

Abstract
Syncytia arising from the fusion of cells expressing a lymphotropic HIV type 1–encoded envelope glycoprotein complex (Env) with cells expressing the CD4/CXC chemokine receptor 4 complex spontaneously undergo cell death. Here we show that this process is accompanied by caspase activation and signs of mitochondrial membrane permeabilization (MMP), including the release of intermembrane proteins such as cytochrome c (Cyt-c) and apoptosis-inducing factor (AIF) from mitochondria. In Env-induced syncytia, caspase inhibition did not suppress AIF- and Cyt-c translocation, yet it prevented all signs of nuclear apoptosis. Translocation of Bax to mitochondria led to MMP, which was inhibited by microinjected Bcl-2 protein or bcl-2 transfection. Bcl-2 also prevented the subsequent nuclear chromatin condensation and DNA fragmentation. The release of AIF occurred before that of Cyt-c and before caspase activation. Microinjection of AIF into syncytia sufficed to trigger rapid, caspase-independent Cyt-c release. Neutralization of endogenous AIF by injection of an antibody prevented all signs of spontaneous apoptosis occurring in syncytia, including the Cyt-c release and nuclear apoptosis. In contrast, Cyt-c neutralization only prevented nuclear apoptosis, and did not affect AIF release. Our results establish that the following molecular sequence governs apoptosis of Env-induced syncytia: Bax-mediated/Bcl-2–inhibited MMP → AIF release → Cyt-c release → caspase activation → nuclear apoptosis.
Keywords: apoptosis-inducing factor, Bcl-2, cell death, cytochrome c, glycoprotein 120
Introduction
Infection by HIV-1 is accompanied by an increased apoptotic turnover of lymphocytes, monocytes, and neurons that can be detected either ex vivo in freshly explanted cells, or in vivo by histocytochemical detection of apoptotic cells. The mechanisms of this increased cellular demise are complex and involve direct effects of the virus and viral products, as well as indirect host-mediated factors 1 2. In vitro, HIV-1 infection can cause apoptosis via a multitude of different mechanisms, including the action of the proapoptotic proteins Tat 3 4 and Vpr 5 6 7 and, perhaps more importantly, via interactions between the glycoprotein (gp)120/gp41 envelope complex (Env) with its receptor (CD4) and a suitable coreceptor (e.g., CXC chemokine receptor [CXCR]4). In cultures of T cells inoculated with lymphotropic HIV-1 strains, Env expressed on the plasma membrane of infected cells interacts with CD4/CXCR4 of uninfected cells, resulting in cell fusion 8 9 10. After several rounds of fusion, syncytia attain volumes equivalent to several dozens or hundreds of individual cells and ultimately lyse, while exhibiting several biochemical characteristics of apoptosis and/or signs of necrosis such as cytoplasmic vacuolization 11 12 13 14. According to several studies, syncytium formation is the principal cause of HIV-1–mediated T cell destruction in vitro 11 15. Env variants interacting with CD4/CXCR4 (rather than those having a preference for CD4/CCR5) are mostly encoded by syncytium-inducing HIV-1 strains, and a strong correlation between CD4+ T cell decline and infection by syncytium-inducing HIV-1 variants has been established by some authors in vitro and in vivo 8 9 15 16.
Apoptosis of single cells is generally associated with the activation of caspases 17, a set of specific proteases which either can serve as molecular switches initiating cell death pathways or, when activated in a massive fashion, can mediate the degradation of essential structural and regulatory proteins, culminating in the activation of the caspase-activated DNase, the enzyme responsible for oligonucleosomal DNA fragmentation 18 19. In addition, apoptosis is accompanied by signs of mitochondrial membrane permeabilization (MMP), including a loss of the inner mitochondrial transmembrane potential (ΔΨm) and the release of soluble intermembrane proteins via the outer mitochondrial membrane 20 21 22 23 24. The functional hierarchy among these events depends on the apoptosis-inducing trigger. In the “extrinsic” pathway, caspase activation is induced as an upstream event, e.g., by recruiting apical caspases to the death-induced signaling complex formed upon the ligation of death receptors expressed on the cell surface (e.g., CD95 and TNFR). Depending on the cell type, receptor-proximal caspase activation then either suffices to set off the caspase activation cascade in an MMP-independent fashion (“type 1 cells”) or must relay to MMP via cleavage of Bid and/or production of ceramide (“type 2 cells” 25 26 27). In contrast, most “intrinsic” apoptosis triggers (e.g., DNA damage, glucocorticoids, and reactive oxygen species) activate caspases in an indirect fashion, namely by first permeabilizing mitochondrial membranes in a Bcl-2–inhibitable fashion 20 21 22 23 28 29. MMP results in the translocation of cytochrome c (Cyt-c) from the mitochondrial intermembrane space to the cytosol, where Cyt-c triggers the assembly of a caspase-9–caspase-3 activation complex, the apoptosome 18 21. In addition to Cyt-c, mitochondria can release a variety of intermembrane proteins 30, including a cell type–specific set of procaspases 31 32 33 and the apoptosis-inducing factor (AIF 34), a flavoprotein oxidoreductase that translocates to the cytosol as well as to the nucleus and stimulates apoptosis via an as yet unknown, caspase-independent mechanism.
As in other models of apoptosis induction, signaling via CD4/CXCR4 induces both caspase activation 35 36 37 38 39 and signs of MMP 14 40. However, the precise mechanisms of Env-mediated apoptosis induction and the molecular order of these events have remained elusive in syncytia. Stimulated by these premises, we studied the role of caspases and MMP in syncytia arising from the fusion between Env- and CD4/CXCR4-expressing cells. Here we report that the death of Env-induced syncytia obeys the rules of the intrinsic pathway of apoptosis induction in the sense that caspase activation occurs downstream of MMP. Our results indicate that MMP is regulated by members of the Bcl-2/Bax family and establish a molecular order of apoptotic signaling in which AIF release occurs upstream of that of Cyt-c, which in turn is required for caspase-dependent nuclear apoptosis.
Materials and Methods
Cells and Culture Conditions.
HeLa cells stably transfected with a vector containing the env gene of HIV-1 LAI (HeLa 243 Env 41) were cultured in complete culture medium (DMEM supplemented with 2 mM glutamine, 10% FCS, 1 mM pyruvate, 10 mM Hepes, and 100 U/ml penicillin/streptomycin) containing 2 μM methotrexate. HeLa cells stably transfected with CD4 (HeLa P4; a gift from P. Charneau, Pasteur Institute, Paris, France 42) were selected in medium containing 500 μg/ml G418. Jurkat cells expressing CD4 and CXCR4 and stably transfected with the human Bcl-2 gene or a neomycin (Neo) resistance vector 43 only were provided by N. Israel (Pasteur Institute, Paris, France). Neo and Bcl-2 U937 cells 44 were a gift from F. Hirsch (Centre National de la Recherche Scientifique, ERS1984, Villejuif, France). Cocultures of different cell types were performed in complete culture medium in the absence of selecting antibiotics by adding trypsinized HeLa cells to adherent HeLa CD4 or HeLa Env cells (density: 1–1.5 × 103 cells/mm2) at an ∼1:1 ratio or by adding Jurkat or U937 cells to adherent HeLa Env cells (National Institutes of Health AIDS Research and Reference Reagent Program, Bethesda, MD). Apoptosis was induced or inhibited by staurosporin (2 μM; Sigma-Aldrich), N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD. fmk), Boc-Asp-fluoromethylketone (Boc-D.fmk), or N-benzyloxycarbonyl-Phe-Ala-fluoromethylketone (Z-FA.fmk) (all used at 100 μM added every 24 h; Enzyme Systems).
HIV-1 Infection.
5 × 106 HeLa CD4 cells were cultured in the presence of 2.5 × 106 chronically HIV-1–infected H9/IIIB cells obtained from Dr. R.C. Gallo (National Institutes of Health, Bethesda, MD) in 5 ml of RPMI 1640 supplemented with 10% heat-inactivated FCS, 2 mM glutamine, and 25 IU/ml recombinant human IL-2. Before the addition of chronically infected cells, target cells were preincubated for 30 min at 37°C in the presence or absence of Z-VAD.fmk (50 μM), and Z-VAD.fmk was readded every 12 h throughout the culture period.
Fluorescence Staining of Live Cells and Immunofluorescence.
For the assessment of mitochondrial and nuclear features of apoptosis, cells cultured on a coverslip were stained with 5,5′,6,6′-tetrachloro-1,1′, 3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1, 2 μM; Molecular Probes) and Hoechst 33342 (2 μM; Sigma-Aldrich) for 30 min at 37°C in complete culture medium. A rabbit antiserum generated against a mixture of three peptides derived from the mouse AIF amino acid (aa) sequence (aa 151–170, 166–185, 181–200, coupled to KLH 34) was used (diluted 1:1,000) on paraformaldehyde (4% wt/vol) and picric acid–fixed (0.19% vol/vol) cells and revealed with a goat anti–rabbit IgG conjugated to PE (Southern Biotechnology Associates, Inc.). Cells were also stained for the detection of Cyt-c (mAb 6H2.B4 from BD PharMingen; revealed by a goat anti–mouse IgG1 FITC conjugate from Southern Biotechnology Associates, Inc.), heat shock protein (hsp)60 (mAb H4149 from Sigma-Aldrich; revealed by a goat anti–mouse IgG1 FITC), Cyt-c oxidase (COX subunit IV, mAb 2038C12 from BD PharMingen; revealed by a goat anti–mouse IgG2a FITC conjugate), Bax (66241A; BD PharMingen), and/or chromatin (Hoechst 33342, 2 μM, 15 min of incubation at room temperature). Several stages of nuclear apoptosis were distinguished by staining with Hoechst 33342: stage I with rippled nuclear contours and a rather partial chromatin condensation, stage IIa with marked peripheral chromatin condensation, and stage IIb with formation of nuclear bodies 45. A rabbit polyclonal antiserum, CM1 (detected as for anti-AIF), which recognizes the p18 subunit of cleaved caspase-3 but not the zymogen 46, was employed to detect the proteolytic activation of caspase-3, followed by detection of the fluorescence intensity by confocal microscopy.
DNA Gel Electrophoresis.
For pulse field gel electrophoresis, DNA was prepared from agarose plugs (2 × 106 nuclei 47), followed by electrophoresis in a Bio-Rad Laboratories CHEF-DR II (1% agarose, TBE, 200 V, 24 h, pulse wave 60 s, 120° angle).
Subcellular Fractionation and Immunoblotting.
Cells were washed once in PBS, resuspended in isotonic HIM buffer (200 mM mannitol, 70 mM sucrose, 1 mM EGTA, 10 mM Hepes, pH 7.5) supplemented with a protease inhibitor mixture (added at a 1:100 dilution; Sigma-Aldrich), and homogenized using a Polytron homogenizer (Brinkmann Instruments) at setting 6.5 for 10 s. Nuclei were separated at 600 g for 10 min as the low speed pellet and washed twice (1,200 g, 10 min). The supernatant was centrifuged at 10,000 g for 10 min to collect the heavy membrane pellet enriched in mitochondria. This supernatant was centrifuged at 100,000 g for 30 min to yield the organelle-free cytosols. Samples were stored at −80°C until analysis by SDS-PAGE under reducing conditions (40 μg protein/lane), Western blot, and immunodetection of AIF and Cyt-c as described 34.
Microinjection.
All syncytia growing on a premarked V-shaped area of a coverslip (>200 per experiment) were microinjected into the cytoplasm (1–3 injections per syncytium depending on their size, 1 injection per ∼4 nuclei) using a computer-controlled microinjector (pressure 200 hPa, 2 s; Eppendorf) with PBS only (pH 7.2), recombinant AIF protein (500 ng/μl 34), horse Cyt-c (12 μg/μl; Sigma-Aldrich), a neutralizing anti-AIF rabbit Ab (titer ∼105 34; diluted 1:1 with PBS) optionally neutralized by preincubation with 10 μM of AIF immunogenic peptides, a preimmune rabbit antiserum, a neutralizing Cyt-c–specific IgG1 mAb (6H2.B4, 250 ng/μl; BD PharMingen), an irrelevant isotype-matched control mAb (anti-hsp60), Koenig's polyanion (2.5 μM; a gift from Dieter Brdizcka, University of Konstanz, Konstanz, Germany), recombinant human Bcl-2 (aa 1–218, 500 ng/μl), Bcl-2Δα5/6 (Bcl-2 Δ143–184, 500 ng/μl), or murine Bax (aa 1–171, 250 ng/μl), BaxΔα5/6 (Δ106–153, 250 ng/μl 48). After microinjection, cells were cultured for 3–24 h and stained for 30 min with the ΔΨm-sensitive dye JC-1 (2 μM) and the DNA-intercalating dye Hoechst 33342 (2 μM), followed by fixation (which removes the JC-1 staining) and immunostaining for Cyt-c and/or AIF as described above.
Data Analysis.
The quantitation of different parameters by fluorescence microscopy was performed on at least 200 syncytia for each data point, and was repeated at least 3 times in independent experiments, as stated in the figure legends. In at least one experiment out of each series, quantitations were performed in a blinded fashion, and in an additional experiment quantitations were performed independently by two individuals. Interexperimental variability was generally <15%.
Results
Caspase-dependent Nuclear Apoptosis of Syncytia Induced by the Interaction between Env- and CD4-expressing Cells.
HeLa cells stably transfected with human CD4 (HeLa CD4) formed syncytia when cocultured with HeLa cells expressing a lymphotropic HIV-1 Env gene (HeLa Env 42; Fig. 1). After 24 h of coculture, several morphologically normal nuclei (detected with Hoechst 33342, blue fluorescence) could be clearly distinguished within a common cytoplasm of HeLa CD4/HeLa Env hybrids (Fig. 1 A). However, after prolonged culture (48–72 h) an increasing percentage of nuclei manifested apoptotic chromatin condensation (Fig. 1A and Fig. B). This chromatin condensation was restricted to syncytia, and inhibition of cell fusion by addition of the anti-CD4 mAb Leu3A at the beginning of coculture prevented all signs of apoptosis (Fig. 1 A). As in other models of apoptosis 45, chromatin condensation rapidly evolved to a stage with strong Hoechst 33342–detectable condensation of most of the chromatin (stage II), without (stage IIa, in a minority of syncytia) or with the formation of nuclear apoptotic bodies (stage IIb, in most syncytia) (Fig. 1 B). Nuclear apoptosis attained all nuclei within the same heterokaryon in a coordinated fashion (Fig. 1 A), and was accompanied by oligonucleosomal DNA degradation (not shown) as well as by “large scale” (∼50 kbp) DNA fragmentation (Fig. 1 D). All morphological and biochemical signs of nuclear apoptosis were strongly reduced by the two pancaspase inhibitors Boc-D.fmk and Z-VAD.fmk, but not by the chemically related cathepsin inhibitor Z-FA.fmk (Fig. 1 A). Only a minority of cells exhibited a partial peripheral chromatin condensation (stage I) in the presence of caspase inhibitors (Fig. 1 B). In conclusion, Env-induced syncytia spontaneously undergo caspase-dependent nuclear apoptosis.
Figure 1.
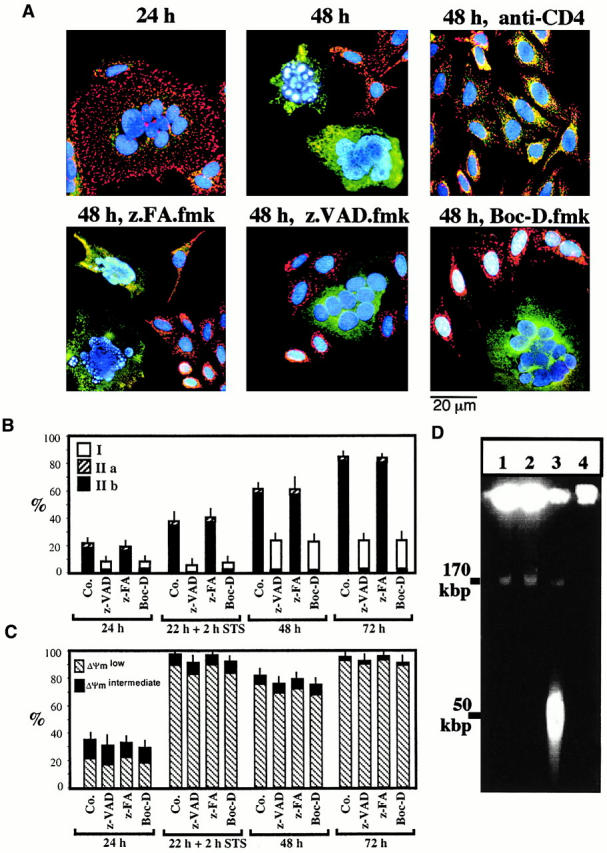
The effect of caspase inhibitors on nuclear apoptosis and ΔΨm collapse induced by the interaction between Env and CD4. (A) Fluorescence micrographs of HeLa Env cells and HeLa CD4 cells cocultured at a 1:1 ratio for the indicated period (24 or 48 h), in the absence or presence of Boc-D.fmk, Z-VAD.fmk, Z-FA.fmk, or anti-CD4 Ab. Cells were stained with Hoechst 33342 (blue fluorescence) and the ΔΨm-sensitive dye JC-1 (red fluorescence of mitochondria with a high ΔΨm, green fluorescence of mitochondria with a low ΔΨm). Syncytia representing the dominant phenotype (>50% of cells) are shown. (B) Frequency of syncytia with chromatin condensation after various periods of coculture (24, 48, or 72 h). As a positive control of apoptosis induction, the apoptosis inducer staurosporin (STS) was added to 22-h-old syncytia for 2 h. The stage of chromatin condensation (I, IIa, or IIb) was determined as described in Materials and Methods and reference 45. Co., control. (C) Kinetic analysis of ΔΨm loss. The frequency of cells exhibiting a homogeneously ΔΨm high, ΔΨm low, or an intermediate phenotype (ΔΨm intermediate, red JC-1 fluorescence in peripheral mitochondria, green fluorescence in perinuclear mitochondria) was determined. Results are the means of 7 independent experiments (mean ± SEM) in which at least 200 cells were counted for each data point. (D) DNA fragmentation induced by the coculture of HeLa CD4 and HeLa Env cells. Cells were either cultured individually (lane 1, HeLa CD4; lane 2, HeLa Env) or cocultured to allow for the generation of syncytia in the absence (lane 3) or presence of Z-VAD.fmk (lane 4). Syncytia were recovered after 72 h of culture, followed by pulse field gel electrophoresis of nuclear DNA.
Caspase-independent Signs of MMP of Env-induced Syncytia.
Staining of Env-induced syncytia with the ΔΨm-sensitive dye JC-1 revealed a progressive ΔΨm loss. Thus, mitochondria from most newly formed syncytia (24 h) possessed a high ΔΨm (red JC-1 fluorescence; Fig. 1 A), whereas mitochondria from aging syncytia (48–72 h) mostly have a low ΔΨm (green JC-1 fluorescence; Fig. 1A and Fig. C). The ΔΨm loss progressed from the perinuclear area to the periphery (not shown), giving rise to a transient intermediate phenotype (Fig. 1 C), and was accompanied by a moderate perinuclear clustering of mitochondria (stained for the matrix protein hsp60, which is not released during apoptosis 30, or the sessile inner membrane protein COX; green fluorescence in Fig. 2 A and Fig. 3 A, respectively). In addition, syncytia progressively manifested signs of outer MMP, as indicated by the translocation of AIF (red fluorescence in Fig. 2 A) from mitochondria to the cytosol and to the nucleus (blue fluorescence in Fig. 2 A and Fig. 3 A), or that of Cyt-c to the cytosol (red fluorescence in Fig. 3 A). The translocation of AIF and Cyt-c from mitochondria to the extramitochondrial compartment was confirmed by subcellular fractionation followed by immunoblot. In single cells or 24-h-old syncytia, AIF and Cyt-c are confined to the mitochondrial compartment. 48 h after initiation of coculture, ectopic AIF becomes detectable in both cytosols and nuclei (Fig. 2 C), whereas ectopic Cyt-c can be detected only in the cytosol (Fig. 3 C). Neither Boc-D.fmk nor Z-VAD.fmk (added every 24 h at a concentration of 100 μM) prevented the mitochondrial manifestations of apoptosis (ΔΨm collapse, AIF and Cyt-c translocation; Fig. 1 Fig. 2 Fig. 3). Hence, MMP proceeds in a caspase-independent fashion in HIV-1–induced syncytia.
Figure 2.
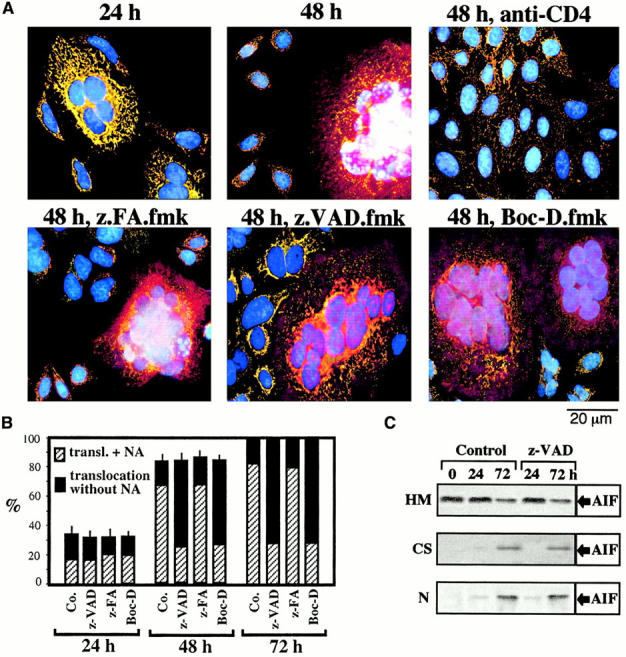
Caspase-independent AIF translocation in Env/CD4-induced syncytia. (A) Syncytia at different stages of coculture (performed as described in the legend to Fig. 1) were fixed and permeabilized, followed by immunostaining with Abs specific for AIF (revealed by PE, red fluorescence) and the mitochondrial matrix protein hsp60 (green fluorescence), as well as counterstaining with Hoechst 33342 (blue fluorescence). Note that AIF and hsp60 are colocalized (yellow punctate fluorescence, blend of green plus red) in 24-h syncytia, as well as in individual cells in which cell fusion was blocked by the anti-CD4 Leu3A mAb. Diffuse red cytoplasmic fluorescence and purple nuclear fluorescence (blend of red plus blue) are indicative of the AIF translocation. As an internal control of caspase inhibition efficacy, nuclear chromatin condensation is inhibited by Boc-D.fmk and Z-VAD.fmk but not by Z-FA. fmk. (B) Kinetic analysis of AIF translocation (transl.) and nuclear apoptosis (NA), as determined by immunofluorescence. Results are the means of 6 independent experiments (mean ± SEM) in which a minimum of 200 syncytia was examined for each data point. Co., control. (C) AIF translocation determined by subcellular fractionation. A mixture of HeLa Env cells and HeLa CD4 cells was cocultured during the indicated intervals (0 = no coculture) in the presence or absence of Z-VAD.fmk, lysed, and separated into nuclei (N), mitochondria-enriched heavy membranes (HM), and organelle-free cytosols (CS), as described in Materials and Methods. Each fraction was subjected to SDS-PAGE, Western blot, and immunodetection of AIF.
Figure 3.

Caspase-independent Cyt-c translocation in syncytia. (A) Syncytia arising from the coculture of HeLa Env and HeLa CD4 cells, treated as described in the legends to Fig. 1 and Fig. 2, were stained with mAbs specific for Cyt-c (revealed by PE) and COX (revealed by FITC) as well as with Hoechst 33342. Representative photomicrographs are shown. Note the diffuse red fluorescence indicative of Cyt-c release from mitochondria occurring in the majority of 48-h-old syncytia. (B) Kinetic analysis of AIF, Cyt-c release, and nuclear apoptosis. Results (obtained as described in A) are the means of six independent experiments (mean ± SEM). NA, nuclear apoptosis; transl., translocation; Co., control. (C) Cyt-c translocation determined by immunoblotting. A mixture of HeLa Env cells and HeLa CD4 cells was cocultured during the indicated intervals in the presence or absence of Z-VAD.fmk, lysed, subjected to subcellular fractionation (HM, heavy membranes; CS, cytosols; N, nuclei), and immunodetection of Cyt-c (same samples as described in the legend to Fig. 2).
Kinetics of AIF and Cyt-c Translocation.
The percentage of cells manifesting ΔΨm dissipation and AIF translocation was higher than that of cells positive for Cyt-c translocation (compare percentage values in Fig. 1 C, 2 B, and 3 B), and double immunofluorescence staining of cells for AIF and Cyt-c confirmed the existence of cells having translocated AIF to the nucleus and still retaining Cyt-c in mitochondria (but not vice versa; not shown), indicating that ΔΨm loss and AIF release occurred before Cyt-c release. AIF and Cyt-c translocation were also observed in heterokaryons generated by coculturing HeLa CD4 cells with a lymphoid cell line chronically infected with a syncytium-inducing HIV-1 isolate. Caspase inhibition with Z-VAD.fmk failed to prevent signs of MMP, although it did inhibit nuclear apoptosis as an internal control of its efficacy (Fig. 4). Kinetic analyses confirmed that mitochondria from HIV-1–induced syncytia translocate AIF before Cyt-c and before caspase-3 activation or nuclear chromatin condensation could be detected (Fig. 4). Hence, immunodetectable translocation of AIF precedes that of Cyt-c.
Figure 4.

Kinetics of AIF translocation, Cyt-c translocation, and caspase-3 activation in HIV-1–infected syncytia. CD4-expressing HeLa cells were cocultured with chronically HIV-1–infected H9/IIIB cells at a 2:1 ratio. At different time points, syncytia were fixed and permeabilized to assess the translocation of AIF (as described in the legend to Fig. 2) or that of Cyt-c (as described in the legend to Fig. 3), as well as the activation of caspase-3, using an antiserum specific for the active caspase-3 p18 subunit. Moreover, the frequency of syncytia exhibiting different stages of nuclear apoptosis was assessed (as described in the legend to Fig. 1). Results are the means of three experiments (mean ± SEM). NA, nuclear apoptosis; transl., translocation.
Bax and Bcl-2 Regulate MMP in Env-induced Syncytia.
Members of the Bcl-2/Bax family regulate apoptosis via their capacity to modulate MMP 21 22 23 49. To address the mechanisms by which MMP occurs in Env-induced syncytia, cells were stained with Abs directed against Bax and Bcl-2. Whereas no changes in Bcl-2 staining were observed upon prolonged culture of syncytia (not shown), Bax (red fluorescence) was found to translocate from a cytoplasmic, preponderantly nonmitochondrial to a punctate, mitochondrial (counterstained with anti-COX, green fluorescence) localization (Fig. 5 A). This finding is reminiscent of other models of apoptosis in which insertion of Bax into mitochondrial membranes causes MMP 50 51 52 53. Microinjection of recombinant Bax (but not microinjection of the mutant BaxΔα5/6 protein lacking the putative membrane insertion domain) induced a rapid (3 h) ΔΨm dissipation, Cyt-c translocation, and nuclear apoptosis (Fig. 5 B). This effect of Bax was reduced by coinjection of Bcl-2 or Koenig's polyanion (Fig. 5 B), an inhibitor of the mitochondrial voltage–dependent anion channel (VDAC) reported to neutralize the effect of Bax on isolated mitochondria 53. Similarly, injection of recombinant Bcl-2 (but not Bcl-2Δα5/6) into freshly generated syncytia inhibited their spontaneous apoptosis, both at the mitochondrial and the nuclear levels (Fig. 5 B). To confirm the Bcl-2–mediated inhibition of apoptosis in a different experimental system, CD4-expressing Jurkat cells or U937 cells were cocultured with HeLa Env cells, a manipulation that resulted in rapid syncytium formation and apoptosis (in the case of Jurkat Neo cells; Fig. 6A–D) or syncytium-independent apoptosis (in the case of U937 Neo cells 54; Fig. 6 E). This latter type of apoptosis requires CD4- and CXCR4-mediated interactions with HeLa Env cells, as it was blocked by the anti-CD4 Ab Leu3a and the natural CXCR4 ligand SDF-1α (Fig. 6 F). Transfection-enforced overexpression of Bcl-2 suppressed the HeLa Env–induced ΔΨm loss and nuclear condensation in both Jurkat and U937 cell types, compared with controls transfected with the Neo resistance vector only (Fig. 6). In conclusion, Bcl-2–regulated MMP is a critical event of Env-induced apoptosis.
Figure 5.
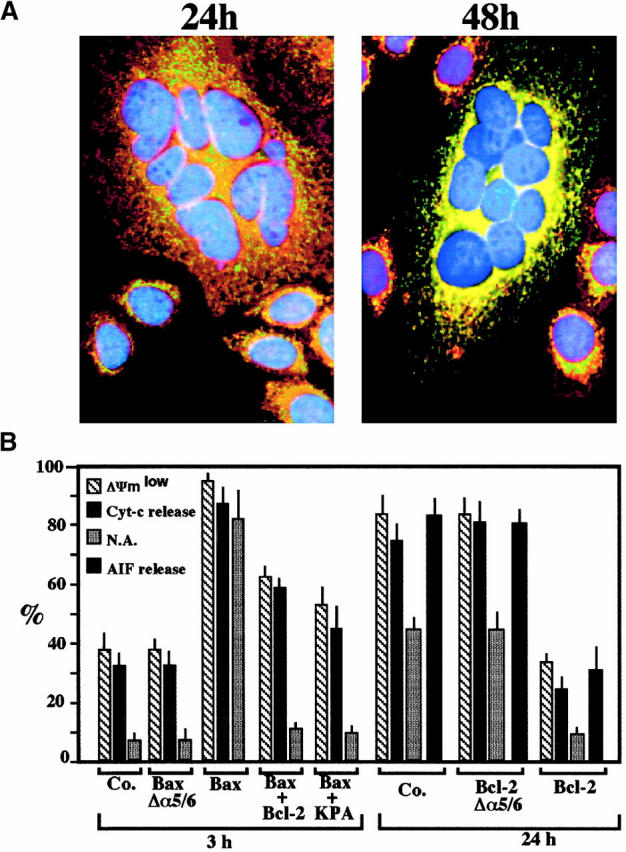
Bax/Bcl-2–mediated control of MMP in Env/CD4-induced syncytia. (A) Subcellular redistribution of Bax during spontaneous apoptosis in syncytia. Syncytia were obtained by the coculture of HeLa Env and HeLa CD4 cells for 24 or 48 h, followed by staining with anti-COX (green fluorescence), an anti-Bax mAb (revealed by PE, red fluorescence), and Hoechst 33342 (blue fluorescence). At 24 h, 82 ± 3% of syncytia manifest a preponderantly nonmitochondrial distribution of Bax, whereas at 48 h, 61 ± 5% cells demonstrate a mainly mitochondrial localization of Bax (88 ± 4% at 72 h, n = 3). (B) Microinjection of recombinant proteins from the Bax/Bcl-2 family. 24-h-old syncytia were microinjected with recombinant Bax, Bcl-2, inactive mutant proteins (BaxΔα5/6, Bcl-2Δα5/6), and/or Koenig's polyanion (KPA), followed by determination of the frequency of cells with a low ΔΨm (green JC-1 fluorescence), Cyt-c translocation, AIF translocation (only determined after 24 h), and nuclear chromatin condensation. Results are the means of three experiments (mean ± SEM). N.A., nuclear apoptosis; Co., control.
Figure 6.

Bcl-2–mediated inhibition of apoptosis induced by the Env–CD4 interaction in a heterologous system. (A) CD4-expressing Jurkat cells transfected with the Bcl-2 gene or an empty vector conferring Neo resistance were fused with HeLa Env cells at a 1:1 ratio. After 18 h of coculture, phase–contrast images of syncytia were obtained. Note that Bcl-2 overexpression does not impede the formation of syncytia, yet prevents cytoplasmic blebbing. White and black arrows indicate normal and apoptotic syncytia, respectively. (B) JC-1/Hoechst 33342 staining patterns of Jurkat/HeLa Env syncytia (same experiment as described in A). (C) Kinetic analysis of apoptosis induction by fusion of Bcl-2 or Neo Jurkat cells to HeLa Env cells. The frequency of cells demonstrating a JC-1–detectable ΔΨm reduction and nuclear apoptosis was determined after the indicated period of coculture (mean ± SEM, n = 3). (D) DNA fragmentation pattern induced by coculture of HeLa Env cells with Neo Jurkat or Bcl-2 cells. Cells were recovered after 18 h and nuclear DNA was analyzed by pulse field gel electrophoresis. (E) Bcl-2–mediated inhibition of HeLa Env–induced apoptosis of U937 cells. U937 cells transfected with Bcl-2 or Neo were cocultured with HeLa Env cells. This manipulation does not lead to the formation of syncytia, although cell-to-cell contacts are established. The frequency of cells acquiring a low ΔΨm and nuclear apoptosis (NA) was assessed at various time points (n = 3, mean ± SEM) for the two cell types involved. (F) Apoptosis of U937 cells interacting with HeLa Env cells involves CD4 and chemokine receptors. U937 Neo cells were cocultured with HeLa Env cells for 24 h, either in the absence (Co) or presence of Leu3a (5 μg/ml) or SDF-1α (10 μg/ml), followed by determination of the frequency of cells with a low ΔΨm and nuclear apoptosis (n = 3, mean ± SEM).
Molecular Ordering of Mitochondrial AIF and Cyt-c Release in Syncytia Undergoing Apoptosis.
To investigate the putative functional relationship between the translocation of AIF and Cyt-c, 24-h-old Env-induced syncytia were microinjected with AIF, Cyt-c, or Abs that neutralize either AIF or Cyt-c, followed by determination of apoptotic parameters after 3 h (Fig. 7 A) or 24 h of culture (Fig. 7 B). Microinjection of both AIF and Cyt-c resulted in the rapid (3 h) induction of nuclear apoptosis, the reduction of ΔΨm, and the translocation of endogenous AIF (induced by Cyt-c) or Cyt-c (induced by AIF; Fig. 7 A). Z-VAD.fmk differentially affected the mitochondrial effects of ectopic (extramitochondrial) AIF and Cyt-c. It suppressed the Cyt-c–induced ΔΨm loss and release of AIF. In contrast, Z-VAD.fmk failed to inhibit the AIF-triggered ΔΨm dissipation and Cyt-c translocation (Fig. 7 A). As an internal control, a neutralizing anti-AIF Ab prevented the acute (3 h) AIF effects when microinjected together with AIF (Fig. 7 A). This Ab also prevents the spontaneous ΔΨm loss, Cyt-c release, and nuclear apoptosis of syncytia (Fig. 7 B). This effect is specific, as it was not observed with a preimmune antiserum nor when the AIF Ab was neutralized by preincubation with an excess of AIF-derived immunogenic peptides (Fig. 7 B). In sharp contrast, neutralization of Cyt-c by microinjection of a specific mAb did not prevent the ΔΨm change nor did it affect the AIF translocation occurring during syncytial aging (Fig. 7 B). However, Cyt-c neutralization did inhibit the Hoechst 33342–detectable chromatin condensation (Fig. 7 B). Altogether, these results suggest that ectopic AIF is sufficient and necessary for caspase-independent mitochondrial Cyt-c release, whereas Cyt-c is critical for caspase-dependent nuclear apoptosis.
Figure 7.
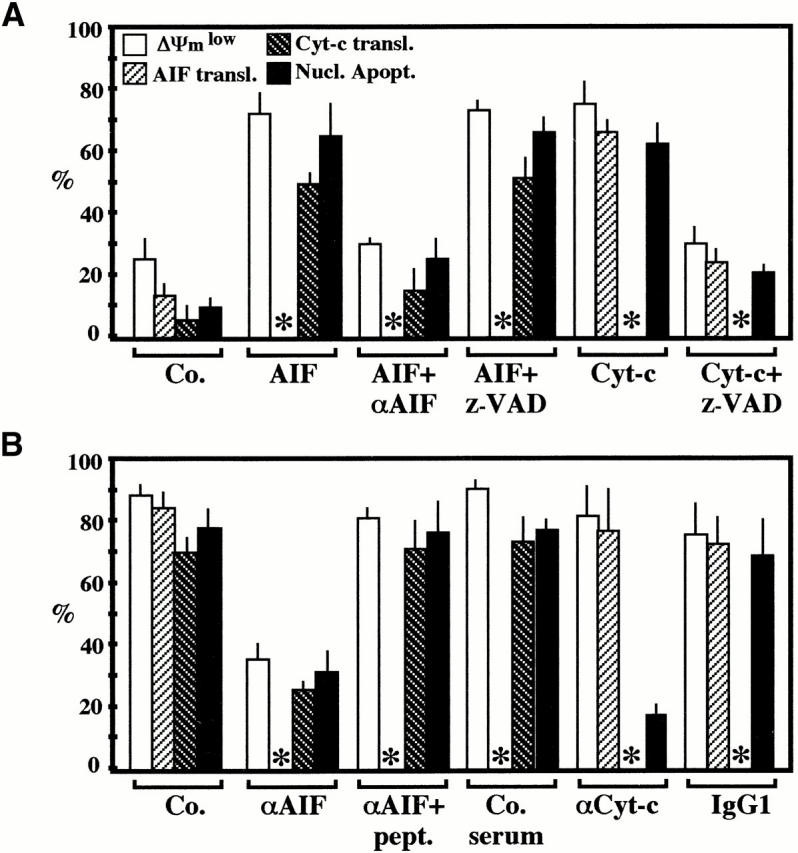
Molecular hierarchy between the mitochondrial release of AIF and Cyt-c. 24-h-old syncytia, generated by coculture of HeLa Env and HeLa CD4 cells, were microinjected with the indicated combination of AIF, Cyt-c and/or anti-AIF, and anti–Cyt-c Abs, followed by an additional culture period of 3 (A) or 24 h (B), optionally in the presence of Z-VAD.fmk, as indicated. Unfixed cells were then stained with Hoechst 33324 and JC-1 or, alternatively, cells were fixed and stained with anti-AIF plus anti-hsp60 or anti–Cyt-c plus anti-COX to determine the nuclear and mitochondrial parameters of apoptosis. After microinjection of anti-AIF or anti–Cyt-c Abs, the subcellular localization of AIF or Cyt-c, respectively, could not be determined (asterisks). Each point represents the mean of at least 200 microinjected syncytia (n = 3, mean ± SEM). Nucl. Apopt., nuclear apoptosis; transl., translocation; Co., control; pept., peptide.
Discussion
The Intrinsic Pathway Governs Apoptosis Triggered by Env.
Here we demonstrate that Env-induced syncytia spontaneously undergo apoptosis and that this apoptotic process obeys the rules of the intrinsic (rather than the extrinsic) cell death pathway. This demonstration is based on several lines of evidence. First, syncytia manifest signs of inner MMP (ΔΨm dissipation; Fig. 1) and outer MMP (release of AIF and Cyt-c; Fig. 2 and Fig. 3), in line with the fact that syncytial mitochondria frequently are dilated 13 14. Swelling of mitochondria is associated with MMP and occurs both in early apoptosis of individual cells (before cell shrinkage 55 56 57) and in necrosis 58. Second, outer and inner MMP occurs well before caspases are activated and before nuclear chromatin is condensed in a caspase-dependent fashion (Fig. 1 Fig. 2 Fig. 3 Fig. 4). Third, inhibition of caspases by oligo- or monopeptidic inhibitors does not prevent MMP, confirming that caspases act downstream of MMP (Fig. 1 Fig. 2 Fig. 3). Fourth, inhibition of MMP by microinjection of recombinant Bcl-2 (Fig. 5 B) or by coculture of Env-positive cells with CD4+ cells expressing a Bcl-2 transgene (Fig. 6) prevents nuclear apoptosis, as this may be expected for the intrinsic (but not the extrinsic) pathway of death induction 25 26 27 36.
Formation of syncytia is a nonphysiological process (with the exception of a few cell types such as syncytiotrophoblasts, spermatogonia, osteoclasts, and myocytes 59), supporting the idea that syncytia could be intrinsically condemned to undergo apoptosis. However, HeLa cells driven to form syncytia by culture with methotrexate or by transfection with fusion-competent proteins from human parainfluenza virus type 4a 60 die more slowly than Env/CD4-induced HeLa syncytia (Ferri, K.F., and G. Kroemer, unpublished observation), suggesting that the receptors involved in the fusion process contribute to the triggering of the intrinsic pathway. Accordingly, engagement of CD4 and CXCR4 can induce lymphocyte death without syncytium formation 38 40 61, and U937 cells die upon contact with Env-transfected cells without prior cell fusion (54 62; Fig. 6 E). Although it remains elusive whether Env-induced syncytium-dependent and syncytium-independent apoptosis are mediated by identical pathways, it appears clear that Bcl-2–regulated, presumably Bax-triggered MMP is a critical event of different types of cell death stimulated via the Env–CD4/CXCR4 interaction (Fig. 5 and Fig. 6).
Cell Type–specific Contribution of AIF and Caspases to Nuclear Apoptosis.
In dying syncytia arising from the fusion of Env- and CD4/CXCR4-expressing cells, mitochondria release both Cyt-c and AIF (Fig. 2 and Fig. 3). Microinjection of Abs neutralizing either AIF or Cyt-c prevents chromatin condensation (Fig. 7 B), suggesting that both factors contribute to nuclear apoptosis: AIF in a caspase-independent fashion, and Cyt-c in a caspase-dependent fashion (Fig. 7 A). In a cell-free system, recombinant AIF causes caspase-independent large scale (∼50 kbp) DNA fragmentation and peripheral chromatin condensation 34 when added to purified nuclei. Paradoxically, however, in Z-VAD.fmk–treated 72-h-old syncytia, AIF is clearly present in the nucleus (Fig. 2), yet no ∼50 kbp DNA fragmentation pattern can be detected (Fig. 1 D) and chromatin condensation is strongly reduced (Fig. 1A and Fig. B). Thus, in contrast to fibroblast cell lines in which large scale DNA fragmentation is caspase independent (and presumably AIF mediated 34 45), HeLa syncytia large scale DNA fragmentation appears to be fully caspase dependent, as this has been reported for Jurkat cells in which the caspase-activated DNase accounts for both oligonucleosomal and large scale DNA fragmentation 63. Of note, microinjection of an excess of exogenous recombinant AIF into freshly formed (24 h) syncytia can trigger acute (3 h) chromatin condensation in a caspase-independent fashion (Fig. 7 A). Thus, in principle, AIF can exert an (presumably direct) effect on nuclei from HeLa syncytia. Nonetheless, in the slow (24–48 h) advancement of chromatin condensation observed in HeLa syncytia spontaneously undergoing apoptosis, AIF clearly acts in a caspase-dependent fashion (Fig. 2). Future work will unravel whether AIF-inhibitory factors and/or the abundance of the nuclear AIF target account for these cell type–specific differences.
Hierarchy of Mitochondrial AIF and Cyt-c Release.
Cyt-c release has been reported to occur in a coordinated, nearly simultaneous fashion in most if not all mitochondria of the same cell 64. Accordingly, syncytia retaining Cyt-c or AIF in a fraction of mitochondria were infrequently observed (<1% of the entire population; Fig. 2 A and 3 A) at any time point, whereas a heterogeneity in the ΔΨm loss was evident in a substantial fraction of cells (Fig. 1 C). Internal feed-forward amplification loops could contribute to the rapid kinetics of Cyt-c release. One such amplification loop has been proposed to be provided by caspases, which, once activated as a consequence of MMP, stimulate MMP 65 66. However, in Env-induced syncytia, caspase inhibition does not affect the kinetics of Cyt-c release (Fig. 3 B). A caspase-independent amplification loop may be provided by AIF based on the observation that the microinjection of an anti-AIF Ab retards all signs of MMP, including ΔΨm collapse and the release of Cyt-c (Fig. 7 B), whereas microinjection of recombinant AIF induces MMP (Fig. 7 A). Intriguingly, the mitochondrial release of AIF precedes that of Cyt-c in Env-induced syncytia by several hours (compare the percentage values in Fig. 2 B and 3 B, and in Fig. 4). This appears counterintuitive because protein-permeant pores formed in the outer mitochondrial membrane should favor the release of small proteins such as Cyt-c (14.5 kD) over that of the much larger AIF (56 kD 53). Cyt-c is known to be associated with inner membrane christae, in electrostatic interaction with cardiolipin 67. It is tempting to speculate that, in addition to outer MMP, (AIF-induced?) changes in inner membrane physicochemistry such as cardiolipin oxidation 44 68 must occur to allow for full Cyt-c release.
Irrespective of the exact mechanism, our results indicate that, at least in the model studied herein, the mitochondrial release of Cyt-c is subordinate to that of AIF. Thus, microinjection of AIF into the cytoplasm of syncytia suffices to cause Cyt-c release in a caspase-independent fashion (Fig. 7 A), mimicking that of the spontaneous (caspase-independent; Fig. 3) Cyt-c release. Moreover, neutralization of AIF by microinjection of a specific Ab prevents the release of Cyt-c as well as nuclear apoptosis, whereas neutralization of Cyt-c has no effect on the AIF translocation and only impedes nuclear condensation (Fig. 7 B). Taken together, these results delineate the following sequence of events: Bax-mediated/Bcl-2–inhibited MMP → AIF release → Cyt-c release → caspase activation → nuclear apoptosis. Future research will unravel whether this hierarchy is only applicable to syncytial apoptosis or whether it can be inscribed into a more general pathway.
This work establishes that MMP is a critical step in Env-induced syncytial apoptosis. Other proapoptotic proteins encoded by HIV-1 (PR, Tat, and Vpr) also favor MMP. Vpr exerts its proapoptotic effect, at least in part, by binding to the mitochondrial adenine nucleotide translocator, thereby directly inducing MMP 7. The HIV-1 protease (PR) can cleave Bcl-2, thereby abolishing its MMP-inhibitory function 69. Tat reduces the expression of the mitochondrial superoxide dismutase 2 isoenzyme 4 70, which is another endogenous MMP inhibitor 71. Tat may also favor apoptosis by physically interacting with mitochondria 72. It thus emerges that HIV-1 employs several independent strategies to induce MMP and apoptosis via the intrinsic pathway. It remains an ongoing conundrum whether these manifold strategies are designed to cooperate among each other in an additive or synergistic fashion, in the same cell, or whether they rather reflect the capacity of HIV-1 to kill a wide array of distinct cell types.
Acknowledgments
We are indebted to Dr. Dominique Piatier-Tonneau for constant support and to Drs. Marc Alizon, Dieter Brdiczka, Pierre Charneau, Nicole Israel, and Anu Srinivasen, as well as the National Institutes of Health AIDS Research and Reference Reagent Program, for gifts of reagents.
This work was supported by a special grant by the Ligue Nationale contre le Cancer, as well as by grants from Association Nationale pour la Recherche sur le SIDA, Fondation pour la Recherche Medicale, the European Commission, the Picasso Program (to G. Kroemer), Fundació irsiCaixa (to J.A. Este), FIS 00/0893 (to J. Blanco), and grants GM 60554 and National Institutes of Health CA69381 (to J.C. Reed). K.F. Ferri receives a fellowship from the French Ministry of Science, and E. Jacotot receives an Association Nationale pour la Recherche sur le SIDA fellowship. J. Blanco is a researcher from the Fundació per a la Recerca Biomèdica Germans Trias i Pujol.
Footnotes
Abbreviations used in this paper: aa, amino acid(s); AIF, apoptosis-inducing factor; COX, Cyt-c oxidase; CXCR, CXC chemokine receptor; Cyt-c, cytochrome c; ΔΨm, mitochondrial transmembrane potential; Env, envelope glycoprotein complex; gp, glycoprotein; hsp, heat shock protein; MMP, mitochondrial membrane permeabilization; Neo, neomycin.
References
- Fauci A.S. Host factors and the pathogenesis of HIV-induced disease. Nature. 1996;384:529–534. doi: 10.1038/384529a0. [DOI] [PubMed] [Google Scholar]
- Gougeon M.L., Montagnier L. Programmed cell death as a mechanism of CD4 and CD8 T cell depletion in AIDS. Molecular control and effect of highly active anti-retroviral therapy. Ann. NY Acad. Sci. 1999;887:199–212. doi: 10.1111/j.1749-6632.1999.tb07934.x. [DOI] [PubMed] [Google Scholar]
- Li C.J., Friedman D.J., Wang C., Metelev V., Pardee A.B. Induction of apoptosis in uninfected lymphocytes by HIV-1 Tat protein. Science. 1995;268:429–431. doi: 10.1126/science.7716549. [DOI] [PubMed] [Google Scholar]
- Westendorp M.O., Shatrov V.A., Schulze-Osthoff K., Frank R., Kraft M., Los M., Krammer P.H., Dröge W., Lehrmann V. HIV-1 Tat potentiates TNF-induced NF-κB activation and cytotoxicity by altering the cellular redox state. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:546–554. doi: 10.1002/j.1460-2075.1995.tb07030.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.A., Poon B., Jowett J.B.M., Chen I.S.Y. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J. Virol. 1997;71:5579–5592. doi: 10.1128/jvi.71.7.5579-5592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S.A., Poon B., Jowett J.B.M., Xie Y., Chen I.S.Y. Lentiviral delivery of HIV-1 Vpr protein induces apoptosis in transformed cells. Proc. Natl. Acad. Sci. USA. 1999;96:12039–12043. doi: 10.1073/pnas.96.21.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacotot E., Ravagnan L., Loeffler M., Ferri K.F., Vieira H.L.A., Zamzami N., Costantini P., Druillennec S., Hoebeke J., Brian J.P. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000;191:33–45. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lifson J.D., Reyes G.R., McGrath M.S., Stein B.S., Engleman E.G. AIDS retrovirus-induced cytopathologygiant cell formation and involvement of CD4 antigen. Science. 1986;232:1123–1127. doi: 10.1126/science.3010463. [DOI] [PubMed] [Google Scholar]
- Sodroski J.G., Goh W.C., Rosen A., Campbell K., Haseltine W.A. Role of the HTLV/LAV envelope in syncytia formation and cytopathicity. Nature. 1986;322:470–474. doi: 10.1038/322470a0. [DOI] [PubMed] [Google Scholar]
- Chirmule N., Pahwa S. Envelope glycoproteins of human immunodeficiency virus type 1profound influences on immune function. Microbiol. Rev. 1996;60:386–406. doi: 10.1128/mr.60.2.386-406.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent-Crawford A.G., Krust B., Riviere Y., Desgranges C., Muller S., Kieny M.P., Dauguet C., Hovanessian A.G. Membrane expression of HIV envelope glycoproteins triggers apoptosis in CD4 cells. AIDS Res. Hum. Retroviruses. 1993;9:761–773. doi: 10.1089/aid.1993.9.761. [DOI] [PubMed] [Google Scholar]
- Sandstrom P.A., Pardi D., Goldsmith C.S., Duan C.Y., Diamond A.M., Folks T.M. Bcl-2 expression facilitates human immunodeficiency virus type 1-mediated cytopathic effects during acute spreading infections. J. Virol. 1996;70:4617–4622. doi: 10.1128/jvi.70.7.4617-4622.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesnitchenko V., King L., Riva A., Tani V., Korsmeyer S.J., Cohen D.I. A major human immunodeficiency virus type 1-initiated killing pathway distinct from apoptosis. J. Virol. 1997;71:9753–9763. doi: 10.1128/jvi.71.12.9753-9763.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plymale D.R., Tang D.S.N., Comardelle A.M., Fermin C.D., Lewis D.E., Garry R.G. Both necrosis and apoptosis contribute to HIV-1-induced killing of CD4 cells. AIDS. 1999;13:1827–1839. doi: 10.1097/00002030-199910010-00004. [DOI] [PubMed] [Google Scholar]
- Sylwester A., Murphy S., Shutt D., Soll D.R. HIV-induced T cell syncytia are self-perpetuating and the primary cause of T cell death in culture. J. Immunol. 1997;158:3996–4007. [PubMed] [Google Scholar]
- Blaak H., van't Wout A.B., Brouwer M., Hoolbrink B., Hovenkamp E., Schuitemaker H. In vivo HIV-1 infection of CD45RA+ CD4+ T cells is established primarily by syncytium-inducing variants and correlates with the rate of CD4+ T cell decline. Proc. Natl. Acad. Sci. USA. 2000;97:1269–1274. doi: 10.1073/pnas.97.3.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson D.W., Thornberry N.A. Caspaseskiller proteases. Trends Biochem. Sci. 1997;22:299–306. doi: 10.1016/s0968-0004(97)01085-2. [DOI] [PubMed] [Google Scholar]
- Budijardjo I., Oliver H., Lutter M., Luo X., Wang X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- Nagata S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999;33:29–55. doi: 10.1146/annurev.genet.33.1.29. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Dallaporta B., Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 1998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Thompson C.B. Bcl-2 proteinsinhibitors of apoptosis or regulators of mitochondrial homeostasis? Nat. Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Reed J.C. Mitochondrial control of cell death. Nat. Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Scaffidi C., Fulda S., Srinivasan A., Friesen C., Li F., Tomaselli K.J., Debatin K.-M., Krammer P.H., Peter M.E. Two CD95 (APO-1/Fas) signaling pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:1675–1687. doi: 10.1093/emboj/17.6.1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi C., Kirchhoff S., Krammer P.H., Peter M.E. Apoptosis signaling in lymphocytes. Curr. Opin. Immunol. 1999;11:277–285. doi: 10.1016/s0952-7915(99)80045-4. [DOI] [PubMed] [Google Scholar]
- Yin X.-M., Wang K., Gross A., Zhao Y., Zinkel S., Klocke B., Rothe K.A., Korsmeyer S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature. 1999;400:886–891. doi: 10.1038/23730. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Hirsch T., Marchetti P., Macho A., Daugas E., Geuskens M., Kroemer G. Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J. Exp. Med. 1996;184:1331–1342. doi: 10.1084/jem.184.4.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Bossy-Wetzel E., Green D.R., Newmeyer D.D. The release of cytochrome c from mitochondriaa primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- Patterson S., Spahr C.S., Daugas E., Susin S.A., Irinopoulos T., Koehler C., Kroemer G. Mass spectrometric identification of proteins released from mitochondria undergoing permeability transition. Cell Death Differ. 2000;7:137–144. doi: 10.1038/sj.cdd.4400640. [DOI] [PubMed] [Google Scholar]
- Mancini M., Nicholson D.W., Roy S., Thornberry N.A., Peterson E.P., Casciola-Rosen L.A., Rosen A. The caspase-3 precursor has a cytosolic and mitochondrial distributionimplications for apoptotic signaling. J. Cell Biol. 1998;140:1485–1495. doi: 10.1083/jcb.140.6.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Larochette N., Alzari P.M., Kroemer G. Mitochondrial release of caspases-2 and -9 during the apoptotic process. J. Exp. Med. 1999;189:381–394. doi: 10.1084/jem.189.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski S., Krajewska M., Ellerby L.M., Welsh K., Xie Z.H., Deveraux Q.L., Salvesen G.S., Bredesen D.E., Rosenthal R.E., Fiskum G., Reed J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA. 1999;96:5752–5757. doi: 10.1073/pnas.96.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Chinnaiyan A.M., Woffendin C., Dixit V.M., Nabel G.J. The inhibition of pro-apoptotic ICE-like proteases enhances HIV replication. Nat. Med. 1997;3:333–337. doi: 10.1038/nm0397-333. [DOI] [PubMed] [Google Scholar]
- Ohnimus H., Heinkelein M., Jassoy C. Apoptotic cell death upon contact of CD4+ T lymphocytes with HIV glucoprotein-expressing cells is mediated by caspases but bypasses CD95 (Fas/Apo-1) and TNF receptor 1. J. Immunol. 1997;159:5246–5252. [PubMed] [Google Scholar]
- Kaul M., Lipton S.A. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc. Natl. Acad. Sci. USA. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco J., Jacotot E., Cabrera C., Cardona A., Clotet B., De Clercq E., Este J.A. The implication of the chemokine receptor CXCR4 in HIV-1 envelope protein-induced apoptosis is independent of the G protein-mediated signalling. AIDS. 1999;13:909–917. doi: 10.1097/00002030-199905280-00006. [DOI] [PubMed] [Google Scholar]
- Cicala C., Arthos J., Rubbert A., Selig S., Wildt K., Cohen O.J., Faus A.S. HIV-1 envelope induces activation of caspase-3 and cleavage of focal adhesion kinase in primary human CD4+ T cells. Proc. Natl. Acad. Sci. USA. 2000;97:1178–1183. doi: 10.1073/pnas.97.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berndt C., Möpps B., Angermüller S., Gierschik P., Krammer P.H. CXCR4 and CD4 mediate a rapid CD95-independent cell death in CD4+ cells. Proc. Natl. Acad. Sci. USA. 1998;95:12556–12561. doi: 10.1073/pnas.95.21.12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz O., Alizon M., Heard J.M., Danos O. Impairment of T cell receptor-dependent stimulation in CD4+ lymphocytes after contact with membrane-bound HIV-1 envelope glycoprotein. Virology. 1994;198:360–365. doi: 10.1006/viro.1994.1042. [DOI] [PubMed] [Google Scholar]
- Dragic T., Charneau P., Clavel F., Alizon M. Complementation of murine cells for human immunodeficiency virus envelope/CD4-mediated fusion in human/murine heterokaryons. J. Virol. 1992;66:4794–4802. doi: 10.1128/jvi.66.8.4794-4802.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aillet F., Masutani H., Elbim C., Raoul H., Chene L., Nugeyre M.T., Paya C., Barre-Sinoussi F., Gougerot-Pocidalo M.A., Israel N. Human immunodeficiency virus induces a dual regulation of Bcl-2, resulting in persistent infection of CD4+ T- or monocytic cell lines. J. Virol. 1998;72:9698–9705. doi: 10.1128/jvi.72.12.9698-9705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamzami N., Marchetti P., Castedo M., Decaudin D., Macho A., Hirsch T., Susin S.A., Petit P.X., Mignotte B., Kroemer G. Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J. Exp. Med. 1995;182:367–377. doi: 10.1084/jem.182.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugas E., Susin S.A., Zamzami N., Ferri K., Irinopoulos T., Larochette N., Prevost M.C., Leber B., Andrews D., Penninger J., Kroemer G. Mitochondrio-nuclear redistribution of AIF in apoptosis and necrosis. FASEB (Fed. Am. Soc. Exp. Biol.) J. 2000;14:729–739. [PubMed] [Google Scholar]
- Srinivasan A., Roth K.A., Sayer R.O., Shindler K.S., Wong A.N., Fritz L.C., Tomaselli K.J. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- Brown D.G., Sun X.M., Cohen G.M. Dexamethasone-induced apoptosis involves cleavage of DNA to large fragments prior to internucleosomal fragmentation. J. Biol. Chem. 1993;268:3037–3039. [PubMed] [Google Scholar]
- Xie Z.H., Schendel S., Matsuyama S., Reed J.C. Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry. 1998;37:6410–6418. doi: 10.1021/bi973052i. [DOI] [PubMed] [Google Scholar]
- Zamzami N., Brenner C., Marzo I., Susin S.A., Kroemer G. Subcellular and submitochondrial mechanisms of apoptosis inhibition by Bcl-2-related proteins. Oncogene. 1998;16:2265–2282. doi: 10.1038/sj.onc.1201989. [DOI] [PubMed] [Google Scholar]
- Wolter K.G., Hsu Y.-T., Smith C.L., Nechushtan A., Xi X.-G., Youle R.J. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jürgensmeier J., Susin S.A., Vieira H.L.A., Prévost M.-C., Xie Z., Mutsiyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Gross A., Jockel J., Wei M.C., Korsmeyer S.J. Enforced dimerization of Bax results in its translocation, mitochondrial dysfunction and apoptosis. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:3878–3885. doi: 10.1093/emboj/17.14.3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Asjo N., Ivhed I., Gidlund M., Fuerstenberg S., Fenyo E., Nilsson M., Wigzell J. Susceptibility to infection by the human immunodeficiency virus correlates with T4 expression in a parental monocytoid cell line and its subclones. Virology. 1987;157:359–365. doi: 10.1016/0042-6822(87)90278-9. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Chandal N.S., Williamson E.K., Schumacker P.T., Thompson C.B. Bcl-XL regulates the membrane potential and volume homeostasis of mitochondria. Cell. 1997;91:627–637. doi: 10.1016/s0092-8674(00)80450-x. [DOI] [PubMed] [Google Scholar]
- Dallaporta B., Marchetti P., de Pablo M., Maisse C., Duc H.T., Metivier D., Zamzami N., Geuskens M., Kroemer G. Plasma membrane potential in thymocyte apoptosis. J. Immunol. 1999;162:6534–6542. [PubMed] [Google Scholar]
- Feldmann G., Haouzi D., Moreai A., Durand-Schneider A.M., Bringuier A., Berson A., Mansouri A., Fau D., Pessayre D. Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology. 2000;31:674–683. doi: 10.1002/hep.510310318. [DOI] [PubMed] [Google Scholar]
- Lemasters J.J., Nieminen A.-L., Qjan T., Trost L.C., Elmore S.P., Nishimura Y., Crowe R.A., Cascio W.E., Bradham C.A., Brenner D.A., Herman B. The mitochondrial permeability transition in cell deatha common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Anderson J.M. Multinucleated giant cells. Curr. Opin. Hematol. 2000;7:40–47. doi: 10.1097/00062752-200001000-00008. [DOI] [PubMed] [Google Scholar]
- Nishio M., Tsurudome M., Komada H., Kawano M., Tabata N., Matsumura H., Ikemura N., Watanabe N., Ito Y. Fusion properties of cells constitutively expressing human parainfluenza virus type 4a hemagglutinin-neuraminidase and fusion glycoproteins. J. Gen. Virol. 1994;75:3517–3523. doi: 10.1099/0022-1317-75-12-3517. [DOI] [PubMed] [Google Scholar]
- Newell M.K., Haughn L.J., Maroun C.R., Julius M.H. Death of mature T cells by separate ligation of CD4 and the T-cell receptor for antigen. Nature. 1990;347:286–289. doi: 10.1038/347286a0. [DOI] [PubMed] [Google Scholar]
- Xiao X., Norwood D., Feng Y.R., Morluchi M., Jones-Trower A., Stantchev T.S., Moriuchi H., Broder C.C., Dimitrov D.S. Inefficient formation of a complex among CXCR4, CD4 and gp120 in U937 clones resistant to X4 gp120-gp41-mediated fusion. Exp. Mol. Pathol. 2000;68:139–146. doi: 10.1006/exmp.1999.2299. [DOI] [PubMed] [Google Scholar]
- Sakahira H., Enari M., Ohsawa Y., Uchiyama Y., Nagata S. Apoptotic nuclear morphological change without DNA fragmentation. Curr. Biol. 1999;9:543–546. doi: 10.1016/s0960-9822(99)80240-1. [DOI] [PubMed] [Google Scholar]
- Goldstein J.C., Waterhouse N.J., Juin P., Evan G.I., Green D.R. The coordinate release of cytochrome c is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Susin S.A., Zamzami N., Castedo M., Daugas E., Wang H.-G., Geley S., Fassy F., Reed J., Kroemer G. The central executioner of apoptosis. Multiple links between protease activation and mitochondria in Fas/Apo-1/CD95– and ceramide-induced apoptosis. J. Exp. Med. 1997;186:25–37. doi: 10.1084/jem.186.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q., Gong B., Almasan A. Distinct stages of cytochrome c release from mitochondriaevidence for a feedback amplification loop linking caspase activation to mitochondrial dysfunction in genotoxic stress induced apoptosis. Cell Death Differ. 2000;7:227–233. doi: 10.1038/sj.cdd.4400629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.M., Shin K.H., Fujiwara T., Akutsu H. The interaction of ferric and ferrous cytochrome c with cardiolipin in phospholipid membranes studied by solid-state H-2 and P-31 NMR. J. Mol. Struct. 1998;441:183–188. [Google Scholar]
- Shidoji Y., Hayashi K., Komura S., Ohishi N., Yagi K. Loss of molecular interaction between cytochrome c and cardiolipin due to lipid peroxidation. Biochem. Biophys. Res. Commun. 1999;264:343–347. doi: 10.1006/bbrc.1999.1410. [DOI] [PubMed] [Google Scholar]
- Strack P.R., Frey M.W., Rizzo C.J., Cordova B., George H.J., Meade R., Jo S.W., Corman J., Tritch R., Korant B.D. Apoptosis mediated by HIV protease is preceded by cleavage of Bcl-2. Proc. Natl. Acad. Sci. USA. 1996;93:9571–9576. doi: 10.1073/pnas.93.18.9571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creaven M., Hans F., Mutskov V., Col E., Caron C., Dimitrov S., Khochbin S. Control of the histone-acetyltransferase activity of Tip60 by the HIV-1 transactivator protein, Tat. Biochemistry. 1999;38:8826–8830. doi: 10.1021/bi9907274. [DOI] [PubMed] [Google Scholar]
- Williams M.D., Van Remmen H., Conrad C.C., Huang T.T., Epstein C.J., Richardson A. Increased oxidative damage is correlated to altered mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J. Biol. Chem. 1998;273:28510–28515. doi: 10.1074/jbc.273.43.28510. [DOI] [PubMed] [Google Scholar]
- Macho A., Calzado M.A., Jimenez-Reina L., Ceballos E., Leon J., Munoz E. Susceptibility of HIV-1-TAT transfected cells to undergo apoptosis. Biochemical mechanisms. Oncogene. 1999;18:7543–7551. doi: 10.1038/sj.onc.1203095. [DOI] [PubMed] [Google Scholar]