

Abstract
A series of cationic porphyrins carrying 1–3 meso-N-pyridinium groups has been synthesised, and their binding to G-quadruplex DNA has been explored by surface plasmon resonance (SPR) and circular dichroism spectroscopy. Two trans substituents appear to be sufficient for tight binding; preferential binding to the anti-parallel intramolecular human telomeric DNA was observed for the A2 trans and A3 porphyrins. The A2 trans is able to induce the formation of an anti-parallel G-quadruplex in a K+ free solution, mimicking the effect of a molecular chaperone.
Introduction
Intramolecular G-quadruplexes are four stranded DNA secondary structures formed by G-rich DNA sequences containing four tracks of at least two consecutive guanines.1 In the presence of monovalent cations (preferentially K+), the tetrads of hydrogen bonded guanines are held together by C=O⋯M+ and π–π or hydrophobic interactions. During the past decade, there has been growing interest in the structure, recognition and function of intramolecular G-quadruplexes. The best studied example is the human telomeric DNA quadruplex that leads to inhibition of telomere extension by telomerase, an enzyme active in most cancer cells.2-4 Recently, a large number of putative quadruplex forming sequences have also been identified throughout the human genome.5 A few examples of quadruplexes located in the promoter regions of oncogenes have been postulated to act as regulator elements controlling the level of expression of these genes.6-9
There is now considerable interest in designing new quadruplex binding ligands as effective therapeutic agents. Most of the molecules reported to date contain extended aromatic surfaces with flat ring systems and are believed to interact with the external tetrad of the quadruplex.10,11 Tetra-N-methylpyridiniumporphyrin, H2-TMPyP4, was previously reported to bind to the intramolecular human telomeric quadruplex with low micromolar affinity.12,13 More recently it was also shown to bind to other quadruplexes with similar affinities.14 A number of tetra-substituted porphyrins have been designed to target quadruplex DNA.15,16 Herein, we describe the synthesis and quadruplex binding affinities of new mono-, bi-, and tri-meso substituted porphyrins in an attempt to define the requisite structural features.
Results and discussion
In order to determine what structural changes to the substitution pattern of H2-TMPyP4 can be tolerated, four porphyrins (A1, A2 cis/trans and A3. Fig. 1) lacking meso substituents were prepared. These porphyrins systematically vary the number and position of N-methylpyridinium groups. The synthesis of A2 trans was previously reported by R. K. Wall et al. in very low yields,17 but A1, A2 cis and A3 have not been previously reported. All porphyrins were synthesised by rational methods, mainly by the MacDonald-type of dipyrromethane coupling,18 as outlined in Scheme 1. The trifluoroacetic acid (TFA) catalysed condensation of 1 with 2,2′-dipyrromethane 2 in THF yielded 4-pyridylporphyrin 3 (7–10% after purification by chromatography). In order to obtain the 5,15-di(4-pyridyl)porphyrin 7, 5-(4-pyridyl)dipyrromethane 5 was reacted with trimethyl orthoformate 6 in the presence of catalytic amount of trichloroacetic acid (TCA) under a nitrogen atmosphere for 4 h, followed by the addition of pyridine and further stirring for 12 h. Purification was carried out by column chromatography giving 9% of porphyrin. The 5,10-di-(4-dyridyl)porphyrin 11 was obtained by the condensation of tripyrromethane 9 with 2,5-bis(hydroxymethyl)pyrrole 10 in DCM, under nitrogen atmosphere catalysed by TFA and purified by column chromatography (3%). The condensation of 5-(4-pyridyl)dipyrromethane 5 with trimethyl orthoformate 6 and with pyridine-3-carboxaldehyde 13, was carried out at room temperature under nitrogen atmosphere and catalysed by TCA, giving after column chromatography purification three porphyrins: 5,15-di(4-pyridyl)porphyrin 7, tetrapyridylporphyrin, and the desired 5,10,15-tri(4-pyridyl)porphyrin 15 with a 0.8% yield.
Fig. 1.

Structure of the four N-methylpyridinium porphyrins used in this study (A1, A2 cis, A2 trans, A3).
Scheme 1.
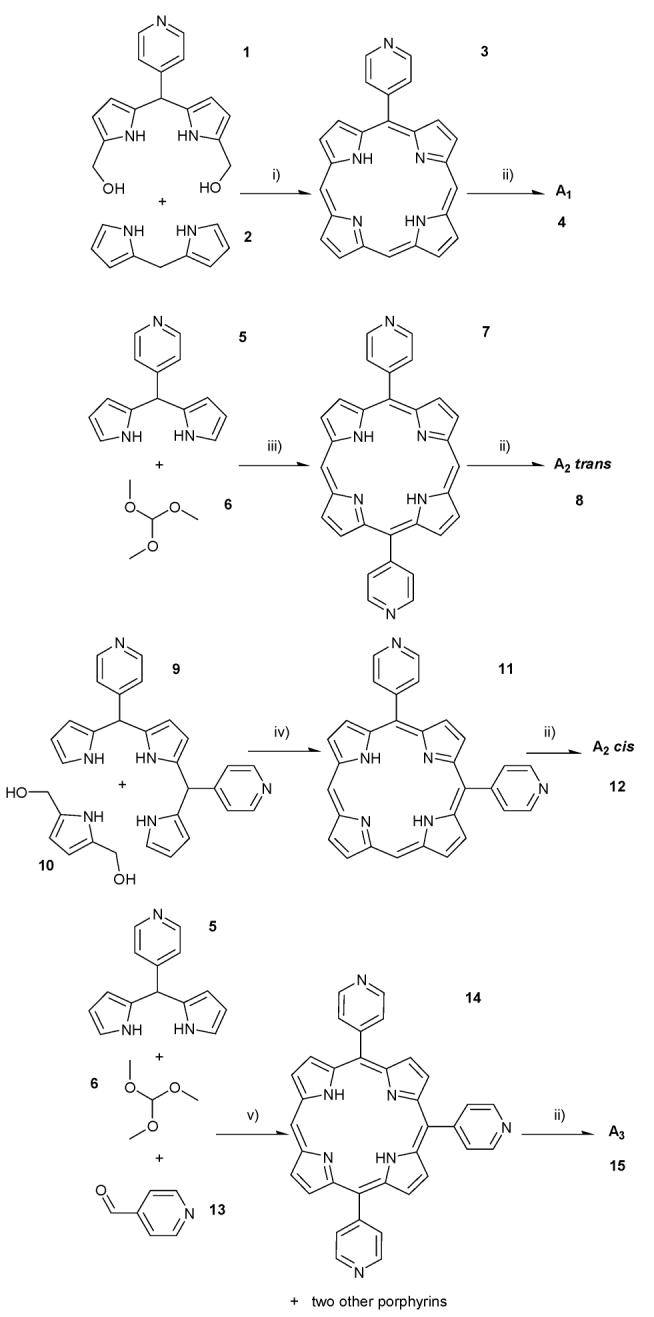
Synthetic pathways for the synthesis of the A1, A2 cis/trans and A3 porphyrins. i) THF, N2(g) and TFA. After 1 h of reaction DDQ was added. ii) CH3I in DMF. iii) DCM, TCA, N2(g) and after 4 h pyridine was added. iv) DCM, TFA under N2(g). v) DCM, TCA under N2(g). After 4 h pyridine was added, followed by DDQ after 3 h.
The quaternisation of the pyridyl groups was achieved by methylation with iodomethane in DMF at 40 °C yielding quantitatively the final A1, A2 cis/trans and A3 porphyrins without need for further purification.
To improve the solubility of porphyrins A1 and A2 cis/trans in water, an anion exchange from iodide to chloride was carried out. While this approach was successful for A2 cis/trans, the water solubility of A1 as either an iodide or chloride salt was too poor to allow DNA binding studies.
The binding of the four porphyrin derivatives A2 cis, A2 trans, A3 and H2-TMPyP4 (control porphyrin) to duplex (5-biotin-[GGCATAGTGCGTGGGCGTTAGC]-3 hybridised with its complementary sequence) and quadruplex DNA (5-biotin-[GTTA(GGGTTA)4GG]-3) was investigated using surface plasmon resonance (SPR), allowing binding events to be monitored in real time, without the use of labels (example shown in Fig. 2).19 The results are summarised in Table 1.
Fig. 2.

Sensorgram overlay obtained for 6 different concentrations of the A2 trans porphyrin (6.25, 3.12, 1.56, 0.78, 0.39, 0.20 μM, top to bottom) binding to the human telomeric quadruplex and the corresponding binding curve obtained using the BIAeval software (BIAcore).
Table 1.
Dissociation constants (KD) of H2-TMPyP4 (control porphyrin), A2 cis, A2 trans and A3 porphyrins to quadruplex DNA. The SPR experiments were carried out in 50 mM Tris·HCl pH 7.4, 100 mM KCl using a streptavidin functionalised chip on a Biacore 2000 SPR biosensor
| Porphyrins | KD/(μM) |
|---|---|
| H2-TMPyP4 | 0.63 ± 0.08 |
| A2 cis | 18.90 ± 2.80 |
| A2 trans | 0.83 ± 0.10 |
| A3 | 5.88 ± 0.90 |
As shown previously with tetrasubstituted porphyrins,19 all new porphyrins we tested have similar affinities for duplex (data not shown) and quadruplex DNA. However the number of peripheral meso pyridinium groups and their location proved to be important for good binding. The A2 trans porphyrin presents the lowest dissociation constant (KD = 0.83 ± 0.10 μM), comparable to that obtained for H2-TMPyP4 (KD = 0.63 ± 0.08 μM). The porphyrins A2 cis and A3 show significantly weaker binding (KD values of 18.9 ± 2.80 μM and 5.88 ± 0.90 μM respectively) (Table 1). These results indicate that two pyridinium substituents can be sufficient for quadruplex recognition as long as they are trans orientated to each other. Any other combination of less than four pyridiniums seems to significantly reduce quadruplex binding. This trans substitution pattern is comparable in geometry to other reported quadruplex binding platforms (e.g. 3,6-disubstituted acridines) that simultaneously target the external guanine and two opposite grooves.20
It is noteworthy that all these porphyrins seem to bind to the quadruplex structure with a high stoichiometry. Similar results have been observed by others using SPR and other techniques.12,16
The concentrations of porphyrin required to run SPR experiments are below those leading to self aggregation (<2 μM). This value was obtained by running Beer–Lambert aggregation experiments (data not shown). Hence, self aggregation of the porphyrins appears to occur after binding to G-quadruplex DNA. This aggregation phenomenon has also been described for H2-TMPyP4 complexes with G-quadruplex; again it was reported that this porphyrin exists as a monomer in water even in the presence of concentrated inorganic salts.21-23
The human telomeric DNA can exist as a mixture of the parallel and anti-parallel G-quadruplex conformations when in a K+ solution.24-26 In order to investigate if the porphyrins were selective for any particular G-quadruplex conformation, circular dichroism (CD) experiments were performed. Those experiments were carried out on the 5-GGATTGGGATTGGGATTGGGATTGGG-3 (Htelo) DNA sequence, which was previously annealed in a 50 mM Tris·HCl pH 7.4, 150 mM KCl buffer. The CD spectra of 10 μM of Htelo quadruplex showed significant changes when in the presence of 15 equivalents of the A2 trans and the A3 porphyrin (Fig. 3). It was observed that those two porphyrins prefer the anti-parallel conformation which is indicated by the characteristic positive peak near 295 nm.24 On the other hand, the A2 cis porphyrin did not seem to alter the CD spectra of the G-quadruplex significantly, suggesting no specific preference for any of the two conformations, supporting the SPR results.
Fig. 3.

CD spectra of 10 μM of Htelo quadruplex (solid line) in the presence of 15 equiv. of: A2 trans (short dash dotted line); A3 (dotted line); and A2 cis (dashed line). Measurements were carried out at 20 °C in a 50 mM Tris·HCl pH 7.4, 150 mM KCl buffer.
It was also of interest to investigate if the porphyrins could induce the formation of a G-quadruplex in a K+ free buffer with non-annealed Htelo DNA. The A2 trans was the only porphyrin that clearly could achieve this: CD spectra show the suppression of the 255 nm positive peak of the non-annealed Htelo (solid line) in favour of the positive peak around 290 nm of the anti-parallel G-quadruplex conformation (dotted line) (Fig. 4). A clear isoelliptic point at around 270 nm suggests a transition between two conformation states upon addition of the porphyrin.
Fig. 4.

CD spectra of 10 μM of non-annealed Htelo in a 50 mM Tris·HCl pH 7.4 buffer (solid line) in the presence of 2.2 equiv. (dashed line) and 12 equiv. (dotted line) of A2 trans.
Conclusions
A series of porphyrins systematically lacking meso pyridinium substituents has been synthesised and their binding to telomeric G-quadruplex was assessed by SPR and CD spectroscopy. According to the SPR results two meso substituents in a trans orientation proved to be sufficient for tight quadruplex binding. Both A2 trans and A3 porphyrins showed preferential binding to the anti-parallel conformation of the intramolecular human telomeric quadruplex, and the A2 trans is capable of acting as a molecular chaperone by inducing the formation of the anti-parallel G-quadruplex in a K+ free buffer.
Experimental
1H NMR spectra were recorded on Bruker DPX 400 and DRX 500 instruments, whilst 13C spectra were collected on a Bruker DRX 500, equipped with a Cryopobe. Chemical shifts are quoted in parts per million; J-values are in Hz. Exact mass spectra were recorded on a Waters-LCT Premier-time of flight mass spectrometer. UV-Vis spectra were recorded on a HP 8452A Diode Array UV-Vis spectrophotometer. Circular dichroism experiments were performed on a JASCO model J-810 circular dichroism spectrapolarimeter equipped with a Peltier temperature controller. Preparative thin layer chromatography were carried out on Kieselgel 60 F254 (Merck) 0.2 mm plates. The porphyrin precursors were visualised by UV-visible absorption (245–365 nm). Flash column chromatography was carried out using Kieselgel 60 (Merk) 230–400 mesh and distilled solvents. Pyrrole was distilled before use and all other chemicals were purchased as reagent grade, being used without any further purification.
1,9-Diformyl-5-(4-pyridyl)dipyrromethane
Vilsmeier reagent was prepared by adding POCl3 (32 mmol, 3.0 mL) drop wise under N2 to DMF (20 mL) at 0 °C. 5-(4-pyridyl)dipyrromethane 527(1.0 g, 4.5 mmol) was dissolved in DMF (15 mL) under N2 and the solution was cooled to 0 °C. To this stirred solution was added drop wise the Vilsmeier reagent (7.7 mL, 10.8 mmol) and the mixture was stirred at 0 °C for 1.5 h. Saturated aqueous sodium acetate solution (50 mL) was carefully added and the mixture was stirred for 4 h at room temperature. The solution was extracted three times with ethyl acetate, the extracts were washed with brine and water, dried over Na2SO4 and evaporated in vacuo to give a brown oil. This was purified by flash chromatography (silica: eluted initially with chloroform, then gradually increasing to chloroform–methanol 10 : 1) to give after evaporation of the solvent in vacuo, 58% (729 mg) of diformylated dipyrromethane. 1H NMR (500 MHz, CDCl3), δ = 11.10 (br s, 2H), 9.16 (s, 2H), 8.51 (d, J = 6.1 Hz, 2H), 7.20 (d, J = 6.1 Hz, 2H), 6.85–6.86 (m, 2H), 6.03–6.04 (m, 2H) and 5.58 (s, 1H) ppm. 13C NMR (500 MHz Cryo, CDCl3), δ = 179.3, 150.1, 148.4, 139.9, 133.0, 123.6, 122.4, 111.9 and 43.8 ppm. Exact mass: calculated: 280.1086; found: 280.1088 (M + H+).
4-Pyridylporphyrin 3
To a stirred solution of 1,9-diformyl-5-(4-pyridyl)dipyrromethane (165 mg, 0.59 mmol) in THF–methanol (10 : 1, 30 mL) was added, under N2, NaBH4 (0.46 g, 11.8 mmol) in small portions (every 2 min). After 40 min at room temperature, the reaction mixture was poured into a mixture of saturated aqueous ammonium chloride and chloroform (1 : 1, 50 mL). The organic phase was isolated, washed with water (2 times) and dried. The solvent was evaporated in vacuo. The residue was immediately dissolved in acetonitrile (56 mL), 2,2′-dipyrromethane 228 (86.2 mg, 0.59 mmol) was added. The resulting mixture was stirred for 5 min at room temperature, then TFA (0.54 mL, 7.1 mmol) was added, and the mixture was stirred for one hour. DDQ (0.4 g, 1.8 mmol) in toluene (10 mL) was added. After mixing for 1 h at room temperature, triethylamine (1 mL, 7.1 mmol) was added. The crude mixture was evaporated to dryness, dissolved in chloroform and purified by preparatory TLC (eluted by chloroform–methanol 10 : 0.5). The fraction containing the porphyrin (as assessed by UV-visible) was collected and extracted from the silica. Evaporation of the solvent in vacuo afforded 7 to 10% of red product (16.0 to 22.8 mg). λmax(CH2Cl2)/nm 400 (log ε/dm3 mol−1 cm−1 4.53), 494 (3.12), 530 (1.67) and 566 (1.49). 1H NMR (500 MHz, acetone-d6), δ = 10.50 (s, 2H), 10.49 (s, 1H), 9.64–9.66 (m, 4H), 9.63 (d, J = 4.6 Hz, 2H), 9.25 (d, J = 6.5 Hz, 2H), 9.17 (d, J = 4.6 Hz, 2H), 8.81 (d, J = 6.5 Hz, 2H) and −3.59 (br s, 2H, NH) ppm. 13C NMR (500 MHz Cryo, acetone-d6), δ = 158.3, 150.2, 150.1, 149.6, 147.7, 145.0, 132.9, 132.7, 132.4, 132.1, 130.7, 113.1, 106.0 and 105.9 ppm. Exact mass: calculated: 388.1562; found: 388.1558 (M + H+).
5,15-Di(4-pyridyl)porphyrin 7
To a stirred solution of 5-(4-pyridyl)dipyrromethane 5 (500 mg, 2.24 mmol) and trimethyl orthoformate 6 (18 mL, 0.17 mol) in dry DCM (630 mL) previously degassed by bubbling with argon, was added drop wise, over 15 min, a solution of TCA (8.83 g, 54 mmol) in dry DCM (227 mL). After the addition was complete, the solution was stirred in the dark and at room temperature, for 4 h, before being quenched with pyridine (15.6 mL) and stirred, again in the dark for further 17 h. The solution was the purged with air for 10 min and stirred, under ambient lighting conditions, for 4 h. Solvent was evaporated in vacuo, first using water aspiration, and then vacuum overnight. The resulting black solid was preadsorbed onto silica and loaded onto the top of a flash chromatography column. The crude product was eluted from the column using a mixture of chloroform–methanol (10 : 1). Evaporation of the eluent afforded 9% of a purple solid (91 mg). 1H NMR (500 MHz, CDCl3), δ = 10.36 (s, 1H), 9.43 (d, J = 4.6 Hz, 4H), 9.05 (d, J = 5.4 Hz, 4H), 9.03 (d, J = 4.6 Hz, 4H), 8.19 (d, J = 5.8 Hz, 4H) and −3.19 (br s, 2H, NH) ppm.
5,10-Di(4-pyridyl)porphyrin 11
5,10-Di(4-pyridyl)tripyrromethane27 (0.45 g, 1.0 mmol) was added to the stirred solvent of dry DCM (200 mL) previously degassed by bubbling with argon at room temperature protected from light. A solution of 2,5-bis(hydroxymethyl)pyrrole 1029 (0.127 g, 1.0 mmol) in methanol (5 mL) was added to the stirred solution. After 10 min, TFA (15.6 μL, 0.2 mmol) was added with a micro syringe, and then the reaction mixture was stirred for 1.5 h. DDQ (0.22 g, 1.0 mmol) was then added, followed by triethylamine (0.03 mL, 0.2 mmol). The crude mixture was evaporated to dryness, dissolved in chloroform and purified by preparative TLC (eluted by chloroform–methanol 10 : 1). The fraction containing the porphyrin (as assessed by UV-visible) was collected and extracted from the silica. Evaporation of the solvent in vacuo afforded 3% purple product (14 mg). λmax(CH2Cl2)/nm 406 (log ε/dm3 mol−1 cm−1 5.18), 500 (4.01), 578 (3.62), 660 (3.20) and 698 (3.16). 1H NMR (500 MHz, CDCl3), δ = 10.28 (s, 2H), 9.46 (s, 2H), 9.39 (d, J = 4.6 Hz, 2H), 9.04 (d, J = 5.8 Hz, 4H), 8.97 (d, J = 4.6 Hz, 2H), 8.90 (s, 2H), 8.77 (d, J = 5.8 Hz, 4H) and −3.45 (br s, 2H, NH) ppm. 13C NMR (500 MHz Cryo, CDCl3), δ = 150.2, 148.3, 130.0–133.0, 129.5, 116.6 and 105.2 ppm. Exact mass: calculated: 465.1828, found 465.1818 (M + H+).
5,10,15-Tri(4-pyridyl)porphyrin 14
To a stirred solution of 5-(4-pyridyl)dipyrromethane 5 (500 mg, 2.24 mmol), 4-pyridinecarboxaldehyde 13 (0.4 mL, 4.1 mmol) and trimethyl orthoformate 6 (9 mL, 82.5 mmol) in dry DCM (630 mL) previously degassed by bubbling with argon, was added drop wise, over 15 min, a solution of TCA (8.83 g, 54 mmol) in dry DCM (227 mL). After the addition was complete, the solution was stirred in the dark and at room temperature, for 4 h, before being quenched with pyridine (15.6 mL) stirred, again in the dark for further 2 h and then DDQ was added (0.3 g, 1.3 mmol). Solvent was evaporated in vacuo, first using water aspiration, and then vacuum overnight. The resulting black solid was preadsorbed onto silica and loaded onto the top of a flash chromatography column. The crude product was eluted from the column using a mixture of chloroform–methanol (10 : 1); the second porphyrinic fraction corresponded to the expected product. Evaporation of the eluent afforded 0.8% of a purple solid (5.1 mg). λmax(CH2Cl2)/nm 414 (log ε/dm3 mol−1 cm−1 4.97), 508 (3.86), 580 (3.38) and 636 (2.64). 1H NMR (500 MHz, CDCl3), δ = 10.40 (s, 1H), 9.44 (d, J = 4.6 Hz, 2H), 9.07 (br s), 9.02 (d, J = 4.8 Hz, 2H), 9.00 (d, J = 4.6 Hz, 2H), 8.92 (d, J = 4.8 Hz, 2H), 8.75 (d, J = 5.6 Hz, 2H), 8.18 (d, J = 5.6 Hz, 4H), 7.65 (dd, J = 6.6, 1.5 Hz, 2H) and −3.06 (br s, 2H, NH) ppm. 13C NMR (500 MHz Cryo, CDCl3), δ = 150.9, 149.2, 148.5, 147.0, 132.8, 131.6, 131.1, 130.1, 129.4, 123.5, 117.5, 114.8 and 107.6 ppm. Exact mass: calculated: 542.2093; found: 542.2079 (M + H+).
General procedure for the methylation of the porphyrins pyridyl groups
The quaternisation of the pyridyl groups was achieved by methylation with a large excess of iodomethane in DMF, at 40 °C for 3 to 7 h, in quantitative yields. The anion-exchange was performed with Dowex 1 × 2–200 in the chloride form, which was shaken together with the previously dissolved porphyrins in 20% acetone 80% water for 2 h. The resin was then filtered off and washed with water, giving the final porphyrins without any need of further purification.
5-(N-Methylpyridinium-4-yl)porphyrin 4
λmax(MeCN)/nm 398 (log ε/dm3 mol−1 cm−1 4.84), 496 (3.89), 538 (3.69) and 580 (3.73). 1H NMR (500 MHz, CD3NO2), δ = 10.59 (s, 2H), 10.54 (s, 1H), 9.83 (d, J = 4.6 Hz, 2H), 9.66 (s, 4H), 9.23 (d, J = 6.5 Hz, 2H), 9.18 (d, J = 4.6 Hz, 2H), 8.98 (d, J = 6.5 Hz, 2H), 4.86 (s, 3H) and −3.77 (br s, 2H, NH) ppm. 13C NMR (500 MHz Cryo, CD3NO2), δ = 160.3, 144.5, 134.4, 133.3, 133.6, 130.6, 112.2, 107.4 and 49.2 ppm. Exact mass: calculated: 402.1719; found: 402.1711 (M).
5,15-Di(N-methylpyridinium-4-yl)porphyrin 8
1H NMR (500 MHz, DMSO), δ = 10.85 (s, 2H), 9.86 (d, J = 4.7 Hz, 4H), 9.53 (d, J = 6.6 Hz, 4H), 9.25 (d, J = 4.7 Hz, 4H), 9.08 (d, J = 6.6 Hz, 4H), 4.74 (s, 6H) and −3.34 (br s, 2H, NH) ppm.
5,10-Di(N-methylpyridinium-4-yl)porphyrin 12
λmax(MeCN)/nm 412 (log ε/dm3 mol−1 cm−1 5.04), 504 (4.01), 574 (3.52) and 632 (2.95). 1H NMR (500 MHz, CD3CN (75%) and CD3OD (25%)), δ = 10.61 (s, 2H), 9.66 (br s, 4H), 9.18 (d, J = 4.5 Hz, 4H), 9.07 (br s, 4H), 8.88 (d, J = 4.5 Hz, 4H) and 4.7 (s, 6H) ppm. 13C NMR (500 MHz Cryo, CD3CN (75%) and CD3OD (25%)), δ = 159.1, 145.0, 134.1, 114.2, 108.3 and 49.4 ppm. Exact mass: calculated: 247.1104; found: 247.1070 (M).
5,10,15-Tri(N-methylpyridinium-4-yl)porphyrin 15
λmax(MeCN)/nm 420 (log ε/dm3 mol−1 cm−1 5.04), 510 (4.21), 548 (4.08) and 580 (4.07). 1H NMR (500 MHz, CD3NO2), δ = 10.73 (s, 1H), 9.72 (d, J = 4.7 Hz, 2H), 9.28 (d, J = 6.5 Hz, 4H), 9.26 (d, J = 6.7 Hz, 2H), 9.18 (d, J = 4.7 Hz, 2H), 9.16 (d, J = 4.9 Hz, 2H), 9.11 (d, J = 4.9 Hz, 2H), 8.97 (d, J = 6.5 Hz, 4H), 8.94 (d, J = 6.7 Hz, 2H) and 4.86 (s, 9H) ppm. 13C NMR (500 MHz Cryo, CD3NO2), δ = 160.0, 159.2, 144.9, 144.8, 134.1, 133.9, 117.0, 115.4, 109.6 and 49.4 ppm. Exact mass: calculated: 195.4234; found: 195.4224 (M).
SPR studies
Surface plasmon resonance was performed on a Biacore 2000 instrument, using degassed Tris·HCl running buffer (50 mM Tris·HCl, 100 mM KCl, pH 7.4). Sensor chips (Type SA, Biacore) were loaded with approximately 600 RU of Htelo (and duplex DNA as a control). Six serial dilutions of compound were injected at a flow rate of 20 μL min−1 and the equilibrium response determined relative to the baseline (blank flow cell). The maximum compound concentrations were 6.25 μM for A2 trans and H2-TMPyP4 and 25 μM for A2 cis and A3. Between injections the sensor surface was refreshed with injections of 1 M KCl and buffer. All experiments were carried out in duplicate.
Acknowledgements
We thank Duncan Howe, Andrew Mason and Brian Crysell for NMR assistance.
The Portuguese Science Foundation (Fundação para a Ciência e a Tecnologia-FCT-Portugal) SFRH/BD/12414/2003 (D. P. N. Gonçalves), the BBSRC and the CRUK (S. Ladame), are also gratefully acknowledged for financial support.
References
- 1.(a) Simonsson T. Biol. Chem. 2001;382:621. doi: 10.1515/BC.2001.073. [DOI] [PubMed] [Google Scholar]; (b) Davis JT. Angew. Chem., Int. Ed. 2004;43:668. doi: 10.1002/anie.200300589. [DOI] [PubMed] [Google Scholar]
- 2.Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins ST, Hurley LH. J. Med. Chem. 1997;40:2113. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 3.Mergny JL, Hélène C. Nat. Med. (N. Y.) 1998;4:1366. doi: 10.1038/3949. [DOI] [PubMed] [Google Scholar]
- 4.Neidle S, Parkinson G. Nat. Rev. Drug Discovery. 2002;1:383. doi: 10.1038/nrd793. [DOI] [PubMed] [Google Scholar]
- 5.(a) Todd AK, Johnston M, Neidle S. Nucleic Acids Res. 2005;33:2901. doi: 10.1093/nar/gki553. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huppert JL, Balasubramanian S. Nucleic Acids Res. 2005;33:2908. doi: 10.1093/nar/gki609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simonsson T, Pecinka P, Kubista M. Nucleic Acids Res. 1998;26:1167. doi: 10.1093/nar/26.5.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siddiqui-Jain A, Grand CL, Bearss DJ, Hurley LH. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11593. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rankin S, Reszka AP, Huppert J, Zloh M, Parkinson GN, Todd AK, Ladame S, Balasubramanian S, Neidle S. J. Am. Chem. Soc. 2005;127:10584. doi: 10.1021/ja050823u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun D, Guo K, Rusche JJ, Hurley LH. Nucleic Acids Res. 2005;33:6070. doi: 10.1093/nar/gki917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haider SM, Parkinson GN, Neidle S. J. Mol. Biol. 2003;326:117. doi: 10.1016/s0022-2836(02)01354-2. [DOI] [PubMed] [Google Scholar]
- 11.Kim MY, Vankayalapati H, Shin-ya K, Wierzba K, Hurley LH. J. Am. Chem. Soc. 2002;124:2098. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]
- 12.Wheelhouse RT, Sun D, Han H, Han FX, Hurley LH. J. Am. Chem. Soc. 1998;120:3261. [Google Scholar]
- 13.Han H, Wheelhouse RT, Hurley LH. J. Am. Chem. Soc. 1999;121:3561. [Google Scholar]
- 14.(a) Han H, Langley DR, Rangan A, Hurley LH. J. Am. Chem. Soc. 2001;123:8902. doi: 10.1021/ja002179j. [DOI] [PubMed] [Google Scholar]; (b) Phan AT, Kuryavyi V, Gaw HY, Patel DJ. Nat. Chem. Biol. 2005;1:167. doi: 10.1038/nchembio723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi DF, Wheelhouse RT, Sun D, Hurley LH. J. Med. Chem. 2001;44:4509. doi: 10.1021/jm010246u. [DOI] [PubMed] [Google Scholar]
- 16.Dixon IM, Lopez F, Estève J-P, Tejera AM, Blasco MA, Pratviel G, Meunier B. ChemBioChem. 2005;6:123. doi: 10.1002/cbic.200400113. [DOI] [PubMed] [Google Scholar]
- 17.Wall RK, Shelton AH, Bonaccorsi LC, Bejune SA, Dubé D, McMillin DR. J. Am. Chem. Soc. 2001;123:11480. doi: 10.1021/ja010005b. [DOI] [PubMed] [Google Scholar]
- 18.Arsenault GP, Bullock E, MacDonald SF. J. Am. Chem. Soc. 1960;82:4384. [Google Scholar]
- 19.Read M, Harrison RJ, Romagnoli B, Tanious FA, Gowan SH, Reszka AP, Wilson WD, Kelland LR, Neidle S. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4844. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Read MA, Wood AA, Harrison JR, Gowan SM, Kelland LR, Dosanjh HS, Neidle S. J. Med. Chem. 1999;42:4538. doi: 10.1021/jm990287e. [DOI] [PubMed] [Google Scholar]
- 21.Pasternack RF, Huber PR, Boyd P, Engasser G, Francesconi L, Gibbs E, Fasella P, Venturo GC, de C. Hinds L. J. Am. Chem. Soc. 1972;94:4511. doi: 10.1021/ja00768a016. [DOI] [PubMed] [Google Scholar]
- 22.Pasternack RF, Gibbs EJ, Goudemer A, Antebi A, Bassner S, De Poy L, Turner DH, Williams A, Laplace F, Lansard MH, Merienne C, Perrée-Fauvet M. J. Am. Chem. Soc. 1985;107:8179. doi: 10.1021/ja00312a061. [DOI] [PubMed] [Google Scholar]
- 23.Kano K, Minamizono H, Kitae T, Negi S. J. Phys. Chem. 1997;101:6118. [Google Scholar]
- 24.Balagurumoorthy P, Brahmachari SK. J. Biol. Chem. 1994;269:21858. [PubMed] [Google Scholar]
- 25.Li J, Correia JC, Wang L, Trent JO, Chaires JB. Nucleic Acids Res. 2005;33:4649. doi: 10.1093/nar/gki782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang C-C, Chu J-F, Kau F-J, Chiu Y-C, Lou P-J, Chen H-C, Chang T-C. Anal. Chem. 2006;78:2810. doi: 10.1021/ac052218f. [DOI] [PubMed] [Google Scholar]
- 27.Gryko D, Lindsey JS. J. Org. Chem. 2000;65:2249. doi: 10.1021/jo9918100. [DOI] [PubMed] [Google Scholar]
- 28.Screen TEO, Lawton KB, Wilson GS, Dolney N, Ispasoiu R, Goodson T, III, Martin SJ, Bradley DDC, Anderson HL. J. Mater. Chem. 2001;11:312. [Google Scholar]
- 29.Taniguchi S, Hasegawa H, Yanagiya S, Tabeta Y, Nakano Y, Takahashi M. Tetrahedron. 2001;57:2103. [Google Scholar]