

Abstract
Chromosomal translocations juxtaposing the MYC protooncogene with regulatory sequences of immunoglobulin (Ig) H chain or kappa (Igκ) or lambda (Igλ) L chain genes and effecting deregulated expression of MYC are the hallmarks of human Burkitt lymphoma (BL). Here we report that lymphomas with striking similarities to BL develop in mice bearing a mutated human MYC gene controlled by a reconstructed Igλ locus encompassing all the elements required for establishment of locus control in vitro. Diffusely infiltrating lymphomas with a typical starry sky appearance occurred in multiple founders and an established line, indicating independence from positional effects. Monoclonal IgM+CD5−CD23− tumors developed from an initially polyclonal population of B cells. These results demonstrate that the phenotype of B lineage lymphomas induced by MYC dysregulation is highly dependent on cooperativity among the regulatory elements that govern expression of the protooncogene and provide a new system for studying the pathogenesis of BL.
Keywords: Burkitt lymphoma, locus control region, mouse lymphoma, MYC, translocation
Introduction
Burkitt lymphoma (BL), first described in 1958 as a disease of African children 1, was later recognized to occur at 10- to 30-fold lesser frequency worldwide. The African form of the disease, termed endemic BL, is associated with EBV in over 95% of cases, while the non-African form, termed sporadic BL, is EBV positive in only 10–20% of cases (for review see reference 2). Regardless of geographic origin, BL is invariably characterized by translocations 3 that juxtapose the MYC gene on chromosome 8 with IgH genes on chromosome 14 or, less often, IgL genes on chromosome 2 (Igκ) or 22 (Igλ) 4 5. The chromosome 8 breakpoints are highly variable, occurring within the MYC transcriptional unit or up to several hundred kilobases upstream or downstream of MYC in cases with t(8;14) translocations and downstream of MYC in the variant t(2;8) and t(8;22) translocations (reference 6). Indeed, the proposed World Health Organization Classification of Hematologic Malignancies indicates that the presence of one of the MYC/Ig translocations should be considered the gold standard for diagnosis of BL 7.
Previous efforts to develop a mouse model of BL centered on generation of transgenic mice that express the MYC gene under the control of IgH or IgL regulatory sequences 8. These constructs induced primarily precursor B cell as well as some surface Ig (sIg)+ B cell lymphomas; however, the histopathologic features of these neoplasms were consistent with the diagnosis of lymphoblastic lymphoma (LL) rather than BL. Tumors with the histologic and phenotypic features of BL have never been described in mice.
We reasoned that the IgH and IgL intronic enhancer–driven MYC transgenes used by others may not have induced BL because they did not fully reflect the mode of MYC activation observed in the context of BL-derived chromosomal translocations. We therefore attempted to reconstruct BL translocation breakpoints and designed assays that test for BL-specific MYC activation in vitro. To this end, we fused the MYC gene derived from a BL-derivative chromosome to elements of the Igκ or Igλ loci. For the Igλ construct, we included a 12-kb genomic fragment including the Igλ enhancer and three additional sites defined by DNaseI hypersensitivity 9. The reconstructed loci were cloned onto EBV-derived episomal vectors and stably introduced into Raji cells 9 10 and the conditionally EBV-immortalized lymphoblastoid cell line ER/EB 11. BL-specific MYC activation was defined using the following criteria: (a) the overall level of MYC expression relative to expression from the endogenous normal (ER/EB cells) or the endogenous translocated allele (Raji cells); (b) the level of MYC expression relative to copy number; (c) promoter shift toward predominant usage of promoter P1 rather than P2, which is preferentially used in normal cells 10 12; (d) absence of transcriptional elongation block 12 13; (e) Epstein-Barr nuclear antigen (EBNA)2- and latent membrane protein (LMP)1-independent proliferation of conditionally EBV-immortalized ER/EB cells 11; and (f) shift in the phenotype and growth behavior of these cells to that of BL cells 11. According to these highly stringent criteria, we succeeded in reconstructing all essential features of MYC activation by the Igκ and Igλ loci in vitro. We hypothesized 9 that the elements we defined act in concert for MYC activation in a BL-specific fashion function as locus control regions (LCRs) 14 that play important roles in the physiologic regulation of IgL expression. In this paper, we addressed whether the described Igλ–MYC construct is able to recapitulate features of BL-specific MYC activation in transgenic mice.
Materials and Methods
Generation of Transgenic Mice.
A translocated MYC gene from the human BL line IARC-BL60 with mutations in the 5′ sequences associated with promoter shift from P2 to P1 12 was chosen for study. The gene was placed under the control of λ chain regulatory sequences as shown in Fig. 1 A. The plasmid inserts from clone BC233A (λ-MYC) 9 were purified for inoculation of C57BL/6N (B6) fertilized eggs. Founder mice were identified by Southern blotting using a human MYC probe. One λ-MYC–transgenic founder was crossed with normal B6 mice to establish a line (B6–λ-MYC) bearing the transgene. The line was maintained by serial crosses with normal B6 mice yielding transgene heterozygotes and normal mice in each litter. Transgenic mice were bred and maintained in a conventional colony.
Figure 1.

Characteristics of MYC-transgenic mice. (A) Construct used for the generation of λ-MYC–transgenic mice. Horizontal arrows indicate the two MYC promoters, P1 and P2, located in the first noncoding exon (open rectangle), with the two coding exons shown by filled rectangles. Vertical arrows indicate the locations of the four previously defined 9 DNaseI-hypersensitive sites (HSS) in the Igλ LCR. The methods used in generating the construct have been described elsewhere 9. (B) Mortality curve for 20 λ-MYC–transgenic mice of the established strain. (C) RT-PCR analyses of mouse and human MYC and mouse GAPDH expression in tissues of λ-MYC–transgenic (TG+) and transgene-negative (TG−) littermates. BL, mouse Burkitt lymphoma; NL, normal; Hu 4937, human breast cancer cell line; ARS6 PCT, mouse PCT cell line; K, kidney; L, liver; S, spleen; B, brain; CL, cell line. (D) Western blot analyses of MYC expression. Nuclear proteins from the RJA BL cell line, spleens of six littermates segregating for the λ-MYC transgene (three negative and three positive), the M1 and C1 cell lines, and a primary splenic λ-MYC lymphoma, 36330, were blotted with polyclonal anti–human MYC Ab. The sizes of protein markers are indicated on the left.
Studies of Mice.
At autopsy, selected tissues were fixed in formalin. Sections from paraffin blocks were examined for histopathologic features. Other samples were frozen for preparation of genomic DNA and Southern blot hybridization analyses of IgH organization. Single-cell suspensions of spleen and bone marrow (BM) were stained for FACS® analyses of cell surface Ag expression using a panel of reagents useful in distinguishing subsets of hematopoietic cells. Cells from lymphomas were also cultured to develop in vitro cell lines. Established cell lines were studied for surface Ag by FACS® and for IgH organization by Southern hybridization. Western analysis of MYC protein expression was performed using a polyclonal Ab (c-Myc [A-14]-G: sc789-G; Santa Cruz Biotechnology, Inc.).
DNA from spleen and tumor cells was examined by Southern hybridization for the organization of IgH using a JH probe, an ecotropic murine leukemia virus envelope–specific probe, or a human MYC exon 2 probe using standard conditions. RNA from these samples was used to examine mouse and human MYC transcripts by RT-PCR. The same 5′ primer was used for both reactions: 5′-CTG-CTG-GTG-GTG-GGC-GGT-GTC-TC-3′. The human MYC-specific 3′ primer was 5′-CGA-GCG-GGC-GGC-CGG-CTA-3′, and the mouse Myc-specific 3′ primer was 5′-TGG-ATT-TCC-TTT-GGG-CGT-TGG-3′.
Results
Characteristics of Transgenic Mice.
Among seven founder mice carrying the λ-MYC transgene, six were killed when moribund with lymphoma between 24 and 36 d of age. One λ-MYC founder that lived 106 d before dying of lymphoma was bred to B6 mice. The progeny of the cross with B6 and members of an established B6 line died between days 38 and 216 (Fig. 1 B) with the same gross findings. In contrast to mouse follicular, splenic marginal zone, and most diffuse large cell lymphomas that seem to originate in spleen 15, these lymphomas appeared to originate in LNs as judged by mice presenting with advanced lymphadenopathy but minimal splenomegaly.
To determine whether the time course for appearance of the lymphomas was related to transgene copy number, DNA from tail biopsies of five λ-MYC founder mice was tested by Southern hybridization using a probe specific for human MYC. Intensities of hybridizing bands were compared with those generated by serially diluted transgene plasmid (data not shown). Two founders with 1 to 2 copies died at days 33 and 106, one with 3 copies died at day 33, and two with 5–10 copies died at days 29 and 37. Latency for lymphoma thus did not correlate with estimated transgene copy number. Because members of an established line were available for testing in only one instance, it is possible that copy number estimates made from tail DNA of founder animals could be misleading because of mosaicism.
Specificity of transgene expression was evaluated by RT-PCR using RNA from various tissues of normal transgenic mice and a mouse with disseminated lymphoma involving the spleen, liver, (Fig. 1 C) and thymus (data not shown) For mice without lymphoma, substantial levels of transcripts were seen in lymphoid tissues, while nonlymphoid tissues had undetectable or very low levels, possibly reflecting the presence of lymphocytes. High levels of transcripts were seen in all involved tissues of the mouse with lymphoma. In addition, FACS®-purified splenic T cells were negative and sorted B cells strongly positive for MYC expression (data not shown). This indicated that the transgene was expressed in a cell type–specific manner.
Expression of nuclear MYC protein in spleens of nonlymphomatous transgenic mice and their control littermates, a primary transgenic lymphoma, and two cell lines established from lymphomas was compared in Western blots with expression in a Burkitt cell line (Fig. 1 D). Consistently low levels of protein were found only in spleens of transgenic mice, with about fivefold higher levels seen in the primary lymphoma and cultured cell lines. The marked differences in expression between the primary lymphoma and cell lines (essentially 100% B cells) and the spleen cells of young transgenic mice (∼40% B cells) suggest there was significant upregulation of MYC expression as part of the transformation process.
Histopathology and FACS® Analyses of Transgenic Mice.
The histologic characteristics of lymphoid tissues from λ-MYC transgene–positive mice (Fig. 2 A) and littermate controls (Fig. 2 B) were indistinguishable through 2–3 mo of age. FACS® analyses showed that the frequencies of B cells in transgenic spleens were ∼70% that of control littermates but that the B cells from mice of both genotypes expressed comparable levels of IgM, IgD, MHC class II, CD23, CD19, and CD45R(B220) (data not shown). T cells were increased in frequency in transgenic spleens but exhibited normal subset distributions and expression of cell surface Ag (data not shown). FACS® analyses of BM cells from young transgenic and control mice (Fig. 3) showed that the compositions of B cell differentiation stages A to F according to the Hardy classification 16 were generally comparable. The reduced frequencies of IgM+IgD− (fraction F) cells in transgenic BM suggests that the balance of processes mediating positive and negative selection of immature/transitional cells 17 18 may be affected by enhanced expression of MYC.
Figure 2.
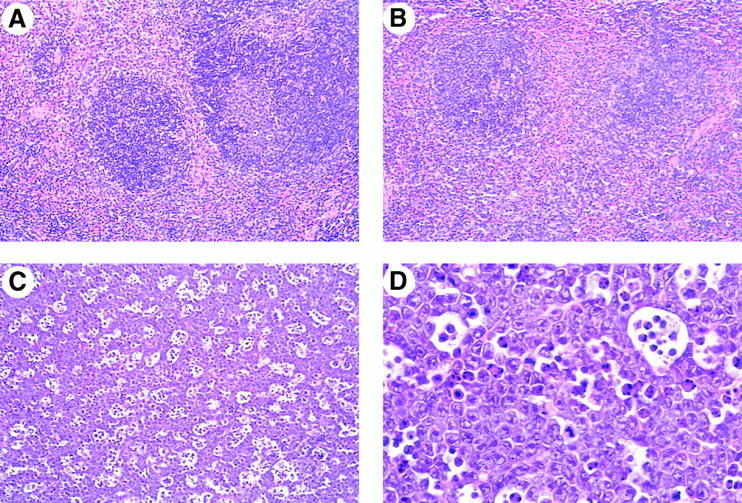
Histologic studies of λ-MYC–transgenic mice. Sections are from spleens of (A) a transgenic mouse, (B) a normal littermate control 7 wk of age, and (C and D) a primary lymphoma from a mouse 70 d of age. Hematoxylin and eosin ×40 (A–C) or ×100 (D).
Figure 3.

FACS® analyses of BM cells from (top panels) a λ-MYC transgene–negative (TG−) and (bottom panels) transgene-positive (TG+) littermate. Cells were stained with Ab to CD43, CD45R(B220), IgM, and IgD. Right panels represent examinations of cells in the D, E, F box in the corresponding panel on the left. The frequencies of cells in the boxed areas as a total of all BM cells is indicated on the left panels. The proportions of the total cells in D, E, F present in each individual fraction is given in the right panels.
In older mice presenting with lymphadenopathy, however, the spleen and LNs were diffusely infiltrated with a highly aggressive lymphoma. Mitotic figures were numerous, and large numbers of metallophilic macrophages containing apoptotic bodies yielded a striking starry sky appearance (Fig. 2 C). The tissues were populated with a monomorphic population of round cells with moderately abundant, deeply blue cytoplasm. Nuclei were round with moderately clumped chromatin, contained one to five large argentophilic nucleoli, and were surrounded by a thick nuclear membrane without convolutions (Fig. 2 D). Infiltration of nonlymphoid tissues including liver, kidneys, lungs and, less often, thymi was often severe.
Other lymphomas that can present with lymphadenopathy in excess of splenomegaly and may exhibit a starry sky appearance have been termed lymphoblastic lymphomas (LLs) 15. LLs are known to be heterogeneous in lineage (T cell versus B cell) and stage of differentiation (precursor B cell versus sIg+ B cell) but indistinguishable by morphologic and cytologic criteria. Comparisons of T cell LLs and B cell LLs with the lymphomas of λ-MYC mice showed the LLs to comprise smaller cells with a lower mitotic index and fewer macrophages containing apoptotic bodies (data not shown).
Studies of lymphomas from the transgenic mice showed that they were positive for IgM, Igκ (not shown), CD45R(B220), CD19, and CD16/CD32 (Fig. 4) but were negative for CD5 (Fig. 4) and T cell markers CD90, CD4, and CD8 (data not shown). IgD was expressed at very low levels or not at all. Cell lines established in culture from tissues of a founder mouse (line C1) and nine backcross mice, including line M1, were typed more extensively. The lines expressed IgM, Igκ, CD19, CD24, CD43, and CD45R(B220) but were negative for CD5, CD23, CD30, CD38, CD80, CD86, and CD95 (data not shown). These phenotypes correspond closely to those characteristic of human BL.
Figure 4.

FACS® analyses of spleen cells from a primary lymphoma of a 75-d-old λ-MYC–transgenic mouse. Cells were stained with the indicated Ab, and the profiles were compared with those of unstained cells.
Characteristics of Ig Loci.
To evaluate clonality, DNA prepared from spleen cells of young λ-MYC–transgenic mice without tumors and tumors from several founder mice, and the two cell lines cultured from primary lymphomas were examined for organization of IgH by Southern hybridization (Fig. 5). The pattern for DNA from spleens of young mice was polyclonal, while DNA from founders exhibited one to three distinct nongermline bands that hybridized with a JH probe, often in association with reduced intensity of the germline band. DNA from cell lines exhibited complete loss of 6.6-kb germline sequences and the presence of prominent nongermline bands (Fig. 5).
Figure 5.
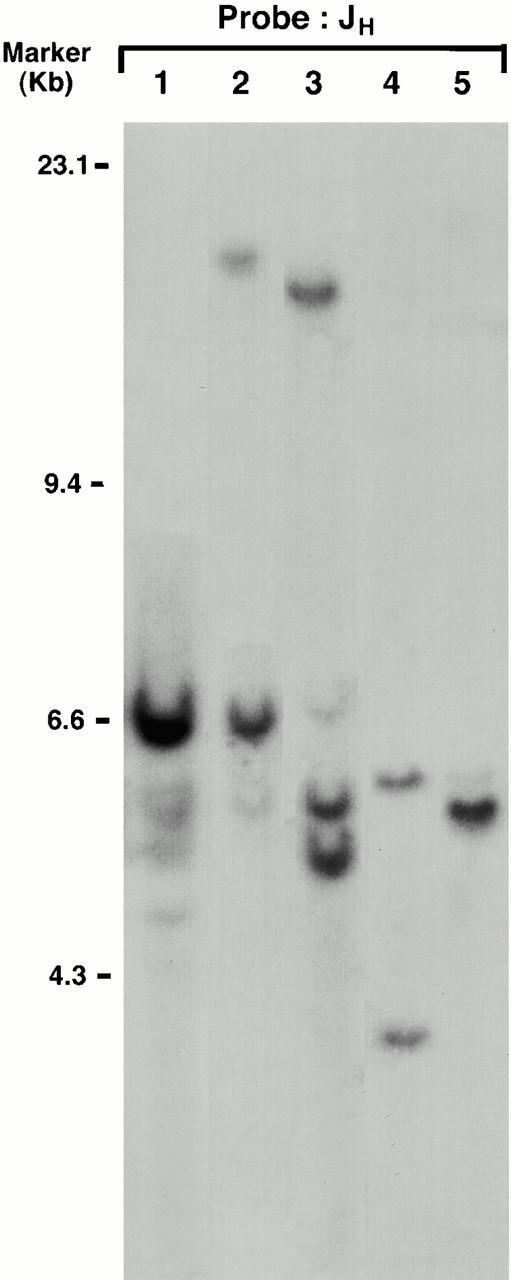
Southern blot analyses of H chain organization in λ-MYC–transgenic mice. DNA from spleen cells of a 6-wk-old transgenic mouse without a lymphoma (lane 1), two founder mice with primary lymphomas (lanes 2 and 3), and the M1 (lane 4) and C1 (lane 5) cell lines generated from a founder mouse and a mouse from the established transgenic line, respectively, was digested with EcoRI separated and hybridized with the J11 JH probe. Size markers are indicated on the left.
Human BL cells are almost universally sIg+, and the Ig variable region sequences of expressed genes are mutated, consistent with the suggestion that the transformed cells have passed through germinal centers 19. To determine whether the Ig genes of mouse BL were mutated, we determined the expressed IgH and Igκ variable sequences of the M1 and C1 cell lines. The IgH V sequences of both lines had single base mutations resulting in amino acid substitutions outside the CDRs. The third CDR of the M1 Igκ gene had a single silent change in CDR3, whereas the C1 sequences were germline (data not shown).
Discussion
The results of this study demonstrate that mice transgenic for a mutated human MYC protooncogene driven by regulatory elements of the Igλ locus develop lymphomas with striking similarities to human BL. Monoclonal tumors with the characteristic starry sky appearance of BL developed from an initially polyclonal population of B cells. LNs and spleens of tumor-bearing mice were populated by uniform populations of IgM+CD19+CD5−CD23− cells, a phenotype similar to that of human BL.
The histopathologic appearance of the lymphomas was consistent for all founder mice and the progeny of the established line, demonstrating that the phenotype cannot be ascribed to specific insertional mutations by the transgenes. The time course of lymphoma development in most founder mice was remarkably homogenous, suggesting that position effects due to random integration of the transgene played a minor if any role in our experiments. Other studies showed that position effects due to random integration of transgenes may give rise to transgene silencing, complicating interpretation of these experiments 14. Position-independent expression of a transgene is recognized as one of the hallmarks of locus control.
Copy number–dependent expression of linked transgenes, established in studies of β globin genes regulated by the homologous LCR, is felt to be a second characteristic of LCR regulation 14. Some studies, however, suggest that inclusion of this standard as an absolute requirement for demonstration of locus control may be unrealistically stringent, particularly if the reporters are genes not normally controlled by the LCR. For example, the β globlin LCR confers neither position-independent nor copy number–dependent expression on a lacZ transgene, even though both features hold if the same LCR is used to regulate a linked β globin gene 20. Parallel studies of the IgH 3′ LCR showed that a linked β globin gene is expressed in a position- but not copy number–independent manner 21. Our studies of MYC regulation by the putative Igλ LCR also indicated a lack of copy number–dependent expression through the indirect readout of latency for lymphoma appearance. This “reporter phenotype” is much less direct than determining β galactosidase activity or β globin levels, because deregulated expression of MYC is not sufficient to induce transformation 8. We therefore tentatively conclude that establishment of locus control suggested by in vitro studies using the λ-MYC construct also occurs in vivo.
Our results differ from those described for mice bearing a series of different MYC transgenes controlled by the intronic IgH and Igκ enhancers 8. Mice with Eμ-Myc transgenes developed lymphomas with near 100% incidence. All tumors had lymphoblastic morphology, and most were sIg− with IgL in germline configuration, features consistent with the diagnosis of precursor B cell LL; however, some tumors were noted to be sIg+. The proportions of pre-B to B cell phenotypes induced by the MYC gene linked to an enhancer on a minigene construct may be affected by transgene copy number, with high numbers of tandem copies and high-level MYC expression favoring immature tumors. This view is supported by the observation that mice carrying single-copy human IgH/c-MYC YAC transgenes developed only IgM+ lymphomas 22; histologic studies of these tumors were not reported.
Features of the latent period for lymphoma development differ markedly for the Eμ-Myc and λ-MYC–transgenic mice. The BM of Eμ-Myc mice 4–7 wk of age is dominated by large pre-B cells, and enlarged spleens were strikingly abnormal in containing ∼25% pre-B cells 23. In contrast, the BMs of λ-MYC–transgenic mice of comparable age were barely distinguishable from those of littermate controls. The slight reductions in the frequency of transgenic BM B cells were reflected in slightly reduced B cell numbers in otherwise normal spleens.
The previously developed Myc transgenics and our new strain also have some features in common. Development of monoclonal disease from a polyclonal background of cells uniformly expressing MYC is characteristic of both Eμ-Myc– and λ-MYC–transgenic mice and is consistent with the concept of a multistep process leading to the malignant transformation of normal cells. It is also in keeping with previous studies demonstrating that Myc translocations occur in preneoplastic B cells in nontransgenic mice prone to plasmacytoma (PCT) development after inoculation of pristane 24.
Development of pre-B cell lymphomas in Eμ-Myc–transgenic mice and mature B cell lymphomas in λ-MYC mice might be related to the fact that the Eμ enhancer is active already in the pre-B cell stage, whereas the L chain enhancers used in the current studies become operative several days later in B cell differentiation. This seems unlikely, because human BL cases with IgH-related t(8;14) translocations are histologically and phenotypically indistinguishable from cases with variant IgL translocations, indicating that the overall activity imposed by the different Ig loci on the translocated MYC gene is very similar if not identical.
One possible explanation for this seeming paradox is that the 3′ Ig enhancers play a decisive role in the development of BL. This concept centers on the fact that the MYC gene is invariably located upstream from the 3′ enhancers of the Ig loci involved in the translocation. For the IgL loci, the activity of single Ig enhancers is weak in comparison to the combination of enhancers that drive Ig expression under physiologic conditions 9 10. While 3′ enhancers function constitutively to interact with 5′ enhancers, they are also responsive to physiologic stimuli, including cross-linking of the BCR 25 or ligation of CD40 26 as well as activation by LPS plus CD40 27. We therefore propose that B cell stimulation leading to activation of Ig 3′ enhancers results in enforced high-level MYC expression as a critical determinant of BL pathogenesis. A requirement for an intact BCR in this signaling matrix could explain why only mature lymphomas with functional Ig receptors are induced.
Expression of a functional BCR as an invariant feature of BL suggests antigenic stimulation as a possible component of pathogenesis. This concept is supported by findings that almost all IgH V region sequences from endemic, sporadic, and AIDS-associated BL cases were mutated to varying extents, ranging from a single point mutation up to 15% of IgH V and J region sequences 19. These data indicate that, for most cases, the cells of origin were Ag-experienced and had passed through germinal centers. The cell lines established from the transgenic mice had single base changes in the productive H chain and L chain alleles, none affecting structure of the CDRs.
Ig hypermutation after antigenic stimulation may be a consistent feature of human BL because it provides the means of accumulating mutations in the coding region of the translocated MYC gene, rendering the MYC protein less regulatable and more oncogenic 28. If so, we would predict that a wild-type MYC gene substituted for the mutant allele in our constructs would be less oncogenic. A second prediction from this model is that lymphomas would develop with longer latency in mice raised under specific pathogen-free conditions. Both of these issues are now being investigated.
Our finding that a reconstructed BL chromosomal breakpoint can recapitulate essential features of the disease in mice strongly supports the notion that there is a link between cytogenetic aberrations and the histopathologic and immunophenotypic presentation of a given tumor and that this link is conserved between mice and humans. Nonetheless, it is noteworthy that the same type of chromosomal translocations observed in human BL are hallmarks of PCT in mice 29. In PCT, the Myc/IgH breakpoints are usually located in the Igα switch region, whereas in human BL, they occur most frequently in the Igμ joining or switch region; however, BL cases with breakpoints in Igα have been reported, as have PCT mice with breakpoints in the Igμ gene. Furthermore, there is no apparent difference in the molecular architecture of variant IgL translocations in BL versus PCT that might explain the different histopathologic presentations of these lymphomas.
The reason that apparently identical chromosomal translocations are associated with different hematologic malignancies such as BL and PCT is not understood. PCT develops in peritoneal granulomas induced by mineral oil and can be prevented by treatment with the antiinflammatory drug indomethacin 30. This suggests that the microenvironment in which a tumor arises impacts on the stage to which the malignant cell will differentiate. According to this model, pristane treatment of our transgenic mice should give rise to PCT, presumably before the onset of BL. Attempts to address this question directly are in progress.
Acknowledgments
We thank Ms. W. Dubois for care of the mice, Dr. Larry Lantz and Mr. Calvin Eigsti for assistance in the bone marrow studies, Dr. J.W. Hartley for review of the manuscript, and Ms. B.R. Marshall for expert editorial assistance.
This work was supported in part by grants to A. Polack and G.W. Bornkamm from the Deutsche Forschungsgemeinschaft, Deutsche Krebshilfe, and Fonds der Chemischen Industrie.
References
- Burkitt D.A. A sarcoma involving the jaws in African children. Br. J. Surg. 1958;46:218–223. doi: 10.1002/bjs.18004619704. [DOI] [PubMed] [Google Scholar]
- Magrath I.T., Bhatia K. Pathogenesis of small noncleaved cell lymphomas (Burkitt's lymphoma) In: Magrath I., editor. The Non-Hodgkin's Lymphomas. Oxford University Press; New York: 1997. pp. 385–409. [Google Scholar]
- Manolov G., Manolov Y. Marker band in one chromosome 14 from Burkitt lymphomas. Nature. 1972;237:33–34. doi: 10.1038/237033a0. [DOI] [PubMed] [Google Scholar]
- Dalla-Favera R., Bregni M., Erikson J., Patterson D., Gallo R.C., Croce C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA. 1982;79:7824–7827. doi: 10.1073/pnas.79.24.7824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taub R., Kirsch I., Morton C., Lenoir G., Swan D., Tronick S., Aaronson S., Leder P. Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and mouse plasmacytoma cells. Proc. Natl. Acad. Sci. USA. 1982;79:7837–7841. doi: 10.1073/pnas.79.24.7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler R., Joos S., Delecluse H.-J., Klobeck G., Vuillaume M., Lenoir G.M., Bornkamm G.W., Lipp M. Breakpoints of Burkitt's lymphoma t(8;22) translocations map within a distance of 300 kb downstream of MYC. Genes Chromosomes Cancer. 1994;9:282–287. doi: 10.1002/gcc.2870090408. [DOI] [PubMed] [Google Scholar]
- Harris N.L., Jaffe E.S., Diebold J., Flandrin G., Muller-Hermelink H.K., Vardiman J., Lister T.A., Bloomfield C.D. The World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissuesreport of the Clinical Advisory Committee meeting, Airlie House, Virginia, November, 1997. J. Clin. Oncol. 1999;17:3835–3849. doi: 10.1200/JCO.1999.17.12.3835. [DOI] [PubMed] [Google Scholar]
- Adams J.M., Harris A.W., Pinkert C.A., Corcoran L.M., Alexander W.S., Cory S., Palmiter R.D., Brinster R.L. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- Gerbitz A., Mautner J., Geltinger C., Hortnagel K., Christoph B., Asenbauer H., Klobeck G., Polack A., Bornkamm G.W. Deregulation of the proto-oncogene c-myc through t(8;22) translocation in Burkitt's lymphoma. Oncogene. 1999;18:1745–1753. doi: 10.1038/sj.onc.1202468. [DOI] [PubMed] [Google Scholar]
- Hortnagel K., Mautner J., Strobl L.J., Wolf D.A., Christoph B., Geltinger C., Polack A. The role of immunoglobulin κ elements in c-myc activation. Oncogene. 1995;19:1393–1401. [PubMed] [Google Scholar]
- Polack A., Hortnagel K., Christoph B., Baier B., Falk M., Mautner J., Geltinger C., Bornkamm G.W., Kempkes B. c-myc activation renders proliferation of Epstein-Barr virus (EBV)-transformed cells independent of EBV nuclear antigen 2 and latent membrane protein 1. Proc. Natl. Acad. Sci. USA. 1996;93:10411–10416. doi: 10.1073/pnas.93.19.10411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobl L.J., Kohlhuber F., Mautner J., Polack A., Eick D. Absence of a paused transcription complex from the c-myc P2 promoter of the translocation chromosome in Burkitt's lymphoma cellsimplications for the c-myc P1/P2 promoter shift. Oncogene. 1993;8:1437–1447. [PubMed] [Google Scholar]
- Cesarman E., Dalla-Favera R., Bentley D., Groudine M. Mutations in the first exon are associated with altered transcription of c-myc in Burkitt lymphoma. Science. 1987;238:1272–1275. doi: 10.1126/science.3685977. [DOI] [PubMed] [Google Scholar]
- Li Q., Harju S., Peterson K.R. Locus control regionscoming of age at a decade plus. Trends Genet. 1999;15:403–408. doi: 10.1016/s0168-9525(99)01780-1. [DOI] [PubMed] [Google Scholar]
- Fredrickson T.N., Harris A.W. Atlas of Mouse Hematopathology 2000. Harwood Academic Publishers; Australia: pp. 84–85 [Google Scholar]
- Hardy R.R., Carmack C.E., Shinton S.A., Kemp J.D., Hayakawa K. Resolution and characterization of pro B and pre-pro B cell stages in normal mouse bone marrow. J. Exp. Med. 1991;173:1213–1225. doi: 10.1084/jem.173.5.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carsetti R., Köhler G., Lamers M.C. Transitional B cells are the target of negative selection in the B cell compartment. J. Exp. Med. 1995;181:2129–2140. doi: 10.1084/jem.181.6.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K., Asano M., Shinton S.A., Gui M., Allman D., Stewart C.L., Silver J., Hardy R.R. Positive selection of natural autoreactive B cells. Science. 1999;285:113–116. doi: 10.1126/science.285.5424.113. [DOI] [PubMed] [Google Scholar]
- Chapman C.J., Wright D., Stevenson F.K. Insight into Burkitt's lymphoma from immunoglobulin variable region gene analysis. Leuk. Lymphoma. 1998;30:257–267. doi: 10.3109/10428199809057539. [DOI] [PubMed] [Google Scholar]
- Guy L.-G., Kothary R., de Repentigny Y., Delvoye N., Ellis J., Wall L. The β-globin locus control region enhances transcription of but does not confer position-independent expression onto the lacZ gene in transgenic mice. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:3713–3721. [PMC free article] [PubMed] [Google Scholar]
- Chauveau C., Jansson E.A., Muller S., Cogne M., Pettersson S. Cutting edgeIg heavy chain 3′ HS1-4 directs correct spatial position-independent expression of a linked transgene to B lineage cells. J. Immunol. 1999;163:4637–4641. [PubMed] [Google Scholar]
- Butzler C., Zou X., Popov A.V., Bruggemann M. Rapid induction of B-cell lymphomas in mice carrying a human IgH/c-myc YAC. Oncogene. 1997;14:1383–1388. doi: 10.1038/sj.onc.1200968. [DOI] [PubMed] [Google Scholar]
- Langdon W.Y., Harris A.W., Cory S., Adams J.M. The c-myc oncogene perturbs B lymphocyte development in Eμ-myc transgenic mice. Cell. 1986;47:11–18. doi: 10.1016/0092-8674(86)90361-2. [DOI] [PubMed] [Google Scholar]
- Muller J.R., Potter M., Janz S. Differences in the molecular structure of c-myc-activating recombinations in murine plasmacytomas and precursor cells. Proc. Natl. Acad. Sci. USA. 1994;91:12066–12070. doi: 10.1073/pnas.91.25.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P.A., Thompson C.B., Pettersson S. IgM receptor-mediated transactivation of the IgH 3′ enhancer couples a novel Elf-1-AP-1 protein complex to the developmental control of enhancer function. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:4501–4513. doi: 10.1002/j.1460-2075.1995.tb00129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant P.A., Andersson T., Neurath M.F., Arulampalam V., Bauch A., Muller R., Reth M., Pettersson S.A. T cell controlled molecular pathway regulating the IgH locusCD40-mediated activation of the IgH 3′ enhancer. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:6691–6700. [PMC free article] [PubMed] [Google Scholar]
- Meyer K.B., Mufti D.A.H. Post-translational regulation of E2A proteins via lipopolysaccharide and CD40 signaling. Eur. J. Immunol. 2000;30:719–724. doi: 10.1002/1521-4141(200002)30:2<719::AID-IMMU719>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Bahrsam F., von der Lehr N., Cetinkaya C., Larsson L.-G. c-Myc hot spot mutations in lymphomas result in inefficient ubiquitination and decreased proteasome-mediated turnover. Blood. 2000;95:2104–2110. [PubMed] [Google Scholar]
- Potter M., Wiener F. Plasmacytomagenesis in micemodel of neoplastic development dependent on chromosomal translocations. Carcinogenesis. 1992;13:1681–1697. doi: 10.1093/carcin/13.10.1681. [DOI] [PubMed] [Google Scholar]
- Potter M., Wax J.S., Anderson A.O., Nordan R.P. Inhibition of plasmacytoma development in BALB/c mice by indomethacin. J. Exp. Med. 1985;161:996–1012. doi: 10.1084/jem.161.5.996. [DOI] [PMC free article] [PubMed] [Google Scholar]