VH3 family is clear because the nucleotides downstream from the embedded heptamer are identical to VH 3–09 and are not found in any VH1 family genes.
DNA Sequence Analyses of VH1-expressing B Cell Clones from a Different Patient Confirm and Extend VH Replacement Events
Based on these findings, we searched for evidence of VH replacement at the genomic DNA level to avoid the possibility that the cDNA results represented artifacts of reverse transcription (RT)-PCR. In addition, these studies were carried out using synovial tissue (ST2) from a different RA patient to assure that this phenomenon was not unique to a single patient.
Fig. 6 shows the three examples of VH replacement found in the VH1-expressing DNA library from sample ST2. Fig. 6A and Fig. B, confirm the occurrence of VH replacement occurring at the 5′ FR3 heptamer and the mid-position FR3 heptamer.
Figure 6.
DNA clones exhibiting VH replacements at the 5′ FR3 heptamer and the mid-position FR3 heptamer and at a heptamer embedded in HCDR2. (A) An example of a replacement that occurred at the 5′ FR3 heptamer. Note that there is another potential heptameric site located 12 bp upstream of the 5′ FR3 heptamer (highlighted in light gray). (B) An example of a replacement that occurred at the mid-position FR3 heptamer. (C) An example of a replacement that occurred at a heptamer located in HCDR2.
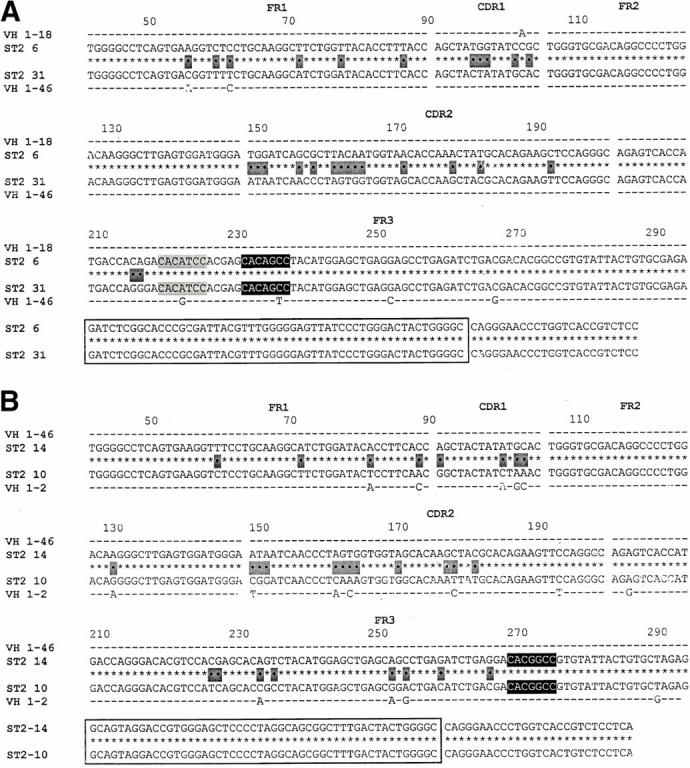

However, Fig. 6 C documents a VH replacement event that we had not detected at the cDNA level. This replacement occurred at a heptamer (CACAGAA) embedded in HCDR2. As in the previous examples, the 2 clones are identical 3′ of the embedded heptamer, but only 82.3% similar 5′ of the heptamer (25 differences). Both clones are identical to their respective germline genes (VH 1–46 and 1–69) 5′ of the heptamer.
Ig VH Gene Diversification among the Clones Undergoing VH Replacement
As indicated in Table , the VH replacement events detected in these synovial tissue samples usually occurred in B cells that had already had significant antigenic exposure and had accumulated V gene mutations. In addition, a comparison of the average numbers of mutations in the original clonal members (236 mutations/22 sequences; 10.73%) and the numbers of mutations in the “replaced” clones (144 mutations/11 sequences; 13.09%) indicates that the diversification process continued in those clones that had undergone VH replacement. Therefore, these B cells remained immunocompetent after the secondary rearrangement event and received and responded to diversification signals.
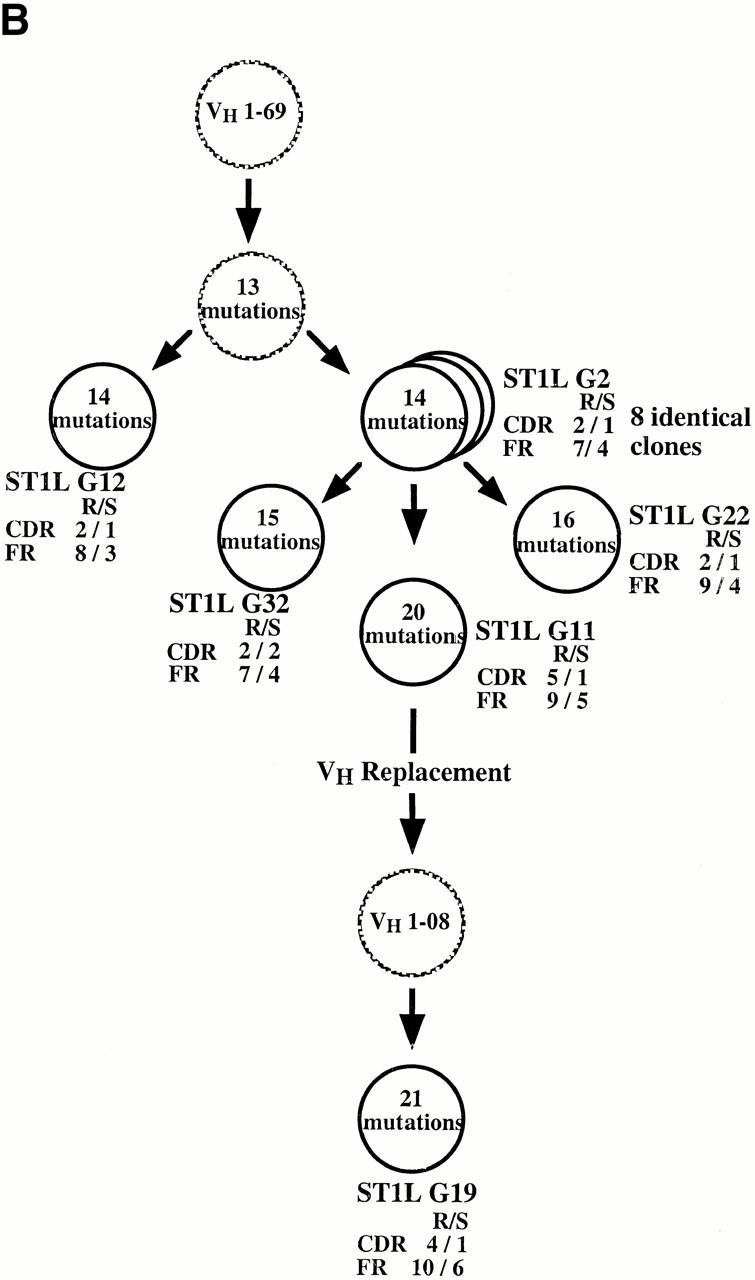
Three genealogical trees that display examples of these clonal diversification events are presented in Fig. 7, along with the numbers, types, and locations of the VH gene mutations that occurred in the various clonal members.
Figure 7.
Representative examples of VH diversification occurring in the B cell clones that exhibit VH replacements. In each instance (A, B, and C), the identified cDNA clones are represented by filled circles and the presumed (unidentified) clones are represented by hashed circles. The numbers of mutations are contained within the circles and their locations (FR vs. CDR) and types (replacement [R] vs. silent [S]) are displayed adjacent to the circles.
Use of LM-PCR to Detect DNA Breaks at the Sites of VH Replacement
We used VH family–specific LM-PCR to probe for blunt-ended dsDNA breaks at the embedded heptamers in the ST2 and ST3 synovial samples. Such products could represent intermediates of VH replacement. Human bone marrow cells were used as positive controls and fibroblasts as negative controls in these studies. VH-containing products were obtained from both the synovial tissue and bone marrow B cell samples, whereas no products were obtained from the fibroblast DNA. For the synovial tissue samples, two products of ∼300 and ∼250 bp were detected (Fig. 8). The DNA sequences of VH1-expressing clones (n = 50) from the PCR products of ST3 revealed the linker ligated at or within the 5′ FR3 heptamer (300-bp product) and the 3′ FR3 heptamer (250-bp product), indicating that blunt dsDNA ends were available at these sites (data not shown).
Figure 8.

LM-PCR identifies dsDNA fragments generated by cleavage at the 5′ and 3′ heptamers. Genomic DNA was prepared from CD19+ cells from two synovial tissue samples (lanes 1 and 2) and from human bone marrow (lane 3) by the agarose plug method (reference 53). LM-PCR was then used to identify blunt-ended dsDNA breaks (reference 55). The VH-containing products of these reactions were visualized with ethidium bromide staining. The 320- and 249-bp bands contained DNA fragments with the linker attached at or in the 5′ (320 bp) and 3′ (249 bp) heptamers. Lane 4 is a template-deprived negative control. M, marker lane.
RAG-1 Gene Expression in the Synovial Tissues of RA Patients
As V(D)J recombination requires RAG proteins, the identification of RAG gene expression would provide a possible mechanism for the identified DNA breaks and the secondary VH gene segment rearrangements. Therefore, we used RT-PCR to search for RAG-1 gene expression in synovial tissue samples.
As shown in Fig. 9, RAG-1 mRNA was readily detected in several synovial tissue samples (lanes 2–5) and from human bone marrow (lane 1). The lack of RAG-1 gene expression in human fibroblasts (lane 6) and direct sequence analyses of these products (data not shown) confirmed the specificity of these reactions. As the primers used for these reactions flank a 5.2-kb intron in the RAG-1 gene, these 180-bp products could not have been generated from genomic DNA templates. Furthermore, preliminary single-cell PCR and cDNA sequencing analyses suggest expression of RAG-1 in a subset (∼20–30%) of CD19+ B cells from ST2 and ST3 (data not shown).
Figure 9.

RAG-1 gene expression in the synovial tissues of RA patients. RAG-1 was readily detected by RT-PCR in the cDNA obtained from several synovial tissue samples (lanes 2–5) and from human bone marrow (lane 1). Lane 6 is a negative control (human fibroblast cDNA) and lane 7 is a template-deprived negative control. PCR products and DNA markers were visualized by ethidium bromide staining after agarose gel electrophoresis. M, marker lane.
Discussion
In this study we identified four types of secondary VH gene rearrangements that occurred among clonally related B cells that were expanded in the synovial tissues of different RA patients. Although chimeric V gene sequences can be generated artifactually by crossover events during PCR and cloning, we believe that it is highly unlikely that our findings represent artifacts because these rearrangements (a) were observed in three different synovial tissue samples from two RA patients, (b) were documented at both the cDNA and genomic DNA levels, (c) occurred only at sites in the VH genes that exhibited heptamer-like sequences, and (d) were always in-frame and coded translatable proteins. (e) Finally, and perhaps most convincingly, is the finding that transcripts from progeny of two different clones displayed replacements of different genes either at the same heptamer (Fig. 5 A) or at distinct heptamers (Fig. 5 B). The possibility that replicates of the same DNA segments would have developed crossover artifacts involving different genes either at the same position or at different positions, each flanked by a cryptic RSS, is remote.
Because these VH replacement events occurred in B cells that exhibited dsDNA breaks at heptamer-like sequences (Fig. 8) and expressed RAG-1 proteins (Fig. 9), we believe that these V gene replacements were RSS specific and RAG mediated. Such a process has been shown to occur in vitro 39 40 41 42 61 and in vivo 6 7 8 9 18 43 44 45 46, and the identification of circular DNA containing the outgoing VH gene joined to the RSS of the incoming VH gene 42 61 suggests that the process involves the RAG proteins and the V(D)J recombination process. However, these rearrangements require the formation of hybrid joints between the coding end of the incoming VH gene and the signal end of the outgoing VH gene segment 5 9 18.
Nevertheless, we cannot exclude the possibility that these VH replacement events were mediated by a non–site-specific, general recombination process such as gene conversion, a mechanism that is involved in the diversification of the λ5/14.1 component of the pre-BCR in humans 62 as well as the BCR of chickens 63 64 and rabbits 65 66. If gene conversion is responsible for these secondary recombination events, the site of cross-strand exchange could be located anywhere downstream of the first mismatch and upstream of the next mismatch between the incoming/donor germline VH gene sequence and the outgoing/recipient rearranged gene segment.
The data that favor a RAG-mediated recombination event include: (a) the size of the fragments being replaced, as gene conversion usually involves smaller segments of DNA; (b) the apparent insertion of the new DNA fragment at specific sites (heptamer-like sequences), as the insertion of DNA via gene conversion is generally viewed as a non–site-specific process; (c) the presence of dsDNA breaks at these heptamer-like sequences, especially those clones with multiple distinct rearrangements (Fig. 5); and (d) the expression of RAG-1 in these B cell populations. The data that are atypical for classical V(D)J recombination include the absence of detectable nonameric sequences 12 or 23 bp from the cryptic heptamers (vide infra) and the lack of coding end processing at the junctions of the new rearrangements (Fig. 2 Fig. 3 Fig. 4 Fig. 5). However, these latter findings are not completely incompatible with a RAG-mediated process, as nonamers are not essential for in vitro rearrangements 67 and in situations in which hybrid joint formation is facilitated (e.g., Ku knockout mice [68]), the hybrid junctions frequently do not display evidence of N-insertions and exonuclease activity 69. Thus, both site-specific and general recombination mechanisms may be responsible for these VH replacement events, and the two mechanisms may not be mutually exclusive. Indeed, a mechanism involving RAG-mediated cleavage at cryptic RSS followed by DNA segment exchange mediated by homologous recombination may not be unreasonable. Clearly, further study will be required to determine the relative contributions of these two mechanisms. Nevertheless, at this point our data suggest a likely role for cryptic heptamers and RAG in these secondary VH gene rearrangements.
In this study we identified four cryptic heptamers that appeared to be involved in these secondary rearrangements. Three of these heptamers were within FR3 (Fig. 2 Fig. 3 Fig. 4 Fig. 5, and Fig. 6A and Fig. B) and one in HCDR2 (Fig. 6 C). The heptamer located at the end of FR3 was in an inverse orientation like that found at the 5′ end of D segments and corresponded exactly to that described previously in murine VH gene replacement events 9 39 40 41. The 5′ FR3 heptamer, the mid-position FR3 heptamer, and the heptamer in HCDR2 were oriented like those found at the 3′ ends of unrearranged VH genes. All of these heptamers displayed the essential 5′ three nucleotides CAC 67, followed by variations on the expected AGTG sequence (e.g., AGCC, GGCC). In vitro studies 67 indicate that these sequences will permit recombination, albeit at reduced efficiencies. It is possible that any heptamer that has a CAC motif at its 5′ end can act as an embedded RSS for secondary rearrangements. In this regard, the genomic DNA clones listed in Fig. 6 A (ST2 6 and ST2 31) exhibit a heptameric sequence (CACATCC; highlighted in light gray in Fig. 6 A) 12 nucleotides upstream of the 5′ FR3 heptamer that could have functioned as an RSS in this instance. If this is the case, then this represents still another FR3 heptamer in addition to the three previously discussed.
Classical V(D)J recombination requires a heptamer, a spacer of either 12 or 23 bp, and a nonamer (ACAAAAACC-3′ [3, 4]). We could not define a standard nonamer sequence 12 or 23 bp from the heptamers described, although the clones listed in Fig. 5 B display a potential nonamer 12 bp downstream of the cryptic heptamer; this sequence spans the FR3–CDR3 junction. Nevertheless, the requirements for effective nonamers are far less stringent than for heptamers 67, and in vitro recombination studies suggest that nonamers may not be necessary 5. The inability to define a classical nonamer is consistent with previous studies of VH replacement 9 and led to the suggestion that there might be a different nonamer sequence specific for V gene replacement 9.
Secondary VH gene recombination events would be expected to involve incoming genes that are located upstream of the originally rearranged (and outgoing) VH gene. In one combination it was impossible to determine which was the original gene (ST1R M17 and M9). However, in 10 instances this determination was clear, either because of critical nucleotide point differences (e.g., ST2 2→ST2 26; see the 9 nucleotides within the common downstream FR3 sequence in Fig. 5 C) or because of an obvious genealogical progression (e.g., ST1L G11→G19; ST1L G27→G29; ST1R M26→M10, M6, and M31; ST1R G1 and G2 [see Fig. 6]). In 5 of the 10 instances in which chronology was clear, the incoming gene was located 5′ of the original outgoing gene. However, in five events this was not the case. Therefore, in these instances the new rearrangement may have involved a VH gene residing on the other chromosome or the original locus may have been inverted (e.g., by a D-JH rearrangement that involved the heptamer at the 3′ end of a D segment [17]).
The VH 1–69 gene was frequently involved in these VH replacement events (6/11 instances), despite the fact that it is not overexpressed in the normal human B cell repertoire 70. This suggests that certain VH genes may be favored in the replacement process. This is consistent with the findings that discrete VH genes were nonstochastically involved in spontaneous 71 and induced 61 VH replacement events in murine B cell lines.
We were surprised that these secondary VH rearrangements occurred so frequently in these synovial tissue B cells. 8/95 VH1-expressing cDNA clones and 3/36 VH1-expressing genomic DNA clones from 3 anatomically distinct synovial tissues exhibited this phenomenon (Table ). This represents a frequency of 8.4%. As our DNA sequencing strategies restricted us to identifying VH replacements that involved VH1 family genes replacing VH1 genes, the frequency of these events might have been higher had we searched for VH replacements that involved VH segments from other families being incorporated into VH1.
It remains to be determined whether this high frequency of VH replacement events represents a feature of normal GC reactions or of the ectopic GC reactions identified in RA 37 38. In this regard, Wilson et al. 18 documented VH replacement occurring at a 5′ FR3 heptamer found in VH4 family genes in a subset of tonsilar B cells that express solely IgD and exhibit an especially high frequency of somatic point mutations 72. As our studies were performed with unfractionated B cells, we cannot determine the precise cellular subset in which these events occurred. However, as all of the cDNA transcripts analyzed in our study were of the IgM and IgG varieties (Fig. 2 Fig. 3 Fig. 4 Fig. 5), VH replacement is not limited to IgD+IgM− B cells. In addition, the appearance of point mutations in every case of VH replacement (Table ) suggests that these B cells maintained a functional BCR that promoted viability 73 and permitted continued clonal expansion and somatic hypermutation in the “replaced” clone. Meffre et al. recently described a subset of human B lymphocytes marked by the coexpression of conventional and surrogate L chains that is enriched in the synovial tissues of certain RA patients 74. These cells express RAG mRNA and show evidence of receptor editing. It will be interesting to see if this unusual B cell subset is a component of the B cell expansions that are characteristic of RA and that can undergo VH replacement.
These surprisingly frequent VH replacement events may have significance for the autoreactivity seen in RA, as they could either maintain or break self-tolerance. RA is characterized classically by the expansion of RF-producing B cells that often increase in affinity as the disease progresses 75 and that correlate with disease severity 76. Furthermore, B cells and plasma cells with this autospecificity are enriched to very high numbers within the synovial tissues of RA patients 77. As it is clear from transgenic mouse models of autoimmunity that secondary rearrangements of either VH or VL can lead to non–self-reactive receptors 6 7 8 9 43, it seems reasonable that VH replacement of autoreactive BCR may be occurring in RA. However, we should stress that we do not know the antigenic reactivities of the B cell clones involved in the VH replacement events reported in this study.
However, it also seems reasonable that because receptor revision may be diversity (not tolerance) driven 56 78, it could lead to autoimmunity. Indeed, several recent studies support this notion 11 79. Most relevant is the study of Brard et al. documenting that anergic B cells from autoimmune prone mice could be activated to produce pathogenic autoantibodies after somatic mutation and receptor revision 11. The fact that the VH 1–69 gene, which appears to be a preferred donor in the VH replacement events detected in synovial tissue B cells, is frequently used to assemble RF 80 may support the notion that revision can inadvertently lead to autoreactivity.
The ectopic GC reactions that take place in the synovial tissues of RA patients may compound this tendency. As these GC reactions occur within a microenvironment that does not normally support B cell diversification, they may not provide the same types and/or relative quantities of diversification signals as natural GCs. This could lead to differences in the degrees that various diversification mechanisms are employed (e.g., somatic mutation versus receptor revision) or in their regulation (e.g., lack of occurrence or selection of replacement mutations at appropriate rates and locations that could lead to dysfunctional BCRs and/or to the initiation of autoreactivity). In the latter regard, the VH gene sequences derived from the synovial tissue B cells of our RA cases in many instances failed to exhibit the types of replacement to silent mutation ratios typically seen in antigen-selected immune reactions (Fig. 7 [81, 82]). Furthermore, they did not localize to RGYW motifs 83 with the expected frequencies (data not shown), a feature also seen in B lymphocytes from some patients with another autoimmune disorder, systemic lupus erythematosus 84. Thus, the nonphysiologic nature of the rheumatoid synovial GC–like environment or a primary defect in the mutation apparatus may support dysregulated B cell diversification events. These dysregulated events, combined with a defect in selection against autoreactivity, either genetic or acquired, and the provision of T cell help, might play an important role in the immunopathogenesis of autoimmune disorders such as RA.
Acknowledgments
The authors thank Drs. Martin Weigert and Michel Nussenzweig for their valuable discussions and Drs. Jack Silver, Charles C. Chu, and Peter K. Gregersen for their critical review of the manuscript.
This work was supported in part by U.S. Public Health Service grant AI10811 from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, by the Leonard Wagner Autoimmunity Research Fund, The Jerry and Cecile Shore Fund for Immunologic Research, and by the Richard and Nancy Leeds Fund of the Department of Medicine of North Shore University Hospital.
Footnotes
Abbreviations used in this paper: ds, double-stranded; FR, framework region; GC, germinal center; LM, ligation-mediated; RA, rheumatoid arthritis; RAG, recombinase activating gene; RF, rheumatoid factor; RSS, recognition signal sequence(s); RT, reverse transcription.
References
- Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Alt F.W., Oltz E.M., Young F., Gorman J., Taccioli G., Chen J. VDJ recombination. Immunol. Today. 1992;13:306–314. doi: 10.1016/0167-5699(92)90043-7. [DOI] [PubMed] [Google Scholar]
- Early P., Huang H., Davis M., Calame K., Hood L. An immunoglobulin heavy chain variable region gene is generated from three segments of DNAVH, D and JH. Cell. 1980;19:981–992. doi: 10.1016/0092-8674(80)90089-6. [DOI] [PubMed] [Google Scholar]
- Sakano H., Maki R., Kurosawa Y., Roeder W., Tonegawa S. Two types of somatic recombination are necessary for the generation of complete immunoglobulin heavy-chain genes. Nature. 1980;286:676–683. doi: 10.1038/286676a0. [DOI] [PubMed] [Google Scholar]
- Gellert M. Recent advances in understanding V(D)J recombination. Adv. Immunol. 1997;64:39–64. doi: 10.1016/s0065-2776(08)60886-x. [DOI] [PubMed] [Google Scholar]
- Gay D., Saunders T., Camper S., Weigert M. Receptor editingan approach by autoreactive B cells to escape tolerance. J. Exp. Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic M.Z., Erikson J., Litwin S., Weigert M. B lymphocytes may escape tolerance by revising their antigen receptors. J. Exp. Med. 1993;177:1165–1173. doi: 10.1084/jem.177.4.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiegs S.L., Russell D.M., Nemazee D. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Nagy Z., Prak E.L., Weigert M. Immunoglobulin heavy chain gene replacementa mechanism of receptor editing. Immunity. 1995;3:747–755. doi: 10.1016/1074-7613(95)90064-0. [DOI] [PubMed] [Google Scholar]
- Bertrand F.E., Golub R., Wu G.E. V(H) gene replacement occurs in the spleen and bone marrow of non-autoimmune quasi-monoclonal mice. Eur. J. Immunol. 1998;28:3362–3370. doi: 10.1002/(SICI)1521-4141(199810)28:10<3362::AID-IMMU3362>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Brard F., Shannon M., Prak E.L., Litwin S., Weigert M. Somatic mutation and light chain rearrangement generate autoimmunity in anti–single-stranded DNA transgenic MRL/lpr mice. J. Exp. Med. 1999;190:691–704. doi: 10.1084/jem.190.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wildt R.M., Hoet R.M.A., van Venrooij W.J., Tomlinson I.M., Winter G. Analysis of heavy and light chain pairings indicates that receptor editing shapes the human antibody repertoire. J. Mol. Biol. 1999;285:895–901. doi: 10.1006/jmbi.1998.2396. [DOI] [PubMed] [Google Scholar]
- Lopez-Macias C., Kalinke U., Cascalho M., Wabl M., Hengartner H., Zinkernagel R.M., Lamarre A. Secondary rearrangements and hypermutation generate sufficient B cell diversity to mount protective antiviral immunoglobulin responses. J. Exp. Med. 1999;189:1791–1798. doi: 10.1084/jem.189.11.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsoe G. Life and death in germinal centers (redux) Immunity. 1996;4:107–111. doi: 10.1016/s1074-7613(00)80675-5. [DOI] [PubMed] [Google Scholar]
- Wilson P., Liu Y.J., Banchereau J., Capra J.D., Pascual V. Amino acid insertions and deletions contribute to diversify the human Ig repertoire. Immunol. Rev. 1998;162:143–151. doi: 10.1111/j.1600-065x.1998.tb01437.x. [DOI] [PubMed] [Google Scholar]
- Goossens T., Klein U., Kuppers R. Frequent occurrence of deletions and duplications during somatic hypermutationimplications for oncogene translocations and heavy chain disease. Proc. Natl. Acad. Sci. USA. 1998;95:2463–2468. doi: 10.1073/pnas.95.5.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemazee D., Weigert M. Revising B cell receptors. J. Exp. Med. 2000;191:1813–1817. doi: 10.1084/jem.191.11.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson P.C., Wilson K., Liu Y.-J., Banchereau J., Pascual V., Capra J.D. Receptor revision of immunoglobulin heavy chain variable region genes in normal human B lymphocytes. J. Exp. Med. 2000;191:1881–1894. doi: 10.1084/jem.191.11.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris E.D., Jr. Rheumatoid arthritis. Pathophysiology and implications for therapy. N. Engl. J. Med. 1990;322:1277–1289. doi: 10.1056/NEJM199005033221805. [DOI] [PubMed] [Google Scholar]
- Feldmann M., Brennan F.M., Maini R.N. Rheumatoid arthritis. Cell. 1996;85:307–310. doi: 10.1016/s0092-8674(00)81109-5. [DOI] [PubMed] [Google Scholar]
- Good R., Mazzitello W. The simultaneous occurrence of rheumatoid arthritis and agammaglobulinemia. J. Lab. Clin. Med. 1957;49:343–357. [PubMed] [Google Scholar]
- Rawson A.J., Hollander J.L., Quismorio F.P., Abelson N.M. Experimental arthritis in man and rabbit dependent upon serum anti-immunoglobulin factors. Ann. NY Acad. Sci. 1969;168:188–194. doi: 10.1111/j.1749-6632.1969.tb43107.x. [DOI] [PubMed] [Google Scholar]
- Holmdahl R., Vingsbo C., Mo J.A., Michaelsson E., Malmstrom V., Jansson L., Brunsberg U. Chronicity of tissue-specific experimental autoimmune diseasea role for B cells? Immunol. Rev. 1995;144:109–135. doi: 10.1111/j.1600-065x.1995.tb00067.x. [DOI] [PubMed] [Google Scholar]
- Zhao Y.X., Abdelnour A., Holmdahl R., Tarkowski A. Mice with the xid B cell defect are less susceptible to developing Staphylococcus aureus-induced arthritis. J. Immunol. 1995;155:2067–2076. [PubMed] [Google Scholar]
- Taylor P.C., Plater-Zyberk C., Maini R.N. The role of the B cells in the adoptive transfer of collagen-induced arthritis from DBA/1 (H-2q) to SCID (H-2d) mice. Eur. J. Immunol. 1995;25:763–769. doi: 10.1002/eji.1830250321. [DOI] [PubMed] [Google Scholar]
- Waaler E. On the occurrence of a factor in human serum activating the specific agglutination of sheep blood corpuscles. Acta Pathol. Microbiol. Scand. 1940;17:172–188. doi: 10.1111/j.1600-0463.2007.apm_682a.x. [DOI] [PubMed] [Google Scholar]
- Rose H.M., Ragan C., Pearce E., Lipman M.O. Differential agglutination of normal and sensitized sheep erythrocytes by sera of patients with rheumatoid arthritis. Proc. Soc. Exp. Biol. Med. 1948;68:1–6. doi: 10.3181/00379727-68-16375. [DOI] [PubMed] [Google Scholar]
- Clague R.B., Morgan K., Reynolds I., Williams H.J. The prevalence of serum IgG antibodies to type II collagen in American patients with rheumatoid arthritis. Br. J. Rheumatol. 1994;33:336–338. doi: 10.1093/rheumatology/33.4.336. [DOI] [PubMed] [Google Scholar]
- Snowden N., Reynolds I., Morgan K., Holt L. T cell responses to human type II collagen in patients with rheumatoid arthritis and healthy controls. Arthritis Rheum. 1997;40:1210–1218. doi: 10.1002/1529-0131(199707)40:7<1210::AID-ART4>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Schellekens G.A., de Jong B.A., van den Hoogen F.H., van de Putte L.B., van Venrooij W.J. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J. Clin. Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips P.E. Evidence implicating infectious agents in rheumatoid arthritis and juvenile rheumatoid arthritis. Clin. Exp. Rheumatol. 1988;6:87–94. [PubMed] [Google Scholar]
- Kouri T., Petersen J., Rhodes G., Aho K., Palosuo T., Heliovaara M., Isomaki H., von Essen R., Vaughan J.H. Antibodies to synthetic peptides from Epstein-Barr nuclear antigen-1 in sera of patients with early rheumatoid arthritis and in preillness sera. J. Rheumatol. 1990;17:1442–1449. [PubMed] [Google Scholar]
- Hajeer A.H., MacGregor A.J., Rigby A.S., Ollier W.E., Carthy D., Silman A.J. Influence of previous exposure to human parvovirus B19 infection in explaining susceptibility to rheumatoid arthritisan analysis of disease discordant twin pairs. Ann. Rheum. Dis. 1994;53:137–139. doi: 10.1136/ard.53.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouskoff V., Korganow A.S., Duchatelle V., Degott C., Benoist C., Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- Korganow A.S., Ji H., Mangialaio S., Duchatelle V., Pelanda R., Martin T., Degott C., Kikutani H., Rajewsky K., Pasquali J.L., Benoist C., Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- Matsumoto I., Staub A., Benoist C., Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- Randen I., Mellbye O.J., Forre O., Natvig J.B. The identification of germinal centres and follicular dendritic cell networks in rheumatoid synovial tissue. Scand. J. Immunol. 1995;41:481–486. doi: 10.1111/j.1365-3083.1995.tb03596.x. [DOI] [PubMed] [Google Scholar]
- Schroder A.E., Greiner A., Seyfert C., Berek C. Differentiation of B cells in the nonlymphoid tissue of the synovial membrane of patients with rheumatoid arthritis. Proc. Natl. Acad. Sci. USA. 1996;93:221–225. doi: 10.1073/pnas.93.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfield R., Hardy R.R., Tarlinton D., Dangl J., Herzenberg L.A., Weigert M. Recombination between an expressed immunoglobulin heavy-chain gene and a germline variable gene segment in a Ly 1+ B-cell lymphoma. Nature. 1986;322:843–846. doi: 10.1038/322843a0. [DOI] [PubMed] [Google Scholar]
- Reth M., Gehrmann P., Petrac E., Wiese P. A novel VH to VHDJH joining mechanism in heavy-chain-negative (null) pre-B cells results in heavy-chain production. Nature. 1986;322:840–842. doi: 10.1038/322840a0. [DOI] [PubMed] [Google Scholar]
- Kleinfield R.W., Weigert M.G. Analysis of VH gene replacement events in a B cell lymphoma. J. Immunol. 1989;142:4475–4482. [PubMed] [Google Scholar]
- Covey L.R., Ferrier P., Alt F.W. VH to VHDJH rearrangement is mediated by the internal VH heptamer. Int. Immunol. 1990;2:579–583. doi: 10.1093/intimm/2.6.579. [DOI] [PubMed] [Google Scholar]
- Cascalho M., Wong J., Wabl M. VH gene replacement in hyperselected B cells of the quasimonoclonal mouse. J. Immunol. 1997;159:5795–5801. [PubMed] [Google Scholar]
- Deane M., Norton J.D. Immunoglobulin heavy chain gene rearrangement involving V-V region recombination. Nucleic Acids Res. 1990;18:1652. doi: 10.1093/nar/18.6.1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokaw J.L., Wetzel S.M., Pollok B.A. Conserved patterns of somatic mutation and secondary VH gene rearrangement create aberrant Ig-encoding genes in Epstein-Barr virus-transformed and normal human B lymphocytes. Int. Immunol. 1992;4:197–206. doi: 10.1093/intimm/4.2.197. [DOI] [PubMed] [Google Scholar]
- Choi Y., Greenberg S.J., Du T.L., Ward P.M., Overturf P.M., Brecher M.L., Ballow M. Clonal evolution in B-lineage acute lymphoblastic leukemia by contemporaneous VH-VH gene replacements and VH-DJH gene rearrangements. Blood. 1996;87:2506–2512. [PubMed] [Google Scholar]
- Itoh K., Furie R., Chartash E., Jain R., Chiorazzi N. Rheumatoid arthritis synovial B cells exhibit novel and unexpected features of the immunoglobulin V gene mutation process. Arthritis Rheum. 1999;42:S85. [Google Scholar]
- Arnett F.C., Edworthy S.M., Bloch D.A., McShane D.J., Fries J.F., Cooper N.S., Healey L.A., Kaplan S.R., Liang M.H., Luthra H.S. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Fais F., Ghiotto F., Hashimoto S., Sellars B., Valetto A., Allen S.L., Schulman P., Vinciguerra V.P., Rai K., Rassenti L.Z. Chronic lymphocytic leukemia B cells express restricted sets of mutated and unmutated antigen receptors. J. Clin. Invest. 1998;102:1515–1525. doi: 10.1172/JCI3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fais F., Sellars B., Ghiotto F., Yan X.J., Dono M., Allen S.L., Budman D., Dittmar K., Kolitz J., Lichtman S.M. Examples of in vivo isotype class switching in IgM+ chronic lymphocytic leukemia B cells. J. Clin. Invest. 1996;98:1659–1666. doi: 10.1172/JCI118961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane M., Norton J.D. Immunoglobulin gene ‘fingerprinting’an approach to analysis of B lymphoid clonality in lymphoproliferative disorders. Br. J. Haematol. 1991;77:274–281. doi: 10.1111/j.1365-2141.1991.tb08570.x. [DOI] [PubMed] [Google Scholar]
- Itoh K., Chiorazzi N. Clonal expansion is a characteristic feature of the B-cell repertoire of patients with rheumatoid arthritis. Arthritis Res. 2000;2:50–58. doi: 10.1186/ar68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse M., Gallagher P., Wilson A., Wu L., Fisicaro N., Miller J.F., Scollay R., Shortman K. Molecular characterization of T-cell antigen receptor expression by subsets of CD4− CD8− murine thymocytes. Proc. Natl. Acad. Sci. USA. 1988;85:6082–6086. doi: 10.1073/pnas.85.16.6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papavasiliou F., Casellas R., Suh H., Qin X.-F., Besmer E., Pelanda R., Nemazee D., Rajewsky K., Nussenzweig M. V(D)J recombination in mature B cellsa mechanism for altering antibody responses. Science. 1997;278:298–300. doi: 10.1126/science.278.5336.298. [DOI] [PubMed] [Google Scholar]
- Schlissel M., Constantinescu A., Morrow T., Baxter M., Peng A. Double-strand signal sequence breaks in V(D)J recombination are blunt, 5′-phosphorylated, RAG-dependent, and cell cycle regulated. Genes Dev. 1993;7:2520–2532. doi: 10.1101/gad.7.12b.2520. [DOI] [PubMed] [Google Scholar]
- Meffre E., Papavasiliou F., Cohen P., de Bouteiller O., Bell D., Karasuyama H., Schiff C., Banchereau J., Liu Y.J., Nussenzweig M.C. Antigen receptor engagement turns off the V(D)J recombination machinery in human tonsil B cells. J. Exp. Med. 1998;188:765–772. doi: 10.1084/jem.188.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges S.L., Jr., Lee S.K., Koopman W.J., Schroeder H.W., Jr. Analysis of immunoglobulin gamma heavy chain expression in synovial tissue of a patient with rheumatoid arthritis. Arthritis Rheum. 1993;36:631–641. [PubMed] [Google Scholar]
- Berek C., Griffiths G.M., Milstein C. Molecular events during maturation of the immune response to oxazolone. Nature. 1985;316:412–418. doi: 10.1038/316412a0. [DOI] [PubMed] [Google Scholar]
- Bridges S.L., Jr., Clausen B.E., Lavelle J.C., Fowler P.G., Koopman W.J., Schroeder H.W., Jr. Analysis of immunoglobulin gamma heavy chains from rheumatoid arthritis synovium. Evidence of antigen-driven selection. Ann. NY Acad. Sci. 1995;764:450–452. doi: 10.1111/j.1749-6632.1995.tb55862.x. [DOI] [PubMed] [Google Scholar]
- Radic M.Z., Zouali M. Receptor editing, immune diversification, and self-tolerance. Immunity. 1996;5:505–511. doi: 10.1016/s1074-7613(00)80266-6. [DOI] [PubMed] [Google Scholar]
- Usuda S., Takemori T., Matsuoka M., Shirasawa T., Yoshida K., Mori A., Ishizaka K., Sakano H. Immunoglobulin V gene replacement is caused by the intramolecular DNA deletion mechanism. EMBO (Eur. Mol. Biol. Organ.) J. 1992;11:611–618. doi: 10.1002/j.1460-2075.1992.tb05093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley M.E., Rapalus L., Boylin E.C., Rohrer J., Minegishi Y. Gene conversion events contribute to the polymorphic variation of the surrogate light chain gene lambda 5/14.1. Clin. Immunol. 1999;93:162–167. doi: 10.1006/clim.1999.4785. [DOI] [PubMed] [Google Scholar]
- Reynaud C.A., Anquez V., Grimal H., Weill J.C. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- Thompson C.B., Neiman P.E. Somatic diversification of the chicken immunoglobulin light chain gene is limited to the rearranged variable gene segment. Cell. 1987;48:369–378. doi: 10.1016/0092-8674(87)90188-7. [DOI] [PubMed] [Google Scholar]
- Becker R.S., Knight K.L. Somatic diversification of immunoglobulin heavy chain VDJ genesevidence for somatic gene conversion in rabbits. Cel. 1990;63:987–997. doi: 10.1016/0092-8674(90)90502-6. [DOI] [PubMed] [Google Scholar]
- Schiaffella E., Sehgal D., Anderson A.O., Mage R.G. Gene conversion and hypermutation during diversification of VH sequences in developing splenic germinal centers of immunized rabbits. J. Immunol. 1999;162:3984–3995. [PubMed] [Google Scholar]
- Hesse J.E., Lieber M.R., Mizuuchi K., Gellert M. V(D)J recombinationa functional definition of the joining signals. Genes Dev. 1989;3:1053–1061. doi: 10.1101/gad.3.7.1053. [DOI] [PubMed] [Google Scholar]
- Bogue M.A., Wang C., Zhu C., Roth D.B. V(D)J recombination in Ku86-deficient micedistinct effects on coding, signal, and hybrid joint formation. Immunity. 1997;7:37–47. doi: 10.1016/s1074-7613(00)80508-7. [DOI] [PubMed] [Google Scholar]
- Han J.O., Steen S.B., Roth D.B. Ku86 is not required for protection of signal ends or for formation of nonstandard V(D)J recombination products. Mol. Cell. Biol. 1997;17:2226–2234. doi: 10.1128/mcb.17.4.2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brezinschek H.P., Foster S.J., Brezinschek R.I., Dorner T., Domiati-Saad R., Lipsky P.E. Analysis of the human VH gene repertoire. Differential effects of selection and somatic hypermutation on human peripheral CD5+/IgM+ and CD5−/IgM+ B cells. J. Clin. Invest. 1997;99:2488–2501. doi: 10.1172/JCI119433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T., Minami Y., Sakato N., Sugiyama H. Biased usage of two restricted VH gene segments in VH replacement. Eur. J. Immunol. 1993;23:517–522. doi: 10.1002/eji.1830230233. [DOI] [PubMed] [Google Scholar]
- Arpin C., de Bouteiller O., Razanajaona D., Fugier-Vivier I., Briere F., Banchereau J., Lebecque S., Liu Y.J. The normal counterpart of IgD myeloma cells in germinal center displays extensively mutated IgVH gene, Cmu-Cdelta switch, and lambda light chain expression. J. Exp. Med. 1998;187:1169–1178. doi: 10.1084/jem.187.8.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K.P., Kuhn R., Rajewsky K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell. 1997;90:1073–1083. doi: 10.1016/s0092-8674(00)80373-6. [DOI] [PubMed] [Google Scholar]
- Meffre E., Davis E., Schiff C., Cunningham-Rundles C., Ivashkiv L.B., Staudt L.M., Young J.W., Nussenzweig M.C. Circulating human B cells that express surrogate light chains and edited receptors. Nat. Immunol. 2000;1:207–214. doi: 10.1038/79739. [DOI] [PubMed] [Google Scholar]
- Randen I., Brown D., Thompson K.M., Hughes-Jones N., Pascual V., Victor K., Capra J.D., Forre O., Natvig J.B. Clonally related IgM rheumatoid factors undergo affinity maturation in the rheumatoid synovial tissue. J. Immunol. 1992;148:3296–3301. [PubMed] [Google Scholar]
- Tuomi T., Aho K., Palosuo T., Kaarela K., von Essen R., Isomaki H., Leirisalo-Repo M., Sarna S. Significance of rheumatoid factors in an eight-year longitudinal study on arthritis. Rheumatol. Int. 1988;8:21–26. doi: 10.1007/BF00541346. [DOI] [PubMed] [Google Scholar]
- Munthe E., Natvig J.B. Immunoglobulin classes, subclasses and complexes of IgG rheumatoid factor in rheumatoid plasma cells. Clin. Exp. Immunol. 1972;12:55–70. [PMC free article] [PubMed] [Google Scholar]
- Hertz M., Kouskoff V., Nakamura T., Nemazee D. V(D)J recombinase induction in splenic B lymphocytes is inhibited by antigen-receptor signalling. Nature. 1998;394:292–295. doi: 10.1038/28419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorner T., Foster S.J., Farner N.L., Lipsky P.E. Immunoglobulin kappa chain receptor editing in systemic lupus erythematosus. J. Clin. Invest. 1998;102:688–694. doi: 10.1172/JCI3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.P., Silverman G.J., Liu M.F., Carson D.A. Idiotypic and molecular characterization of human rheumatoid factors. Chem. Immunol. 1990;48:63–81. [PubMed] [Google Scholar]
- Jukes T.H., King J.L. Evolutionary nucleotide replacements in DNA. Nature. 1979;281:605–606. doi: 10.1038/281605a0. [DOI] [PubMed] [Google Scholar]
- Shlomchik M.J., Marshak-Rothstein A., Wolfowicz C.B., Rothstein T.L., Weigert M.G. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805–811. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- Rogozin I.B., Sredneva N.E., Kolchanov N.A. Somatic hypermutagenesis in immunoglobulin genes. III. Somatic mutations in the chicken light chain locus. Biochim. Biophys. Acta. 1996;1306:171–178. doi: 10.1016/0167-4781(95)00241-3. [DOI] [PubMed] [Google Scholar]
- Manheimer-Lory A.J., Zandman-Goddard G., Davidson A., Aranow C., Diamond B. Lupus-specific antibodies reveal an altered pattern of somatic mutation. J. Clin. Invest. 1997;100:2538–2546. doi: 10.1172/JCI119796. [DOI] [PMC free article] [PubMed] [Google Scholar]