

Abstract
Viral protein R (Vpr), an apoptogenic accessory protein encoded by HIV-1, induces mitochondrial membrane permeabilization (MMP) via a specific interaction with the permeability transition pore complex, which comprises the voltage-dependent anion channel (VDAC) in the outer membrane (OM) and the adenine nucleotide translocator (ANT) in the inner membrane. Here, we demonstrate that a synthetic Vpr-derived peptide (Vpr52-96) specifically binds to the intermembrane face of the ANT with an affinity in the nanomolar range. Taking advantage of this specific interaction, we determined the role of ANT in the control of MMP. In planar lipid bilayers, Vpr52-96 and purified ANT cooperatively form large conductance channels. This cooperative channel formation relies on a direct protein–protein interaction since it is abolished by the addition of a peptide corresponding to the Vpr binding site of ANT. When added to isolated mitochondria, Vpr52-96 uncouples the respiratory chain and induces a rapid inner MMP to protons and NADH. This inner MMP precedes outer MMP to cytochrome c. Vpr52-96–induced matrix swelling and inner MMP both are prevented by preincubation of purified mitochondria with recombinant Bcl-2 protein. In contrast to König's polyanion (PA10), a specific inhibitor of the VDAC, Bcl-2 fails to prevent Vpr52-96 from crossing the mitochondrial OM. Rather, Bcl-2 reduces the ANT–Vpr interaction, as determined by affinity purification and plasmon resonance studies. Concomitantly, Bcl-2 suppresses channel formation by the ANT–Vpr complex in synthetic membranes. In conclusion, both Vpr and Bcl-2 modulate MMP through a direct interaction with ANT.
Keywords: ADP/ATP translocase, HIV, Vpr, mitochondria, Bcl-2
Introduction
Mitochondrial membrane permeabilization (MMP) is a key event of apoptotic cell death associated with the release of caspase activators and caspase-independent death effectors from the intermembrane space, dissipation of the inner mitochondrial transmembrane potential (ΔΨm), and a perturbation of oxidative phosphorylation 1 2 3 4 5 6 7. Pro- and antiapoptotic members of the Bcl-2 family regulate inner and outer MMP through interactions with the adenine nucleotide translocator (ANT) in the inner membrane (IM), the voltage-dependent anion channel (VDAC) in the outer membrane (OM), and/or through autonomous channel-forming activities 5 6 7 8 9 10. ANT and VDAC are major components of the permeability transition pore complex (PTPC), a polyprotein structure organized at sites at which the two mitochondrial membranes are apposed 7 11. It is a matter of debate whether Bcl-2 primarily regulates MMP through its interaction with VDAC or ANT.
The HIV-1 viral protein of regulation, viral protein R (Vpr), is abundant in virions 12 13 14 and is detectable in the serum of HIV-1 carriers, correlating with the viral load 15. Vpr has pleiotropic effects on viral replication and cellular proliferation, differentiation, cytokine production, nuclear factor κB–mediated transcription, and apoptosis 13 16 17 18. In HIV-1–infected cultures, Vpr is found in the nuclear and membrane fractions as well as the conditioned medium 19. Since Vpr can cross the plasma membrane 20 21 22, it can gain access to intracellular compartments not only as a consequence of the viral life cycle, but also independently from the infection process (i.e., via a “bystander effect”). In addition, Vpr can localize to mitochondria to kill cells by apoptosis 20 22 23 24 25. Full-length (Vpr1-96) or truncated synthetic forms of Vpr act on the PTPC to induce all mitochondrial hallmarks of apoptosis, including ΔΨm loss and the release of cytochrome c and apoptosis-inducing factor (AIF) 22. The MMP-inducing activity of Vpr resides in its COOH-terminal moiety (Vpr52-96), within an α-helical motif of 12 amino acids (Vpr71-82) containing several critical arginine (R) residues (R73, R77, R80) which are strongly conserved among different pathogenic HIV-1 isolates 22 23 24.
In this study, we used Vpr and Vpr-derived peptides as a tool to dissect the molecular events of MMP and its control by Bcl-2. Here we demonstrate that Vpr52-96 physically and functionally interacts with ANT to form large conductance channels, closure of VDAC abrogates Vpr52-96 binding to mitochondria, Vpr triggers inner MMP through its interaction with ANT before outer MMP, and Bcl-2 prevents the Vpr52-96–induced MMP through a direct interaction with ANT.
Materials and Methods
Plasmon Resonance.
NH2-terminally biotinylated peptides (Vpr52-96, Vpr52-96[R73A,80A], or an irrelevant control peptide, HWWRAESDEARRCYNDPKCCDFVTNR) were immobilized on biosensor chips of a BIAcore™ apparatus (Amersham Pharmacia Biotech), and 140 nM highly purified ANT from rat heart mitochondria 26 was added (time 0, on) in the mobile phase (5 μl/min) for real time measurement (in resonance units, RU). When required, the surface was regenerated using 1 M NaCl containing 0.05% P20 detergent (BIAcore). The data were analyzed using the v3.0 software and fitted to a 1:1 Langmuir binding model with separate k on and k off determination. The association constant (K A) was determined as k off/k on, and the dissociation constant (K D) as 1/K A. Control experiments revealed no specific binding of Vpr52-96 to recombinant VDAC (a gift from C. Mannella, Wadsworth Center, Albany, NY; reference 27), without any difference between Vpr52-96 and Vpr52-96[R73A, 80A] and without detectable saturation for VDAC concentrations up to 10 μM.
ANT Purification and Reconstitution in Liposomes.
ANT was purified from rat heart mitochondria as described previously 8. After mechanical shearing, mitochondria were suspended in 220 mM mannitol, 70 mM sucrose, 10 mM Hepes, 200 μM EDTA, 100 mM dithiothreitol, 0.5 mg/ml subtilisin, pH 7.4, kept on ice for 8 min, and sedimented twice by differential centrifugations (5 min, 500 g; 10 min, 10,000 g). Mitochondrial proteins were solubilized by 6% (vol/vol) Triton X-100 (Boehringer) in 40 mM K2HPO4, 40 mM KCl, 2 mM EDTA, pH 6.0, for 6 min at room temperature (RT), and solubilized proteins were recovered by ultracentrifugation (30 min, 24,000 g, 4°C). Then, 2 ml of this Triton X-100 extract was applied to a column filled with 1 g of hydroxyapatite (BioGel HTP; Bio-Rad Laboratories), eluted with previous buffer, and diluted (vol/vol) with 20 mM MES, 200 μM EDTA, 0.5% Triton X-100, pH 6.0. Subsequently, the sample was separated with a Hitrap SP column using an FPLC system (Amersham Pharmacia Biotech) and a linear NaCl gradient (0–1 M). Protein concentration was determined using micro-BCA assay (Pierce Chemical Co.). Purified ANT and/or recombinant Bcl-2 was reconstituted in phosphatidylcholine (PC)/cardiolipin liposomes. In brief, to prepare liposomes, 45 mg PC and 1 mg cardiolipin were mixed in 1 ml of chloroform, and the solvent was evaporated under nitrogen. Dry lipids were resuspended in 1 ml liposome buffer (125 mM sucrose plus 10 mM Hepes, pH 7.4) containing 0.3% n-octyl-β-d-pyranoside and mixed by continuous vortexing for 40 min at RT. ANT (0.1 mg/ml) or recombinant Bcl-2 (0.1 mg/ml) was then mixed with liposomes (vol/vol) and incubated for 20 min at RT. Proteoliposomes were finally dialyzed overnight at 4°C.
Vpr Binding to Liposomes.
Proteoliposomes or plain liposomes were exposed to different concentrations of FITC-labeled Vpr52-96 for 30 min in liposome buffer, washed (290,000 g, 45 min, 10°C), and analyzed in a FACS Vantage™ (Becton Dickinson) cytofluorometer (X ± SD, n = 3).
Pore-opening Assay.
ANT proteoliposomes were sonicated in the presence of 1 mM 4-methylumbelliferylphosphate (4-MUP) and 10 mM KCl (50 W, 22 s, sonifier model 250; Branson) on ice as described previously 28. Then, liposomes were separated on Sephadex G-25 columns (PD-10; Amersham Pharmacia Biotech) from unencapsulated products. 25-μl aliquots of liposomes were diluted to 3 ml in 10 mM Hepes, 125 mM saccharose, pH 7.4, mixed with various concentrations of the proapoptotic inducers, and incubated for 1 h at RT. Potential inhibitors of MMP such as BA, ATP, and ADP were added to the liposomes 30 min before treatment. After the addition of 10 μl of alkaline phosphatase (5 U/ml; Boehringer) diluted in liposome buffer plus 0.5 mM MgCl2, samples were incubated for 15 min at 37°C under agitation, and the enzymatic conversion of 4-MUP in 4-methylumbelliferone (4-MU) was stopped by the addition of 150 μl stop buffer (10 mM Hepes-NaOH, 200 mM EDTA, pH 10). The 4-MU–dependent fluorescence (360/450 nm) was subsequently quantitated 28 using a PerkinElmer spectrofluorimeter. Atractyloside (Atr), a proapoptotic permeability transition inducer, was used in each experiment as a standard to determine the 100% response. The percentage of 4-MUP release induced by Vpr-derived peptides was calculated as follows: [(fluorescence of liposomes treated by Vpr peptide − fluorescence of untreated liposomes)/(fluorescence of liposomes treated by Atr − fluorescence of untreated liposomes)] × 100.
Planar Lipid Bilayer Experiments.
The pore-forming activities of ANT, Vpr52-96, Bcl-2, and/or Bax were investigated at the single channel (patch clamp) levels, following published protocols 29. Virtually solvent-free planar lipid bilayers were formed by the method of Montal and Mueller (1972) by the apposition of two monolayers on a 125-μm-diameter hole in 10-μm thin Teflon film pretreated with hexadecane/hexane (1:40, vol/vol). Measurement compartments were glass cells. Membrane currents under applied voltage were measured using a BLM 120 amplifier (Biologic) and Ag/AgCl electrodes. The current fluctuations were stored on a DTR 1202 (Biologic) and transferred to a computer where different measurements (current and histogram amplitude) were performed using Satori v3.01 software (Intracel Ltd.). The lipid mixture palmitoyloleoy-PC/dioleoylphosphatidylethanolamine (POPC/DOPE, 7:3, wt/wt) supplemented with 3% cardiolipin (Avanti Polar Lipids, Inc.) was dissolved in 1 or 0.1% hexane, and then was added to both sides of the bilayer in the macroscopic configuration and to the bath in the “tip-dip” configuration (2.5 μg protein/ml). Recordings were performed in symmetric conditions (100 mM KCl, 2 mM MgCl2, 10 mM Hepes, pH 7.4). Lipid bilayers were preformed at the tip of patch clamp glass micropipettes by the tip-dip method 30. Bilayer formation was monitored by the capacitance response and, before protein addition, bare membranes were checked under applied potentials for electrical silence. Before addition to the bilayer, proteins were incubated with 1 mM CaCl2, 40 μM Atr, or control electrolyte (10 mM Hepes, 100 mM KCl, 2 mM MgCl2, pH 7.4) for 30 min at RT, then mixed with evaporated lipid mixture (60 μg protein/mg lipid) and treated with Biobeads SM-2 (Bio-Rad Laboratories) to eliminate Triton X-100. In some experiments, Vpr52-96 was added directly to the aqueous subphase after bilayer formation. Single channel recordings are representative of the most frequently observed events. ANT was preincubated or not with Bcl-2 or Bax and added to one compartment, then Vpr52-96 was added to the same compartment.
Binding Assays and Western Blot Analysis.
Mouse liver mitochondria were isolated as described 22. For the determination of cytochrome c release, supernatants from Vpr52-96–treated mitochondria (6,800 g for 15 min; 20,000 g for 1 h, both at 4°C) were frozen at −80°C until immunodetection of cytochrome c (mouse monoclonal antibody clone 7H8.2C12; BD PharMingen). For binding assays, purified mitochondria were incubated (250 μg of protein in 100 μl swelling buffer) for 30 min at RT 5 μM (binding assay) Vpr52-96 or biotin–Vpr52-96. Mitochondria were lysed either after incubation with biotinylated Vpr52-96 or before with 150 μl of a buffer containing 20 mM Tris-HCl, pH 7.6, 400 mM NaCl, 50 mM KCl, 1 mM EDTA, 0.2 mM PMSF, 100 U/ml aprotinin, 1% Triton X-100, and 20% glycerol. Such extracts were diluted with 2 vol of PBS plus 1 mM EDTA before the addition of 150 μl avidin-agarose (ImmunoPure; Pierce Chemical Co.) to capture the biotin-labeled Vpr52-96 complexed with its mitochondrial ligand(s) (2 h at 4°C in a roller drum). The avidin-agarose was washed batchwise with PBS (five times with 5 ml; 1,000 g, 5 min, 4°C), resuspended in 100 μl of twofold concentrated Laemmli buffer containing 4% SDS and 5 mM β-mercaptoethanol, incubated for 10 min at RT, and centrifuged (1,000 g, 10 min, 4°C). Finally, the supernatants were heated at 95°C for 5 min and analyzed by SDS-PAGE (12%), followed by Western blot analysis and immunodetection with a rabbit polyclonal antiserum against human ANT (provided by Dr. Heide H. Schmid, The Hormel Institute, University of Minnesota, Austin, MN; reference 31).
Cell Culture and Microinjection Experiments.
COS cells (cultured in DMEM Glutamax medium supplemented with Hepes, antibiotics, and 10% FCS) that were growing on a premarked V-shaped area of a coverslip (>200 per experiment) were microinjected into the cytoplasm using a computer-controlled microinjector equipment (Eppendorf; pressure 200 hPa, 0.5 s) with PBS only, pH 7.2, recombinant human Bcl-2 (amino acids 1–218; 10 μM, purified as described in reference 32), 2 μM König's polyanion (PA10; a gift of Dieter Brdiczka, University of Konstanz, Konstanz, Germany). After microinjection, cells were cultured in the absence (Co.) or presence of 1 μM Vpr52-96 for 3 h and stained for 30 min with 2 μM ΔΨm-sensitive dye 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) (red fluorescence shows mitochondria with a high ΔΨm, green fluorescence shows mitochondria with a low ΔΨm; reference 33).
Flow Cytometric Analysis of Purified Mitochondria.
Purified mitochondria were resuspended in PT buffer (200 mM sucrose, 10 mM Tris-MOPS, pH 7.4, 5 mM Tris-succinate, 1 mM Tris-phosphate, 2 μM rotenone, and 10 μM EGTA). Cytofluorometric (FACS Vantage™; Becton Dickinson) detection was restricted to mitochondria by gating on the forward scatter (FSC)/side scatter (SSC) parameters and on the main peak of the FSC-W parameter. Confirmation a posteriori of the validity of this double gating was obtained by the labeling of mitochondria with ΔΨm-insensitive mitochondrial dye MitoTracker® Green (75 nM, green fluorescence; Molecular Probes). To determine the percentage of mitochondria having a low ΔΨm, the ΔΨm-sensitive fluorochrome JC-1 (200 nM, 570–595 nm) was added 10 min before carbonylcyanide m-chlorophenylhydrazone (CCCP) or Vpr-derived peptides. The percentage of mitochondria having a low ΔΨm was determined in dot plot FSC/FL-2 (red fluorescence) windows.
Polarographic Studies.
Purified mitochondria were incubated at 37°C in a thermostated cell fitted with a Clark electrode (Hansatech) and containing 250 ml of 0.3 M mannitol, 10 mM KCl, 5 mM MgCl2, 1 mg/ml BSA, and 10 mM KH2PO4, pH 7.4, as described 34. Cytochrome c oxidase activities were spectrophotometrically measured 34.
Results
Physical Interaction between Vpr and ANT.
Surface plasmon resonance measurements indicate that purified detergent-solubilized ANT protein binds to the immobilized Vpr COOH-terminal moiety biotin–Vpr52-96 (but to a far lesser extent to mutated biotin–Vpr52-96[R73,80A]) with an affinity in the nanomolar range (Fig. 1A and Fig. B). This interaction was modulated by two ANT ligands which differentially affect ANT conformation 35, namely the PTPC activator (opener) Atr, which favored Vpr binding, and the PTPC inhibitor (closer) bongkrekic acid (BA), which reduced Vpr binding (Fig. 1 C). Bcl-2–like proteins bind to a motif of ANT (amino acids 105–155; reference 8) whose implication in apoptosis control has been confirmed by deletion mapping 36. This motif partially overlaps with the second ANT loop (amino acids 92–116), a regulatory domain exposed to the intermembrane space 37 38. A peptide corresponding to the overlap between the Bcl-2 binding motif and this loop (ANT104-116) inhibited the ANT–Vpr interaction (Fig. 1 C), presumably via direct association with Vpr52-96 (Fig. 1 D). Neither a topologically related peptide motif derived from the human phosphate carrier nor mutated and scrambled versions of ANT104-116 (control peptides in Fig. 1 C) had such inhibitory effects. Vpr also interacted with ANT incorporated into liposomal membranes. Indeed, Vpr52-96 binding to membranes was greatly facilitated in liposomes in which ANT has been reconstituted compared with protein-free liposomes (Fig. 2 A). The ANT-facilitated incorporation of Vpr into membranes was inhibited by BA (Fig. 2 B). In conclusion, Vpr binds to ANT, at least in part via an interaction with a domain of ANT (ANT104-116) that is exposed to the intermembrane side of this protein, coinciding with the apoptogenic portion of ANT.
Figure 1.
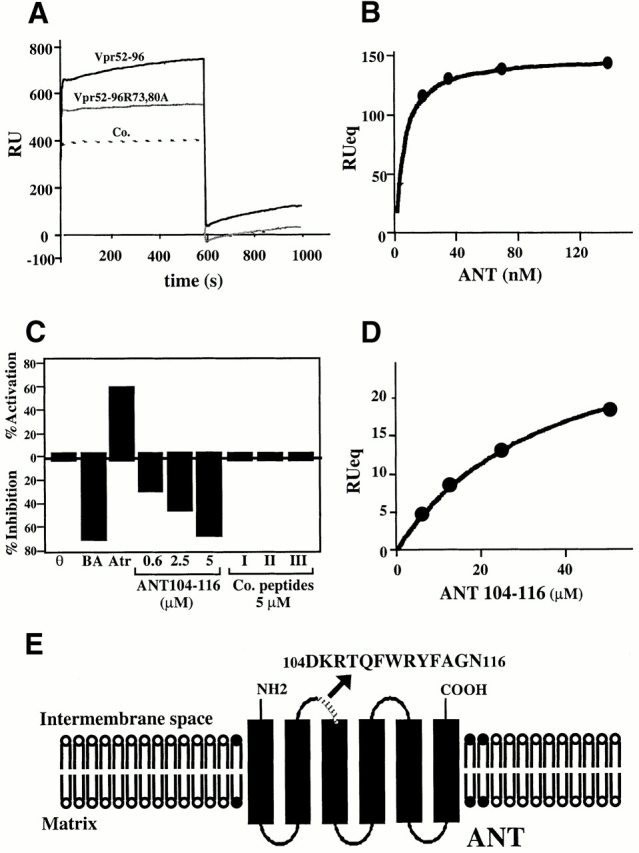
Physical and functional interaction between Vpr and ANT. (A) Plasmon surface resonance sensorgrams of the interaction of ANT with Vpr52-96, Vpr52-96[R73A,80A], or an irrelevant control (Co.). Only the sensorgram of the interaction with Vpr52-96 exhibits an increase of binding as a function of time and a positive signal at the start of the dissociation phase (off). The calculated K D (K D= k on/k off) of the interaction is 9.7 ± 6.4 nM (X ± SD, n = 5). (B) Langmuir isotherm determined at different concentrations of ANT on sensorgrams corrected by substraction of the blank (sensorgrams obtained with Vpr52-96[R73A,80A]). (C) Modulation of the Vpr52-96–ANT interaction by ANT ligands and ANT-derived peptides. Measurements were performed as in A, in the absence (θ) or presence of BA (250 μM), Atr (50 μM), the ANT104-116 peptide, or three control peptides (all at 5 μM). ANT-2–derived peptide ANT104-116 (DKRTQFWRYFAGN) and control peptides (Co.I: scrambled ANT104-116 [FQNYWGHKRFRDA]; Co.II: mutated ANT104-116 [DGHKQFWGYFAGN]; Co.III: topologically equivalent peptide [amino acids 149–161]) from the ANT-related human phosphate carrier protein (SNMLGEENTYLWR). Activation or inhibition was calculated as (1 − k 0a/k 0) × 100, in which k 0a and k 0 are the initial velocity in the presence or absence of the agent, respectively. (D) Langmuir isotherm for the binding of ANT104-116 to biotinylated Vpr52-96 (as determined in A). The calculated K D of the interaction is 35 μM. (E) Schematic diagram showing the topology of ANT and the sequence of the ANT-2–derived peptide ANT104-116.
Figure 2.


Physical (A and B) and functional (C) interaction between Vpr and liposomes containing ANT. (A) Dose–response curve of FITC-labeled Vpr52-96 binding on ANT liposomes and plain liposomes. (B) Binding of FITC–Vpr52-96 (2 μM) to plain liposomes, ANT proteoliposomes, in the presence or absence of BA (50 μM). (C) Permeabilization of ANT proteoliposomes by Vpr (X ± SD, n = 3). Liposomes were loaded with 4-MUP and exposed for 60 min to Atr (200 μM) or the indicated Vpr-derived peptides (1 μM), in the presence or absence of BA (50 μM), ADP (800 μM), and/or the indicated peptides (same as in B, 0.5 μM, preincubated with Vpr52-96 for 5 min). Then, alkaline phosphatase was added to convert liposome-released 4-MUP into the fluorochrome 4-MU, and the percentage of 4-MUP release induced by Vpr-derived peptides was calculated as described in Materials and Methods.
Cooperative Channel Formation by Vpr and ANT in Synthetic Membranes.
Vpr52-96 (but not the NH2-terminal moiety of Vpr [Vpr1-51] or mutated Vpr52-96, in which arginine 77 is replaced by alanine, Vpr52-96[R77A]) caused permeabilization of ANT proteoliposomes (Fig. 2 C), yet had no effect on plain liposomes (data not shown). ANT104-116 (but not the control peptides) prevented the Vpr52-96–induced membrane permeabilization of ANT proteoliposomes (Fig. 2 C), indicating that in the context of the lipid bilayer, the effect of Vpr involved a direct interaction with ANT. In planar lipid bilayers, low doses of Vpr52-96 (<1 nM) were incapable of forming channels, unless ANT was present. ANT and Vpr52-96 cooperated to form discrete channels whose conductance (190 ± 2 picosiemens [pS]; Fig. 3) was much larger than those formed by high doses (80 nM) of Vpr52-96 alone (55 ± 2 pS) (Fig. 3; reference 39), yet was in the range of those formed by Ca2+-treated ANT 26 40. These biophysical experiments demonstrate that ANT and Vpr directly interact in membranes to form functionally competent channel-forming hetero(poly)mers.
Figure 3.

Electrophysiological properties of Vpr52-96 and ANT in planar lipid bilayers. Current fluctuations of Vpr52-96 (80 nM, +150 mV), Vpr52-96 (0.4 nM, +100 mV), ANT (1 nM, +110 mV), and Vpr52-96 plus ANT (0.4:1 nM, +115 mV), and associated histograms (right) of conductance levels are shown. (A) Cooperative effect between ANT and Vpr52-96 at the single channel level. Current fluctuations of Vpr52-96 (80 nM, +150 mV), Vpr52-96 (0.4 nM, +100 mV), ANT (1 nM, +110 mV), and Vpr52-96 plus ANT (0.4:1 nM, +115 mV) after incorporation into synthetic membranes. Single channel recordings were performed using the tip-dip technique. The recordings shown are representative of at least three independent determinations. (B) Statistical analysis of conductances obtained in A. Results were expressed as current distributions at different voltages. Conductances (γ; in pS) are calculated by division of current by voltage.
Vpr-induced IM Permeabilization Studied in Isolated Mitochondria.
Compared with untreated organelles (Fig. 4 A, trace a), purified mitochondria preincubated with Vpr52-96 (Fig. 4 A, trace b) exhibited a gross deficiency in respiratory control (RC). Vpr increased succinate oxidation preceding ADP addition and abolished both the inhibitory effect of oligomycin (a specific ATPase inhibitor) and the stimulatory effect of uncoupling by the protonophore CCCP. Thus, Vpr52-96 (but not Vpr1-51) reduced the RC (ratio of oxygen consumption with oligomycin versus CCCP) to a value of 1.1, compared with 5.3 in control mitochondria (Table ). The entire Vpr protein (Vpr1-96), and a short peptide corresponding to the minimum “mitochondriotoxic” motif of Vpr (Vpr71-82) 22 23, also reduced the RC values (Table ). Noticeably, the Vpr-induced loss of RC was not associated with a significant decrease of the oxidation rate (Fig. 4 A), suggesting that no major loss of membrane-bound cytochrome c occurred on short-term incubation with Vpr52-96. Accordingly, adding cytochrome c to Vpr52-96–treated mitochondria oxidizing succinate did not stimulate the rate of oxygen uptake (Fig. 4 A, trace b). The observation of Vpr-mediated uncoupling of the respiratory chain prompted us to test its capacity to induce IM permeabilization. The IM being essentially impermeable to NADH 41, no significant oxygen uptake could be measured when NADH was added to control mitochondria (Fig. 5 A, trace a). However, addition of Vpr52-96 prompted a significant, NADH-stimulated oxygen consumption (Fig. 5 A, trace b). This indicates that Vpr permeabilized the IM both to protons (leading to uncoupling; Fig. 4 A, trace b) and NADH (Fig. 5 A, trace b). Moreover, addition of ADP (the physiological substrate of ANT) strongly inhibited the Vpr-dependent, NADH-stimulated oxygen consumption (Fig. 5 A, trace c), suggesting that the ANT–Vpr interaction is essential for inner MMP by Vpr.
Figure 4.

Oxidative properties of purified mitochondria exposed to Vpr. Oxygen consumption curves after addition of the indicated agents. Trace a: control mitochondria (no pretreatment). Trace b: mitochondria pretreated for 10 min with 1 μM Vpr52-96. Numbers along the traces are nanomoles of O2 consumed per minute per milligram of protein.
Table 1.
RC Values
| Peptide | RC |
|---|---|
| Vpr-51 | 5.1 |
| Vpr52-96 | 1.1 |
| Vpr1-96 | 2.0 |
| Vpr71-82 | 2.6 |
| None | 5.3 |
RC values were calculated by dividing oxygen consumption in the presence of CCCP by that measured with oligomycin (determined as in the legend to Fig. 4), 10 min after the addition of 1 μm Vpr-derived peptides (mean values of three determinations).
Figure 5.


Inner versus outer MMP. (A) Respirometry performed after addition of NADH and Vpr52-96 (1 μM). Numbers along the traces are nmol of O2 consumed per minute per milligram protein. Note that the Vpr-stimulated, NADH-dependent O2 consumption was fully sensitive to rotenone. (B) Kinetics of Vpr52-96–induced IM permeabilization to NADH and OM permeabilization to reduced cytochrome c. Oxygen consumption was determined in the presence of 2 mM NADH (▪) as in A (trace c) and cytochrome c (15 μM) oxidation (○) was spectrofluorometrically measured, as described (reference 34). The 100% value of cytochrome c oxidation was determined by addition of 2.5 mM laurylmaltoside. (C) Kinetics of Vpr52-96–induced ΔΨm loss and cytochrome c release. Purified mitochondria were treated with 1 μM Vpr52-96 subjected to cytofluorometric determination of the percentage of mitochondria having a low ΔΨm using the ΔΨm-sensitive fluorochrome JC-1. In parallel, cytochrome c was immunodetected in the supernatant of mitochondria.
Vpr-induced IM Permeabilization Precedes OM Permeabilization.
To determine the primary site of action of Vpr52-96 on mitochondria, we studied the differential kinetics of inner and outer MMP to NADH and cytochrome c, respectively (Fig. 5 B). NADH oxidation by mitochondria incubated with Vpr52-96 was found maximal 10 min after addition of Vpr52-96 (Fig. 5 B). Under similar conditions, Vpr52-96 only induced a marginal access of cytochrome c to cytochrome c oxidase (Fig. 5 B). Moreover, the ΔΨm loss occurred before cytochrome c release can be detected by immunoblot (Fig. 5 C). Hence, Vpr52-96 causes inner MMP well before OM becomes permeable to cytochrome c. Accordingly, at the ultrastructural level (see also Fig. 6 C), Vpr52-96–treated mitochondria exhibited matrix swelling before OM rupture became apparent.
Figure 6.
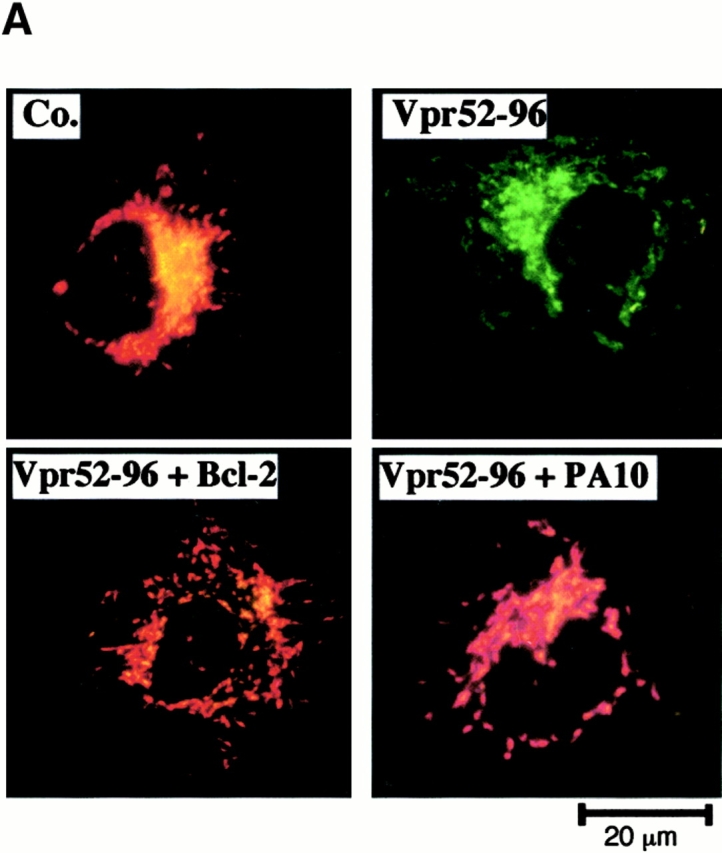
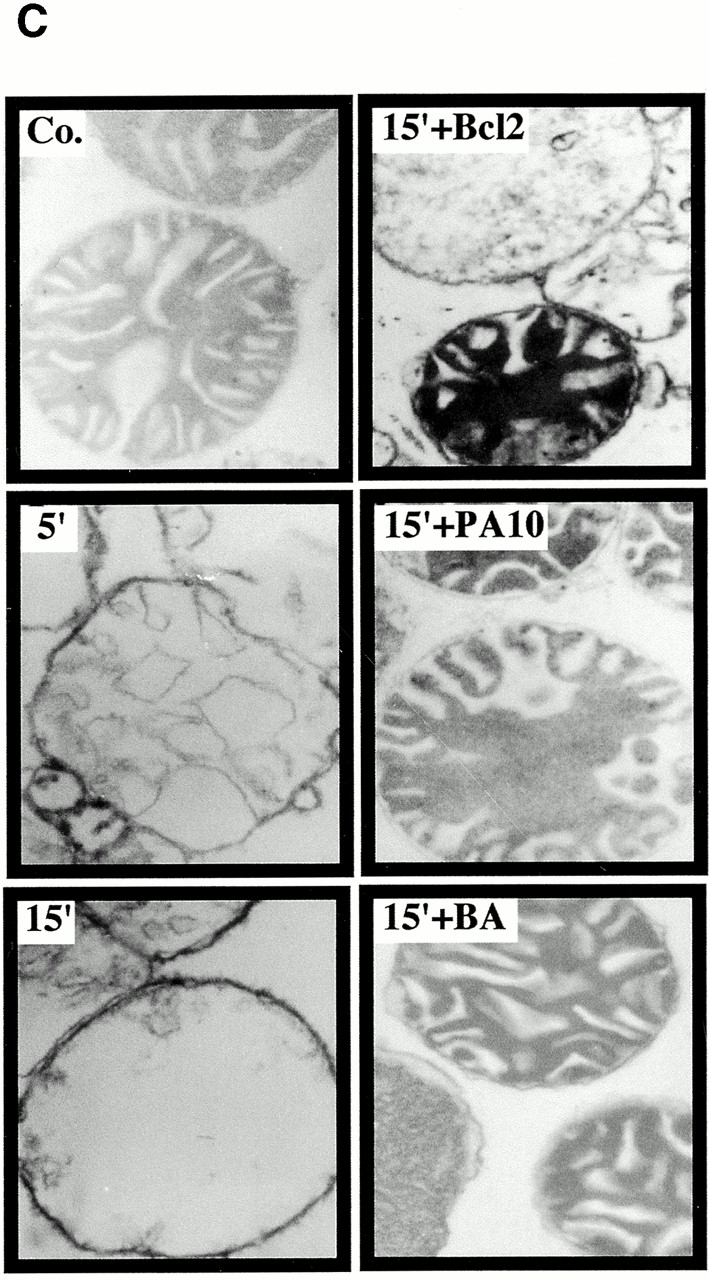

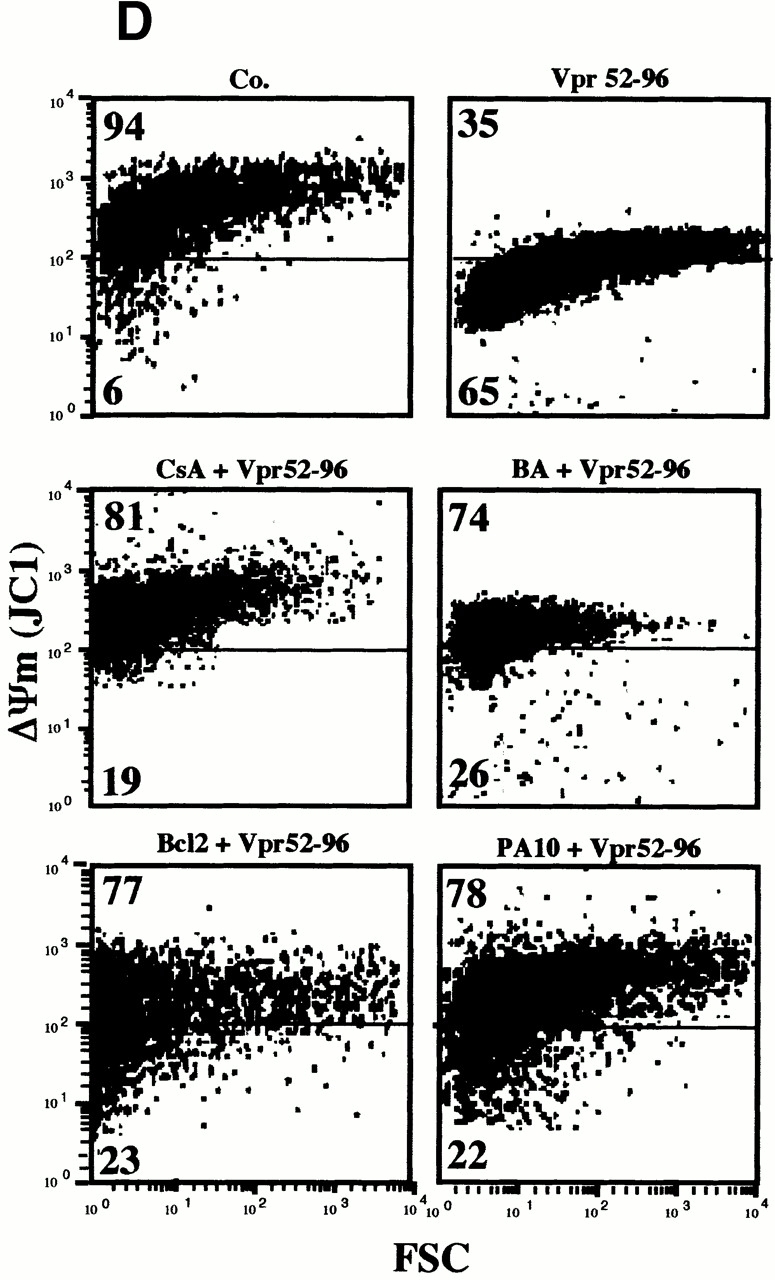
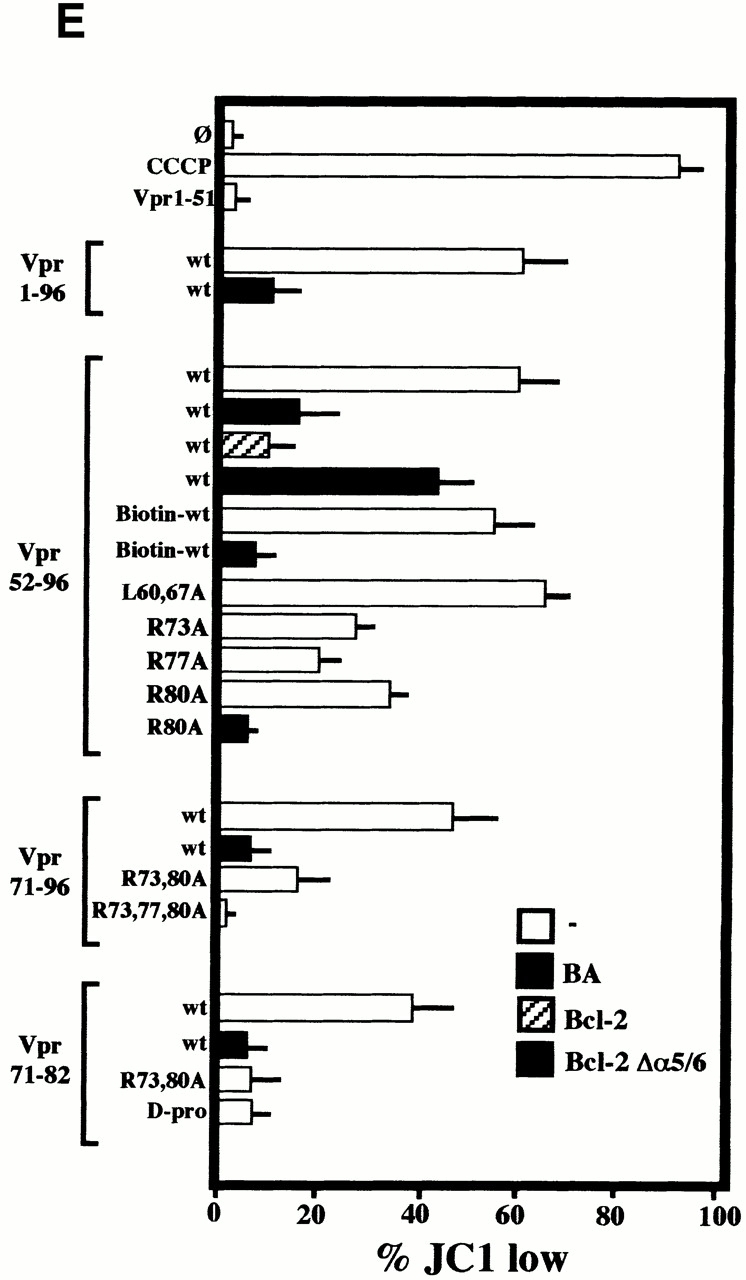
Bcl-2–mediated inhibition of Vpr effects on mitochondria. (A) Vpr52-96–induced ΔΨm dissipation induced in intact cells. COS cells were microinjected with 10 μM recombinant human Bcl-2, 2 μM PA10, or PBS only, then incubated in the absence (Co.) or presence of 1 μM Vpr52-96 for 3 h, and stained with 2 μM ΔΨm-sensitive dye JC-1 (red fluorescence shows mitochondria with a high ΔΨm, green fluorescence shows mitochondria with a low ΔΨm). (B) Effect of Bcl-2 on the Vpr-induced inner MMP to NADH. Mitochondria were left untreated (Co.) or were pretreated (10 min) with Bcl-2 (0.8 μM) or BA (10 μM). Oxygen consumption of purified mitochondria was measured as in the legend to Fig. 5 after addition of succinate plus CCCP or NADH, as indicated. (C) Ultrastructural effects of Vpr on isolated mitochondria. Electron micrographs were obtained after incubation of mitochondria for 5 or 15 min with 3 μM Vpr52-96, after a 5-min preincubation with 0.8 μM Bcl-2 or 2 μM PA10. The percentage of swollen mitochondria was <5% in the control, 82 ± 4% (n = 3, X ± SEM) 5 min after Vpr addition, 99 ± 1% 15 min after Vpr, 52 ± 4% after treatment with Bcl-2 plus 15 min Vpr, and 24 ± 3% after treatment with BA plus 15 min Vpr. (D) Effect of Bcl-2 and PA10 on Vpr52-96–induced ΔΨm dissipation in purified mitochondria. Isolated mitochondria (200 μg protein/ml) were preincubated with the indicated inhibitors (5 μM CsA, 50 μM BA, 0.8 μM Bcl-2, 2 μM PA10; 5–10 min), washed (10 min, 6,800 g, 20°C), incubated with the ΔΨm-sensitive dye JC-1 (200 nM for 10 min), exposed to 3 μM Vpr52-96 for 5 min, and subjected to flow cytometric determination of the fluorescence (570–595 nm) and the particle size (FSC). Numbers indicate the percentage of JC-1high and JC-1low mitochondria among ∼104 events. (E) Quantitation of the frequency of JC-1low mitochondria (X ± SD, n = 5) after incubation with different Vpr-derived peptides. Purified mitochondria were preincubated 10 min with or without 0.8 μM Bcl-2, 0.8 μM Bcl-2Δα5/6, or 10 μM BA in PT buffer, incubated with the ΔΨm-sensitive dye JC-1 (200 nM for 10 min), and then treated for 5 min with 3 μM Vpr52-96 (wild-type [wt], biotinylated, or modified as indicated), or 10 min with 5–10 μM Vpr1-96, Vpr1-51, Vpr71-96, Vpr71-82 (wild-type or modified as indicated), and finally subjected to flow cytometric analysis as in D.
Bcl-2 and PA10 Both Prevent MMP Induced by Vpr.
Extracellular addition of Vpr to intact cells induced a rapid ΔΨm loss before nuclear condensation occurred. This ΔΨm loss could be fully prevented (≥95% of inhibition) by microinjection of recombinant Bcl-2 into the cytoplasm (Fig. 6 A). Less than 5% of Bcl-2–injected cells manifested a ΔΨm reduction, whereas 61 ± 8% (n = 3) of vehicle-microinjected cells exhibited a complete ΔΨm dissipation in response to Vpr.
Preincubation of purified mitochondria with recombinant Bcl-2 also inhibited the Vpr-mediated inner MMP to NADH (Fig. 6 B). Concomitantly, Bcl-2 preincubation inhibited both the Vpr-induced matrix swelling (Fig. 6 C; ∼50% inhibition) and ΔΨm loss (Fig. 6d and Fig. e; ∼70% inhibition). No such protective effect was observed when Bcl-2 was replaced by Bcl-2Δα5/6, a deletion mutant lacking the putative pore-forming α5 and α6 helices 42. Two pharmacological inhibitors of the PTPC (the ANT ligand BA and the cyclophilin D ligand cyclosporin A [CsA]), as well as the specific VDAC inhibitor (closer) PA10 43, also inhibited the manifestations of inner MMP (Fig. 6b Fig. c Fig. d Fig. e). Microinjected PA10 also inhibited the effect of Vpr52-96 on intact cells (Fig. 6 A). In conclusion, both Bcl-2 and PA10 protect mitochondria against Vpr-induced MMP.
Differential Effects of Bcl-2 and PA10 on Vpr Binding to Mitochondria.
When added to purified mitochondria, FITC-labeled Vpr52-96 rapidly bound to intact mitochondria and then provoked ΔΨm loss (Fig. 7 A). Binding of Vpr52-96 to purified mitochondria was completely abolished by preincubation of the organelles with PA10, partially reduced by BA, but not affected by CsA (Fig. 7 B). Thus, Vpr must access mitochondria through VDAC. Bcl-2 may be expected to prevent Vpr from crossing the OM via VDAC (based on the Bcl-2–mediated closure of VDAC; references 9 and 44) and/or to inhibit the Vpr effect on ANT (based on its physical and functional interaction with ANT; references 8, 26, 45). Although recombinant Bcl-2 strongly reduced the Vpr52-96–induced matrix swelling (Fig. 6 C) and ΔΨm loss (Fig. 6d and Fig. e), it failed to impair the binding of Vpr52-96 to purified mitochondria (Fig. 7 B). The differential inhibitory effects of PA10 and Bcl-2 on the Vpr–ANT interaction was confirmed in a distinct experimental system. PA10 fully abolished the affinity-mediated purification of ANT using biotinylated Vpr52-96 (Fig. 7 C), provided that its effect was assessed on intact mitochondria (in which Vpr52-96 has to cross the OM to reach ANT). In contrast, PA10 did not affect the Vpr52-96–mediated purification of ANT from Triton-solubilized mitochondria (in which ANT is readily accessible to Vpr52-96). In the same conditions, Bcl-2 reduced the Vpr52-96–mediated recovery of ANT, irrespective of its addition to intact or solubilized mitochondria (Fig. 7 C). Thus, Bcl-2 does not interfere with the (PA10-inhibited) VDAC-mediated conduit, allowing Vpr52-96 to pass the OM.
Figure 7.
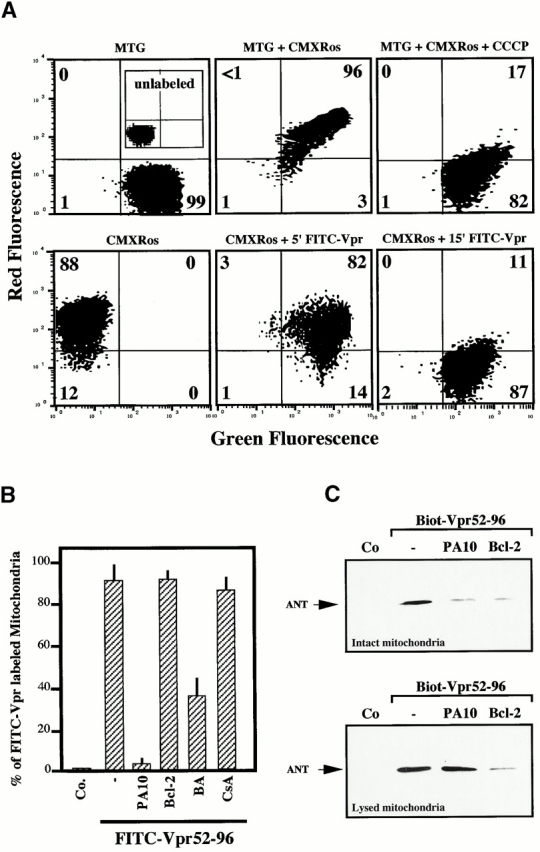
Differential effect of Bcl-2 and PA10 on Vpr52-96 binding to mitochondria. (A) Vpr52-96 binds mitochondria before inducing ΔΨm loss. Mitochondria were left unstained (inset in control, top left panel) or exposed to the ΔΨm-insensitive mitochondrial dye MitoTracker® Green (75 nM), alone (MTG) or with 0.5 μM FITC–Vpr52-96 (green fluorescence) in combination with the ΔΨm-sensitive mitochondrial dye MitoTracker® Red (chloromethyl-X-rosamine; red fluorescence) followed by cytofluorometric two-color analysis. Numbers indicate the percentage of mitochondria in each quadrant. (B) PA10, but not Bcl-2, inhibits Vpr52-96 binding to mitochondria. Mitochondria were preincubated for 10 min with the indicated inhibitors, and the percentage of FITC–Vpr52-96–labeled mitochondria was determined as in A. (C) Inhibitory effect of Bcl-2 on affinity purification of ANT by biotinylated Vpr52-96. Mitochondria were incubated with the indicated inhibitors, and then exposed for 30 min at RT with 5 μM biotinylated Vpr52-96. Mitochondria were lysed either after incubation with biotinylated Vpr52-96 (top) or lysed before (bottom) with Tris-HCl as described in Materials and Methods. Biotinylated Vpr52-96 complexed with its mitochondrial ligands was retained on avidin-agarose and subjected to immunoblot detection of ANT. Co, control.
Bcl-2–mediated Inhibition of the Physical and Functional Interaction between Vpr and ANT.
A further series of experiments indicated that Bcl-2 modulated the physical and functional interaction of Vpr with ANT. Recombinant Bcl-2 (but not Bcl-2Δα5/6) reduced Vpr52-96 binding to soluble (Fig. 8 A) or membrane-associated (Fig. 8 B) ANT. In an independent series of control experiments, Vpr53-96 was found not to interact with recombinant Bcl-2, as determined by surface plasmon resonance. Hence, inhibition of the Vpr–ANT binding is likely attributable to a direct Bcl-2–ANT interaction 8 26. Accordingly, Bcl-2 abolished the formation of Vpr52-96–induced channels in ANT-containing lipid bilayers (Fig. 8 C). In contrast, in the same conditions, Bax exacerbated the conductance of Vpr52-96–ANT channels to a mean value of 245 ± 2 pS (compared with 190 ± 2 pS for Vpr52-96–ANT without any further addition; Fig. 8 C). In conclusion, Bcl-2 specifically prevents cooperative channel formation by Vpr–ANT, presumably by disrupting their interaction.
Figure 8.


Bcl-2–mediated inhibition of the Vpr–ANT interaction. (A) Plasmon surface resonance determination of the Bcl-2–mediated inhibition of interaction between Vpr52-96 and native purified ANT. The interaction was measured after the addition of the indicated concentrations of recombinant Bcl-2, Bcl-2Δα5/6, or recombinant Bid, and data (X ± SD, n = 3) were calculated as in the legend to Fig. 1. (B) Effect of Bcl-2 on Vpr binding to ANT proteoliposomes. The retention (X ± SD, n = 3) of FITC-labeled Vpr52-96 on ANT proteoliposomes preincubated with 800 nM Bcl-2 or 2 μM PA10 (followed by washing) was assessed as in the legend to Fig. 2 A. Co, control. (C) Effect of Bcl-2 on the formation of Vpr–ANT channels in planar lipid bilayers. Single channel recordings (+75 mV) of Vpr52-96 plus ANT plus Bax (0.4:1:0.3 nM) and Vpr52-96 plus ANT plus Bcl-2 (0.4:1:1 nM), and corresponding amplitude histograms are displayed. Control values for Vpr52-96 plus ANT alone are similar as in Fig. 1 E (data not shown). C, closed; O, open.
Discussion
The mechanism by which endogenous end stage effectors achieve permeabilization of the IM and/or OM is a matter of debate. Depending on the apoptotic stimulus, permeabilization may affect the OM and IM in a variable fashion and may or may not be accompanied by matrix swelling 1 2 5 6 7 46. In vitro experiments performed on purified mitochondria or proteins reconstituted into artificial membranes suggest at least two competing models of MMP. On the one hand, pore formation by ANT has been proposed to account for IM permeabilization, osmotic matrix swelling, and consequent OM rupture, resulting because the surface area of the IM with its folded cristae exceeds that of the OM. In support of this hypothesis, proapoptotic molecules such as Bax, Atr, Ca2+, and thiol oxidants cause ANT (which normally is a strictly specific ADP/ATP antiporter) to form a nonspecific pore 8 26 28 40. On the other hand, VDAC has been suggested to account for a primary OM permeabilization not affecting the IM 9 44. In favor of this hypothesis, the permeabilization of VDAC-containing liposomes to sucrose or cytochrome c is enhanced by Bax and inhibited by Bcl-2 in vitro 9 44.
The data presented here indicate that the HIV-1 protein Vpr52-96 primarily affects IM and not OM permeability in vitro. Vpr52-96 binds ANT in an ANT conformation–dependent fashion (Fig. 1) and cooperates with it to form channels (Fig. 2 and Fig. 3) which permeabilize the IM before the OM becomes permeable to cytochrome c (Fig. 4 and Fig. 5). Bcl-2 antagonizes this effect (Fig. 6), but clearly does so independently of VDAC, based on two independent observations. First, its mode of action clearly differs from that of the VDAC inhibitor PA10 (Fig. 7). Second, Bcl-2 can affect the physical and functional ANT–Vpr interaction in a synthetic, VDAC-free system (Fig. 8). It should be noted that although Bcl-2 is generally considered to be preferentially associated with the mitochondrial OM, some reports also indicate an IM localization 47 48 49. This suggests that Bcl-2 may adopt different submitochondrial localizations possibly depending on the state of the cell. Although our data do not exclude the possibility that Bcl-2 modulates the permeability of VDAC to relatively large, globular proteins (14.5 kD for cytochrome c, as opposed to the linear, mostly α helical structure of Vpr52-96 resolved by nuclear magnetic resonance; reference 50), they indicate that, at least in this particular model, Bcl-2 exerts its membrane-protective mitochondrial effect via ANT.
The fact that PA10-mediated closure of VDAC completely blocks the binding of Vpr52-96 to intact mitochondria suggests that Vpr52-96 must cross the OM via VDAC to reach its mitochondrial target(s). Bcl-2 inhibits binding of Vpr52-96 to ANT in three experimental systems: plasmon resonance (Fig. 8 A), ANT proteoliposomes (Fig. 8 B), and affinity purification of ANT by biotinylated Vpr added to intact or Triton-solubilized mitochondria (Fig. 8 C). Conversely, Vpr52-96 binding to intact mitochondria was not affected by Bcl-2 (Fig. 7 B). One possible explanation for this result may be that Vpr52-96 has a second mitochondrial target different from ANT. The ANT ligand BA inhibits the ANT–Vpr52-96 binding (Fig. 1 C) and reduces the binding of Vpr to mitochondria to some extent (Fig. 7 B). One explanation to reconcile the differential inhibitory action of Bcl-2 and PA10 is to assume that BA (but not Bcl-2) affects the Vpr target which is different from ANT. Alternatively, BA (but not Bcl-2) might modify the general conformation of the PTPC (including that of VDAC), and thus reduce the (VDAC-mediated) Vpr entry into mitochondria.
Irrespective of this latter possibility, our data are compatible with a scenario in which Vpr affects mitochondrial membrane integrity in four steps (Fig. 9). First, Vpr crosses the OM via VDAC (inhibition by PA10). Second, the COOH-terminal half of Vpr (Vpr52-96) interacts with the ANT104-116 site in ANT (and possibly with other ANT domains). Third, as a consequence of its association with Vpr, ANT is converted into a nonspecific pore, leading to inner MMP. Fourth, inner MMP triggers matrix swelling resulting in OM rupture.
Figure 9.

Model of the Vpr–PTPC interactions. Vpr crosses the OM through VDAC, which is inhibited by PA10. Vpr then interacts with ANT. Bcl-2 and the ANT ligand BA (Bongkrekate) inhibit the binding of Vpr to ANT, whereas CsA indirectly affects the pore forming function of ANT via its effect on cyclophilin D (Cyp-D).
Recent studies have revealed the existence of several viral apoptosis inhibitors acting on mitochondria. For example, adenovirus, Epstein-Barr virus, herpesvirus saimiri, and Kaposi sarcoma–associated human herpesvirus 8 produce apoptosis-suppressive Bcl-2 homologues 51 52 53 54. In addition, several viruses encode PTPC-interacting proteins without any obvious homology to the Bcl-2/Bax family. The cytomegalovirus apoptosis inhibitor pUL37X 55 and Vpr, an HIV-1–encoded apoptosis inducer, selectively bind to ANT. The proapoptotic p13 (II) protein derived from the X-II open reading frame of HTLV-1 is also targeted to mitochondria via a peptide motif that bears structural similarities to the mitochondriotoxic domain of Vpr 56. Moreover, the proapoptotic, MMP-inducing hepatitis virus B protein X interacts with VDAC 57. Thus, both VDAC and ANT emerge as major targets of viral apoptosis regulation and, perhaps, as targets for pharmacological intervention on viral pathogenesis.
Acknowledgments
This work has been supported by grants from the Agence Nationale de Recherches sur le Sida, European Community, Fondation pour la Recherche Médicale, and Lique Nationale contre le Cancer (to G. Kroemer), and by National Institutes of Health grant GM60554 (to J.C. Reed). E. Jacotot received a fellowship from the Agence Nationale de Recherches sur le Sida, H.L.A. Vieria from the Portuguese Government, A.S. Belzacq from the Association pou la Recherche sur le Cancer, K.F. Ferri from the French Ministry of Sciences, and M. Loeffler from the Austrian Science Foundation.
Footnotes
Abbreviations used in this paper: ANT, adenine nucleotide translocator; Atr, atractyloside; BA, bongkrekic acid; CCCP, carbonylcyanide m-chlorophenylhydrazone; CsA, cyclosporin A; ΔΨm, mitochondrial transmembrane potential; FSC, forward scatter; IM, inner membrane; MMP, mitochondrial membrane permeabilization; OM, outer membrane; PA10, König's polyanion; PC, phosphatidylcholine; PTPC, permeability transition pore complex; RC, respiratory control; RT, room temperature; VDAC, voltage-dependent anion channel; Vpr, viral protein R.
References
- Kroemer G., Zamzami N., Susin S.A. Mitochondrial control of apoptosis. Immunol. Today. 1997;18:44–51. doi: 10.1016/s0167-5699(97)80014-x. [DOI] [PubMed] [Google Scholar]
- Green D.R., Reed J.C. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- Lemasters J.J., Nieminen A.-L., Qjan T., Trost L.C., Elmore S.P., Nishimura Y., Crowe R.A., Cascio W.E., Bradham C.A., Brenner D.A., Herman B. The mitochondrial permeability transition in cell deatha common mechanism in necrosis, apoptosis and autophagy. Biochim. Biophys. Acta. 1998;1366:177–196. doi: 10.1016/s0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- Wallace D.C. Mitochondrial diseases in mouse and man. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- Vander Heiden M.G., Thompson C.B. Bcl-2 proteinsinhibitors of apoptosis or regulators of mitochondrial homeostasis? Nat. Cell Biol. 1999;1:E209–E216. doi: 10.1038/70237. [DOI] [PubMed] [Google Scholar]
- Gross A., McDonnell J.M., Korsmeyer S.J. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 1999;13:1899–1911. doi: 10.1101/gad.13.15.1899. [DOI] [PubMed] [Google Scholar]
- Kroemer G., Reed J.C. Mitochondrial control of cell death. Nat. Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- Marzo I., Brenner C., Zamzami N., Jürgensmeier J., Susin S.A., Vieira H.L.A., Prévost M.-C., Xie Z., Mutsiyama S., Reed J.C., Kroemer G. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science. 1998;281:2027–2031. doi: 10.1126/science.281.5385.2027. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Narita M., Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- Desagher S., Osen-Sand A., Nichols A., Eskes R., Montessuit S., Lauper S., Maundrell K., Antonsson B., Martinou J.-C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999;144:891–901. doi: 10.1083/jcb.144.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Cullen B.R. HIV-1 auxiliary proteinsmaking connections in a dying cell. Cell. 1998;93:685–692. doi: 10.1016/s0092-8674(00)81431-2. [DOI] [PubMed] [Google Scholar]
- Emerman M., Malim M.H. HIV-1 regulatory/accessory geneskeys to unraveling viral and host cell biology. Science. 1998;280:1880–1884. doi: 10.1126/science.280.5371.1880. [DOI] [PubMed] [Google Scholar]
- Singh S.P., Lai D., Cartas M., Serio D., Murali R., Kalyanaraman V.S., Srinivasan A. Epitope-tagging approach to determine the stoichiometry of the structural and nonstructural proteins in the virus particlesamount of Vpr in relation to Gag in HIV-1. Virology. 2000;268:364–371. doi: 10.1006/viro.2000.0191. [DOI] [PubMed] [Google Scholar]
- Levy D.N., Refaeli Y., MacGregor B.R., Weiner D.B. Serum Vpr regulates productive infection and latency of human immunodefiency virus type 1. Proc. Natl. Acad. Sci. USA. 1994;91:10873–10877. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A.D., Young J.A.T. HIV-1fifteen proteins and an RNA. Annu. Rev. Biochem. 1998;67:1–25. doi: 10.1146/annurev.biochem.67.1.1. [DOI] [PubMed] [Google Scholar]
- Bukrinsky M., Adzhubei A. Viral protein R of HIV-1. J. Med. Virol. 1999;9:39–49. doi: 10.1002/(sici)1099-1654(199901/03)9:1<39::aid-rmv235>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Stewart S.A., Poon B., Song J.Y., Chen I.S.Y. Human immunodeficiency virus type 1 Vpr induces apoptosis through caspase activation. J. Virol. 2000;74:3105–3111. doi: 10.1128/jvi.74.7.3105-3111.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y.L., Spearman P., Ratner L. Human immunodeficiency virus type 1 viral protein R localization in infected cells and virions. J. Virol. 1993;67:6542–6550. doi: 10.1128/jvi.67.11.6542-6550.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunagiri C., Macreadie I., Hewhish D., Azadi A. A C-terminal domain of HIV-1 accessory protein Vpr is involved in penetration, mitochondrial dysfunction and apoptosis of human CD4+ lymphocytes. Apoptosis. 1997;2:69–76. doi: 10.1023/a:1026487609215. [DOI] [PubMed] [Google Scholar]
- Kichler A., Pages J.C., Leborgne C., Druillenec S., Lenoir C., Coulaud D., Delain E., Le Cam E., Roques B.P., Danos O. Efficient DNA transfection mediated by the C-terminal domain of human immunodeficiency virus type 1 viral protein R. J. Virol. 2000;74:5424–5431. doi: 10.1128/jvi.74.12.5424-5431.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacotot E., Ravagnan L., Loeffler M., Ferri K.F., Vieira H.L.A., Zamzami N., Costantini P., Druillennec S., Hoebeke J., Brian J.P. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000;191:33–45. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macreadie I.G., Castelli L.A., Hewish D.R., Kirkpatrick A., Ward A.C., Azad A.A. A domain of human immunodeficiency virus type 1 Vpr containing repeated H(S/F)RIG amino acid motifs causes cell growth arrest and structural defects. Proc. Natl. Acad. Sci. USA. 1995;92:2770–2774. doi: 10.1073/pnas.92.7.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macreadie I.G., Thorburn D.R., Kirby D.M., Castelli L.A., Derozario N.L., Azad A.A. HIV-1 protein Vpr causes gross mitochondrial dysfunction in the yeast Saccharomyces cerevisiae . FEBS Lett. 1997;410:145–149. doi: 10.1016/s0014-5793(97)00542-5. [DOI] [PubMed] [Google Scholar]
- Muthumani K., Montaner L.J., Ayyavoo V., Weine D.B. Vpr-GFP virion particle identifies HIV-infected targets and preserves HIV-1 Vpr function in macrophages and T-cells. DNA Cell Biol. 2000;19:179–188. doi: 10.1089/104454900314564. [DOI] [PubMed] [Google Scholar]
- Brenner C., Cardiou H., Vieira H.L.A., Zamzami N., Marzo I., Xie Z., Leber B., Andrews D., Duclohier H., Reed J.C., Kroemer G. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene. 2000;19:329–336. doi: 10.1038/sj.onc.1203298. [DOI] [PubMed] [Google Scholar]
- Song J.M., Midson C., Blachly-Dyson E., Forte M., Colombini M. The topology of VDAC as probed by biotin modification. J. Biol. Chem. 1998;273:24406–24413. doi: 10.1074/jbc.273.38.24406. [DOI] [PubMed] [Google Scholar]
- Costantini P., Belzacq A.-S., Vieira H.L.A., Larochette N., de Pablo M., Zamzami N., Susin S.A., Brenner C., Kroemer G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene. 2000;19:307–314. doi: 10.1038/sj.onc.1203299. [DOI] [PubMed] [Google Scholar]
- Brullemans M., Helluin O., Dugast J.-Y., Molle G., Duclohier H. Implications of segment S45 in the permeation pathway of voltage-dependent sodium channels. Eur. Biophys. J. 1994;23:39–49. doi: 10.1007/BF00192204. [DOI] [PubMed] [Google Scholar]
- Hanke W., Methfessel C., Wilmsen U., Boheim G. Ion channel reconstitution into planar lipid bilayers on glass pipettes. Bioelectrochem. Bioenerg. 1984;12:329–339. [Google Scholar]
- Giron-Calle J., Schmid H.H. Peroxidative modification of a membrane protein. Conformation-dependent chemical modification of adenine nucleotide translocase in Cu2+/ter-butylhydroperoxide treated mitochondria. Biochemistry. 1996;35:15440–15446. doi: 10.1021/bi960840j. [DOI] [PubMed] [Google Scholar]
- Xie Z.H., Schendel S., Matsuyama S., Reed J.C. Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry. 1998;37:6410–6418. doi: 10.1021/bi973052i. [DOI] [PubMed] [Google Scholar]
- Ferri K.F., Jacotot E., Blanco J., Esté J.A., Zamzami A., Susin S.A., Brothers G., Reed J.C., Penninger J.M., Kroemer G. Apoptosis control in syncytia induced by the HIV-1–envelope glycoprotein complex. Role of mitochondria and caspases. J. Exp. Med. 2000;192:1081–1092. doi: 10.1084/jem.192.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P., Chretien D., Bourgeron T., Gerard B., Rotig A., Saudubray J.M., Munnich A. Biochemical and molecular investigations in respiratory chain deficiencies. Clin. Chem. Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. The ADP-ATP translocation in mitochondria, a membrane potential controlled transport. J. Membr. Biol. 1980;56:97–105. doi: 10.1007/BF01875961. [DOI] [PubMed] [Google Scholar]
- Bauer M.K.A., Schubert A., Rocks O., Grimm S. Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J. Cell Biol. 1999;147:1493–1501. doi: 10.1083/jcb.147.7.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandolin G., Le-Saux A., Trezeguet V., Lauquinn G.J., Vignais P.V. Chemical, immunological, enzymatic, and genetic approaches to studying the arrangement of the peptide chain of the ADP/ATP carrier in the mitochondrial membrane. J. Bioenerg. Biomembr. 1993;25:493–501. doi: 10.1007/BF01108403. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. Dialectics in carrier researchthe ADP/ATP carrier and the uncoupling protein. J. Bioenerg. Biomembr. 1993;25:447–457. doi: 10.1007/BF01108402. [DOI] [PubMed] [Google Scholar]
- Piller S.C., Ewart G.D., Premkumar A., Cox G.B., Gage P.W. Vpr protein of human immunodeficiency virus type 1 forms cation-selective channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA. 1996;93:111–115. doi: 10.1073/pnas.93.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N., Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+ . Biochemistry. 1996;35:8483–8488. doi: 10.1021/bi960833v. [DOI] [PubMed] [Google Scholar]
- Rustin P., Parfait B., Chretien D., Bourgeon T., Djouadi F., Bastin J., Rötig A., Munnich A. Fluxes of nicotinamide adenine dinucleotides through mitochondrial membranes in human cultured cells. J. Biol. Chem. 1996;271:14785–14790. doi: 10.1074/jbc.271.25.14785. [DOI] [PubMed] [Google Scholar]
- Schendel S., Montal M., Reed J.C. Bcl-2 family proteins as ion channels. Cell Death Differ. 1998;5:372–380. doi: 10.1038/sj.cdd.4400365. [DOI] [PubMed] [Google Scholar]
- Stanley S., Dias J.A., D'Arcangelis D., Mannella C.A. Peptide-specific antibodies as probes of the topography of the voltage-gated channel in the mitochondrial outer membrane of Neurospora crassa . J. Biol. Chem. 1995;270:16694–16700. doi: 10.1074/jbc.270.28.16694. [DOI] [PubMed] [Google Scholar]
- Shimizu S., Konishi A., Kodama T., Tsujimoto Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl. Acad. Sci. USA. 2000;97:3100–3105. doi: 10.1073/pnas.97.7.3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M., Shimizu S., Ito T., Chittenden T., Lutz R.J., Matsuda H., Tsujimoto Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA. 1998;95:14681–14686. doi: 10.1073/pnas.95.25.14681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluck R.M., Esposti M.D., Perkins G., Renken C., Kuwana T., Bossy-Wetzel E., Goldberg M., Allen T., Barber M.J., Green D.R., Newmeyer D.D. The pro-apoptotic proteins, Bid and Bax, cause a limited permeabilization of the mitochondrial outer membrane that is enhanced by cytosol. J. Cell Biol. 1999;147:809–822. doi: 10.1083/jcb.147.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotow T., Shibata M., Kanamori S., Tokuno O., Ohsawa Y., Sato N., Isahara K., Yayoi Y., Watanabe T., Leterrier J.F. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell Death Differ. 2000;7:666–674. doi: 10.1038/sj.cdd.4400694. [DOI] [PubMed] [Google Scholar]
- Motoyama S., Kitamura M., Saito S., Minamiya Y., Suzuki H., Saito R., Terada K., Ogawa J., Inaba H. Bcl-2 is located predominantly in the inner membrane and crista of mitochondria in rat liver. Biochem. Biophys. Res. Commun. 1998;249:628–636. doi: 10.1006/bbrc.1998.9205. [DOI] [PubMed] [Google Scholar]
- Monaghan P., Robertson D., Amos T.A., Dyer M.J., Mason D.Y., Greaves M.F. Ultrastructural localization of bcl-2 protein. J. Histochem. Cytochem. 1992;40:1819–1825. doi: 10.1177/40.12.1453000. [DOI] [PubMed] [Google Scholar]
- Schüler W., Wecker K., de Rocquigny H., Baudat Y., Sire J., Roques B.P. NMR structure of the (52-96) C-terminal domain of the HIV-1 regulatory protein Vprmolecular insights into its biological functions. J. Mol. Biol. 1999;285:2105–2117. doi: 10.1006/jmbi.1998.2381. [DOI] [PubMed] [Google Scholar]
- Cheng E.H.-Y., Nicholas J., Bellows D.S., Hayward G.S., Guo H.-G., Reitz M.S., Hardwick J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc. Natl. Acad. Sci. USA. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J.H., Modha D., White E. Interaction of E1B 19K with Bax is required to block Bax-induced loss of mitochondrial membrane potential and apoptosis. Oncogene. 1998;17:2993–3005. doi: 10.1038/sj.onc.1202215. [DOI] [PubMed] [Google Scholar]
- Derfuss T., Fickenscher H., Kraft M.S., Henning G., Fleckenstein B., Meinl E. Antiapoptotic activity of the herpesvirus saimiri-encoded Bcl-2 homologstabilization of mitochondria and inhibition of caspase-3-like activity. J. Virol. 1998;72:5897–5904. doi: 10.1128/jvi.72.7.5897-5904.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall W.L., Yim C., Gustafon E., Graf T., Sage D.R., Hanify K., Williams L., Fingeroth J., Finberg R.W. Epstein-Barr virus encodes a novel homolog of the bcl-2 oncogene that inhibits apoptosis and associates with Bax and Bak. J. Virol. 1999;73:5181–5185. doi: 10.1128/jvi.73.6.5181-5185.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldmacher V.S., Bartle L.M., Skletskaya S., Dionne C.A., Kedersha N.L., Vater C.A., Han J.W., Lutz R.J., Watanabe S., McFarland E.D.C. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA. 1999;96:12536–12541. doi: 10.1073/pnas.96.22.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciminale V., Zotti L., D'Agostini D.M., Ferro T., Casareto L., Franchini G., Bernardi P., Chieco-Bianchi L. Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I) Oncogene. 1999;18:4505–4514. doi: 10.1038/sj.onc.1203047. [DOI] [PubMed] [Google Scholar]
- Rahmani Z., Huh K.W., Lasher R., Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 2000;74:2840–2846. doi: 10.1128/jvi.74.6.2840-2846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]