

Abstract
Interferon (IFN)-γ and macrophages (Mϕ) play key roles in acute, persistent, and latent murine cytomegalovirus (MCMV) infection. IFN-γ mechanisms were compared in embryonic fibroblasts (MEFs) and bone marrow Mϕ (BMMϕ). IFN-γ inhibited MCMV replication in a signal transducer and activator of transcription (STAT)-1α–dependent manner much more effectively in BMMϕ (∼100-fold) than MEF (5–10-fold). Although initial STAT-1α activation by IFN-γ was equivalent in MEF and BMMϕ, microarray analysis demonstrated that IFN-γ regulates different sets of genes in BMMϕ compared with MEFs. IFN-γ inhibition of MCMV growth was independent of known mechanisms involving IFN-α/β, tumor necrosis factor α, inducible nitric oxide synthase, protein kinase RNA activated (PKR), RNaseL, and Mx1, and did not involve IFN-γ–induced soluble mediators. To characterize this novel mechanism, we identified the viral targets of IFN-γ action, which differed in MEF and BMMϕ. In BMMϕ, IFN-γ reduced immediate early 1 (IE1) mRNA during the first 3 h of infection, and significantly reduced IE1 protein expression for 96 h. Effects of IFN-γ on IE1 protein expression were independent of RNaseL and PKR. In contrast, IFN-γ had no significant effects on IE1 protein or mRNA expression in MEFs, but did decrease late gene mRNA expression. These studies in primary cells define a novel mechanism of IFN-γ action restricted to Mϕ, a cell type key for MCMV pathogenesis and latency.
Keywords: interferon γ, cytomegalovirus, signal transducer and activator of transcription 1, microarray analysis, macrophage
Introduction
Murine CMV (MCMV) provides an excellent small animal model for the study of the immune control of β-herpes virus infection. Although many studies show the importance of the IFN system, and specifically IFN-γ, in controlling MCMV infection both in vivo 1 2 3 4 5 6 7 8 9 10 and in vitro 7 9 11 12 13 14 15, the mechanisms by which IFN-γ regulates CMV infection remain largely undefined. In addition to studies consistent with IFN-γ inhibiting MCMV replication in vivo, we showed that IFN-γ inhibits reactivation of MCMV from latently infected explants of spleen and lung 7, which is consistent with the finding that reactivation in vivo is enhanced by treating with Abs to IFN-γ 10. Mice lacking the IFN-γ receptor develop chronic MCMV-associated large vessel vasculitis, demonstrating the importance of IFN-γ in regulating chronic CMV disease 7. Effects on reactivation from latency in tissue explants are due at least in part due to a blockade of growth from low levels of virus in primary cells 7, suggesting that defining mechanisms of IFN-γ antiviral action in primary cells will be critical to understanding how IFN-γ plays such a pivotal role in multiple stages of MCMV infection.
Identification of specific stages in viral replication blocked by IFNs has historically been critical to defining the molecular mechanisms of IFN action. Despite the importance of IFN-γ in vivo, there is some conflict in the literature regarding the stage in the viral life cycle at which IFN-γ blocks MCMV replication in fibroblasts, and other cell types have not been investigated. One study showed that IFN-γ inhibits expression of MCMV immediate early transcripts (IE) in 3T3 cells 13, but another study in primary embryonic fibroblasts found that IFN-γ blocked a late phase of MCMV infection 11. In addition to our lack of understanding of the specific targets of IFN-γ action against MCMV in primary cells, it is not known how currently defined mechanisms involving protein kinase RNA activated (PKR), RNaseL, Mx1, the IFN-α/β receptor, TNF-α receptors (TNFR1 or TNFR2), inducible nitric oxide synthase (iNOS), or Mx1 (for reviews, see references 16 17 18) influence IFN-γ action against MCMV.
To define the molecular mechanisms of IFN-γ action, we studied its effects on two primary cell types, murine embryonic fibroblasts (MEFs) and bone marrow–derived macrophages (BMMϕ). MEFs have traditionally been the cell type in which MCMV is propagated in vitro 19. Macrophages (Mϕ) are an important cell in the pathogenesis of MCMV infection during both acute and latent infection, both stages at which IFN-γ has an important role. Mϕ play a key role during acute infection as mediators of spread of the virus 20 21 22 23, as a predominant cell in inflammatory infiltrates occurring during MCMV infection 9 24, and as a cell type that can determine the outcome of in vivo infection 25 26. Mϕ are critical to MCMV latency as a site of latent infection 27 28 29.
We found that IFN-γ has an Mϕ-restricted, IFN-γ receptor and signal transducer and activator of transcription (STAT)-1α–dependent, mechanism of action independent of PKR, RNaseL, Mx1, IFN-α/β receptor, TNFR1, TNFR2, and iNOS. In BMMϕ, IFN-γ targets the MCMV IE1 gene product, a key regulator of CMV gene expression 30 31 32.
Materials and Methods
Cells, Virus, and Viral Assays.
BMMϕ and MEFs were differentiated and maintained in low endotoxin medium containing 10% FCS, and used at passage one or two, respectively 24 27. The differentiation state of these cells has been described previously (33, 34, and references therein). Unless otherwise indicated, BMMϕ and MEFs were derived from BALB/c mice. MCMV Smith strain was obtained from the American Type Culture Collection (no. VR-194, Lot 10), grown in 3T12 cells, and quantitated by plaque assay 9. MEFs and BMMϕ were plated in 6-well tissue culture–treated plates (Falcon) at 2.5 × 105 cells/well with or without IFN-γ (Genzyme). After 48 h, cells were infected at a multiplicity of infection (MOI) of 0.001 to 30 for 1 h at 37°C, washed twice with medium, and cultured in 1.5 ml of medium. At the times indicated, plates were frozen at −70°C and titered by plaque assay. For limiting dilution analysis, immunofluorescence, and Northern and Western analysis, cells were treated with or without 100 U/ml IFN-γ for 48 h, washed with PBS-EDTA (Sigma-Aldrich), and scraped (BMMϕ) or detached with trypsin (MEF), counted, centrifuged, and resuspended in a minimal volume of medium (generally 1 ml/107 cells) before infection at an MOI of 1 for 2 h on ice.
Limiting Dilution Analysis.
Infected BMMϕ or MEFs were cultured for 2 h at 37°C to allow for internalization; washed 2–3 times with 10 ml media, harvested, and serially diluted twofold from 2 × 103 cells/well into 96-well plates (24 wells/dilution/experiment). MEFs were added (5 × 103 cells/well) as indicator cells for cytopathic effect (cpe). Cultures were scored for cpe at 2 and 3 wk after infection. This assay has a sensitivity of about 1 PFU/well 7 35.
Immunofluorescence for IE1 Expression.
BMMϕ were plated onto 8-well Lab-Tek Glass Chamber slides (Nunc), cultured on the slides for 12 h, washed with PBS, and fixed with 1% paraformaldehyde in PBS for 20 min at room temperature. Cells were incubated overnight in blocking buffer containing 2% BSA (Sigma-Aldrich), and 10% each normal rabbit serum (Sigma-Aldrich) and goat serum (GIBCO BRL) in PBS. Immunofluorescence was performed using culture supernatant derived from the anti-IE1–specific mAb line Croma (donated by M. Messerle, Ludwig-Maximilians-Universitat Munchen, Munich, Germany) or an isotype-matched IgG1 control mAb HI-gamma-1-109.3 specific for DNP (0.01 μg/ml; reference 36) for 1 h at 4°C. Cells were then stained with FITC-conjugated goat anti–mouse IgG and IgM at 1:200 (Tago) in blocking buffer for 1 h at 4°C, and counterstained with bisbenzimide. Cells were coverslipped with PBS-glycerol and kept in the dark at 4°C until viewing. Slides were viewed on a ZEISS Axiophot Microscope and pictures taken with a Spot CCD camera using Northern Eclipse software (Empix Imaging). Blue bisbenzimide staining was converted to the red plane for ease of visualization of dual stained slides.
Northern Blot Analysis.
RNA was harvested at the indicated times as described 37 38, size fractionated on a 1.5% denaturing gel, and transferred to nylon membranes (Hybond-N; Amersham Pharmacia Biotech). Probes for IE1, DNA polymerase (DNApol), or glycoprotein B (gB) were generated by PCR, using plasmid pAMB25 for IE1 39 and the HindIII D fragment of the MCMV genome for DNApol and gB as template DNA 40. Primers for PCR were: IE1, GGGGAATGATAACAGCGACA/ATCCAGACTCTCTTTTCTGAGG; DNApol, GTGGCGTGTTGTATGATGGT/CTGGTCGTAGGTGTGGAAGC; gB (nested PCR), outer primers, CAGCCTGGAC-GAGATCAT/TCCTCGCAGCGTCTCCAATC, inner primers, CGTGTATCTCATCTTCACGAG/AGTGTCCATGTC-GGCCGTCA 23. Each PCR reaction contained: 50 mM KCl, 10 mM Tris-HCl (pH 9.0), 0.1% Triton X-100, 2.5 mM MgCl2 (for IE1 and gB) or 1.5 mM MgCl2 (for DNApol), 0.2 mM nucleotides, 0.15 μM each primer, and 1 U Taq (Thermus aquaticus) DNA polymerase. PCR was performed on a Temp Tronic thermocycler (Barnstead/Thermolyne) for 35 cycles. Annealing temperatures were: 59.5°C for IE1 and 55°C for DNApol and gB. The probe for E1 was a 484-bp Pst1/Xho1 fragment from a 5.5-kb Pst1 fragment from the MCMV HindIII F region 40. The probe for cyclophilin was a 680-bp BamH1/HindIII fragment of pSP65 containing the rat cyclophilin gene 41. Probes were radiolabeled with [32P]dCTP (MegaPrime DNA labeling system; Amersham Pharmacia Biotech), and hybridization signals were quantified by PhosphorImager (ImageQuant; Molecular Dynamics) and normalized to cellular cyclophilin.
Western Blot Analysis.
Western analysis was performed as described 42. Generally, 2–3 × 106 cells were lysed directly in 0.5 ml of Laemmli sample buffer, frozen at −70°C, boiled for 5 min, and then 5 μl was run on a 12.5% polyacrylamide gel with a 5% stacking gel. Gels were transferred and then blotted using supernatant derived from the Croma cell line producing anti-IE1 mAB, or the anti-E1 mAb 20-234-28 31, or anti–β-actin mAb AC-74 (used at 1:1,000; Sigma-Aldrich). IE1 has two isoforms (pp89 and pp72; references 43 and 44), both of which are detected by the Croma anti-IE1 mAB. A horseradish peroxidase–conjugated goat anti–mouse secondary was used (1:1,000; Tago), and membranes were developed using ECL Western Blotting Detection Reagents (Amersham Pharmacia Biotech). Protein quantitation was performed by comparing net intensity of predominant bands using Kodak ds1D software after photography on a DC120 camera (Eastman Kodak Co.).
Mice Used in These Studies.
All mice were housed in a Biosafety Level 2 facility at Washington University in accordance with all federal and university policies. BALB/c mice were from the National Cancer Institute, and C57BL/6 mice were from the The Jackson Laboratory. All other strains were bred at Washington University. STAT-1α–deficient mice (STAT-1α−/−) and TNF double receptor knockout mice (TNFR1R2−/−) were obtained from Dr. R. Schreiber (Washington University School of Medicine, St. Louis, MO; reference 45 46 47). 129 Sv/Ev mice and mice with null mutations in the IFN-γ receptor (IFN-γR−/−) or IFN-α/β receptor (IFN-α/βR−/−) were obtained from Dr. M. Aguet (Swiss Institute for Experimental Cancer Research, Epalinges, Switzerland; reference 48). Mice deficient in iNOS (iNOS−/−) were obtained from Dr. C. Nathan (Cornell University, New York, NY; reference 49). Mice deficient in RNase L (RNaseL−/−) were obtained from Dr. R. Silverman (Cleveland Clinic Foundation, Cleveland, OH; reference 50). Mice deficient in PKR (PKR−/−) were obtained from Dr. B. Williams (Cleveland Clinic Foundation, Cleveland, OH; reference 51).
Electrophoretic Mobility Shift Assays for STAT-1α Activation.
1.5–2 × 106 cells per sample were treated with concentrations of IFN-γ noted in text for 15 min at 37°C, and nuclear extracts were prepared as described 34. Samples were normalized according to cell number. 20% of each extract was assayed by electrophoretic mobility shift assay (EMSA) against a probe derived from the IFN-γ–activating sequence of the FcγR1 promoter 52. After acrylamide gel electrophoresis (6%), gels were dried and quantified by PhosphorImager (ImageQuant). To combine data from four independent experiments, PhosphorImage values were normalized by setting the highest value to one and lowest value to zero (GraphPad Prism) and results were averaged.
Microarray Analysis.
BMMϕ and MEF from IFN-α/βR−/− mice were mock infected for 1 h followed by treatment with or without 100 U/ml of IFN-γ for 48 h. RNA was harvested as above. Poly A purification, generation of cDNA, fluorescent labeling, and hybridization to gene chip were performed by Genome Systems essentially as described 53. 8,717 mouse genes were analyzed on Mouse GEM 1 (Incyte Pharmaceuticals, Inc.) microarrays using the software GEMtool 2.4.1.
Results
IFN-γ Pretreatment Inhibits Growth of MCMV in Mϕ and MEFs.
Pretreatment for 48 h with IFN-γ caused a dose-dependent inhibition of MCMV growth in both BMMϕ (Fig. 1 A) and MEFs (Fig. 1 B). Growth inhibition was more significant in BMMϕ than MEFs. BMMϕ showed a >100-fold decrease in viral titer by 72 h after infection (for example, at 100 U/ml of IFN-γ), whereas MEFs showed a maximum decrease of 5–10-fold even at 1,000 U/ml of IFN-γ. These results are consistent with other studies of IFN-γ effects on the growth of MCMV in fibroblasts 11 13 and demonstrate an antiviral effect of IFN-γ in primary BMMϕ.
Figure 1.


IFN-γ pretreatment inhibits growth of MCMV more effectively in BMMϕ than MEFs. (A and B) BMMϕ (A) or MEFs (B) were treated with or without IFN-γ for 48 h, infected at an MOI of 1, and cultured for the indicated times before freeze-thawing and plaque assay. Data shown is representative of two (BMMϕ) or three (MEF) independent experiments. Analysis of data in MEFs (B) revealed that the effects of IFN-γ were significant at 100 U/ml (48 h, P = 0.0231; 72 h, P = 0.02), and 1,000 U/ml (72 h, P = 0.0091), but that all other differences were insignificant (P > 0.05). (C) To determine the effect of MOI on IFN-γ treatment, BMMϕ or MEFs were treated with or without 100 U/ml IFN-γ for 48 h, infected at the MOIs shown, and cultured for 72 h before freeze-thawing and plaque assay (mean ± SEM from two independent experiments).
As lower final titers of virus were produced in untreated BMMϕ than untreated MEF at an MOI of 1 (Fig. 1A and Fig. B), we tested the hypothesis that the difference in effectiveness of IFN-γ treatment between the two cell types was due to inefficient MCMV replication in Mϕ by treating BMMϕ or MEFs with IFN-γ, and then infecting at a variety of MOIs (0.001–30; Fig. 1 C and data not shown). As we increased the MOI in untreated BMMϕ, the yield of MCMV increased to levels equivalent to or higher than those seen in untreated MEFs, without a decrease in the effect of IFN-γ (Fig. 1 C). The efficacy of IFN-γ in fibroblasts increased somewhat at an MOI of 0.1, a finding consistent with previous literature 7 11 13. However, we examined the relative efficacy of IFN-γ in BMMϕ and MEFs at MOIs of 0.01 and 0.001 and found that IFN-γ was consistently more effective at controlling MCMV growth in BMMϕ than MEFs (two- to fivefold in three independent experiments, not shown). We also considered the possibility that IFN-γ might be more effective in BMMϕ via induction of cell death, but found no decrease in BMMϕ viability with or without IFN-γ in the presence or absence of MCMV at 8 and 24 h after infection (data not shown, three independent experiments). These data showed cell type specificity of IFN-γ action, with IFN-γ being more effective at inhibiting MCMV growth in BMMϕ than in MEFs across a broad range of IFN doses, times after infection, MOIs, and regardless of the final yield of virus.
Mediators of IFN-γ Inhibition of MCMV Replication in BMMϕ.
Mϕ secrete molecules which may synergize with IFN-γ or provide their own protective effects including: (a) IFN-α/β, (b) TNF-α, and (c) nitric oxide (NO; references 11 and 54 55 56 57 58 59 60). These molecules were not required for IFN-γ action in BMMϕ, as IFN-γ efficiently inhibited MCMV growth in BMMϕ derived from mice with null mutations in genes encoding the IFN-α/β receptor (IFN-α/βR−/−), both TNF-α receptors (TNFR1/R−/−), or iNOS (Fig. 2).
Figure 2.

TNF-α, iNOS, IFN-α/β, PKR, and RNase L are not required for IFN-γ inhibition of MCMV replication in BMMϕ. BMMϕ were prepared from BALB/c (A), 129 (B), C57/Bl6 (C), TNFR1/TNFR2−/− (D), iNOS−/− (E), IFN-α/βR−/− (F), PKR−/− (G) or RNaseL−/− (H) mice. Cells were treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1, and cultured for the indicated times before freeze-thawing and plaque assay (mean ± SEM from two to six independent experiments).
To assess the possible role of novel IFN-γ–induced soluble mediators, we first demonstrated that IFN-γ action requires expression of the IFN-γ receptor (Fig. 3 A), and then determined whether IFN-γ treated wild-type BMMϕ secrete a mediator that inhibits growth of MCMV in IFN-γR−/− BMMϕ. Supernatant from IFN-γ–treated wild-type Mϕ (derived from 129 or BALB/c mice) did not protect IFN-γR−/− BMMϕ, although wild-type Mϕ were protected (Fig. 3B and Fig. C). Thus, IFN-γ treatment did not induce a stable molecule that can inhibit MCMV replication in IFN-γR−/− cells. To rule out contributions of unstable mediators, we determined whether wild-type BMMϕ can protect IFN-γR−/− cells when the cells are cocultured. Wild-type BMMϕ were not able to protect IFN-γR−/− BMMϕ in these cultures (Fig. 3 D). This experiment argues against a secretion of a IFN-γ–induced mediator responsible for the antiviral state of BMMϕ, but must be caveated with the possibility that such a mediator was induced, but also requires IFN-γ induction of its receptor. Together with data in BMMϕ lacking known secretion-dependent antiviral mechanisms (Fig. 2), these data argue that IFN-γ acts in BMMϕ by inducing intracellular changes that result in decreased viral yield.
Figure 3.

Secreted mediators from IFN-γ–treated wild-type BMMϕ do not protect IFN-γ unresponsive BMMϕ. (A) BMMϕ from IFN-γR−/− mice and wild-type mice were treated with or without 100 U/ml of IFN-γ, infected at an MOI of 1, and cultured for the indicated times before freeze-thawing and plaque assay (mean ± SEM from three independent experiments). Data is shown for IFN-γR−/− BMMϕ only. IFN-γ efficiently inhibited MCMV growth in wild-type BMMϕ (not shown, see Fig. 1). (B and C) BMMϕ from wild-type (Balb or 129; B) or IFN-γR−/− (C) mice were treated for 48 h with medium, medium plus 100 U/ml IFN-γ, or supernatant from IFN-γ (100 U/ml)–treated wild-type BMMϕ. BMMϕ were then infected at an MOI of 1 and cultured for the indicated times before freeze-thawing and plaque assay (mean ± SEM from three independent experiments). (D) Wild-type (wt; BALB/c or 129), IFN-γR−/− or a 50:50 mix of wild-type and IFN-γR−/− BMMϕ were plated and treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1 and cultured for the indicated times before freeze-thawing and plaque assay (mean ± SEM from two independent experiments). Results from the IFN-γ–treated cultures are shown. Results from cultures lacking IFN-γ were superimposable to the IFN-γ–treated IFN-γR−/− BMMϕ and are not shown.
Three effector mechanisms for IFN action that involve changes in intracellular molecules have been identified: 2′,5′ oligoadenylate synthetase (OAS), PKR, and Mx1 (for reviews, see references 16 and 17). BALB/c mice, the strain used for the majority of the experiments above, are deficient in Mx1 61 62, and therefore this pathway was not responsible for IFN-γ effects in BMMϕ. To test the other pathways, we used BMMϕ deficient in RNaseL 50, the only known downstream effector of activated OAS, or in PKR 51. IFN-γ effectively controlled MCMV replication in PKR−/− and RNaseL−/− BMMϕ (Fig. 2G and Fig. H).
STAT-1α Activation Is Essential for IFN-γ Effects in BMMϕ, but STAT-1α Activation Is Similar in BMMϕ and MEFs.
The latent cytoplasmic transcription factor STAT-1α is a proximal signaling molecule essential for IFN-γ antiviral effects against vesicular stomatitis virus (VSV) in cell lines and in vivo 45 63. Using BMMϕ from mice with a null mutation in the STAT-1α gene 45, we found that the antiviral effect of IFN-γ was completely dependent on STAT-1α (Fig. 4 A). We therefore tested the hypothesis that lack of effective STAT-1α activation in MEF explains the lack of effective IFN-γ action against MCMV in these cells (Fig. 1) using EMSA (Fig. 4B and Fig. C). We used a probe derived from the IFN-γ–activated sequence of the FcγRI promoter shown by supershift analysis to be STAT-1α specific in primary BMMϕ 34 52. STAT-1α activation was similar between MEF and BMMϕ across a range of IFN-γ doses, demonstrating that the proximal signaling cascade for IFN-γ is intact in both cell types (Fig. 4 B). Near maximal STAT-1α activation was seen with IFN-γ doses of 10 U/ ml, and STAT-1α activation was maximal in both MEF and BMMϕ at a dose of 100 U/ml. Thus, the difference in effectiveness of IFN-γ at inhibiting MCMV replication in MEFs and BMMϕ observed between 10 and 1,000 U/ml of IFN-γ (Fig. 1A and Fig. B) cannot be explained by different proximal signaling events in these two cell types.
Figure 4.
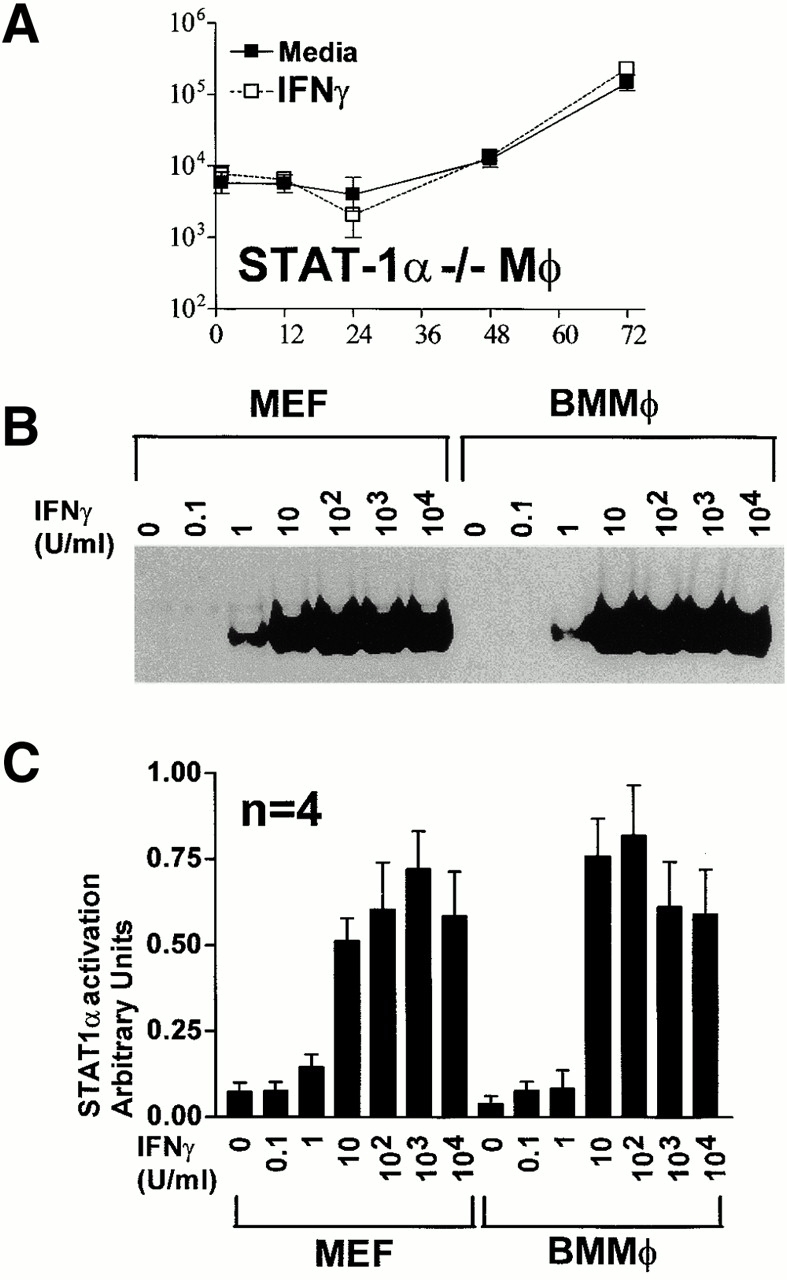
STAT-1α activation is required for IFN-γ inhibition of MCMV growth, but differences in early STAT-1α activation does not explain the cell type specificity of IFN-γ action. (A) STAT-1α−/− BMMϕ were treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1, and cultured for the indicated times before freeze-thawing and plaque assay (mean ± SEM from two independent experiments). IFN-γ efficiently inhibited MCMV replication in concurrent cultures of wild-type 129 BMMϕ (not shown). (B and C) Cells were stimulated with IFN-γ at the indicated doses for 15 min at 37°C, nuclear lysates were prepared, and EMSA performed using a probe derived from the FcγRI gamma response region. One representative (of four) experiment is shown in (B). Quantitation of STAT-1α activation by PhosphorImage analysis is shown in C (mean ± SEM for four independent experiments).
Cell Type–specific Regulation of Genes in MEFs Versus BMMϕ Defined by Microarray Analysis.
Although STAT-1α activation was similar in BMMϕ and MEF (Fig. 4), the differences in IFN-γ effectiveness at inhibiting MCMV growth suggested that IFN-γ effects differ between MEFs and BMMϕ. We therefore performed microarray analysis to compare levels of 8,717 mouse genes 48 h (the time at which we infect with MCMV) after IFN-γ treatment of MEFs and BMMϕ (Fig. 5). IFN-γ had less than twofold effects on the vast majority of genes in either MEFs or BMMϕ at the 48-h time point (Fig. 5A and Fig. B). However, 49 genes, many previously known to be induced by IFN-γ, were changed more than fivefold in BMMϕ and/or MEF (Fig. 5C and Fig. D, and Table ).
Figure 5.
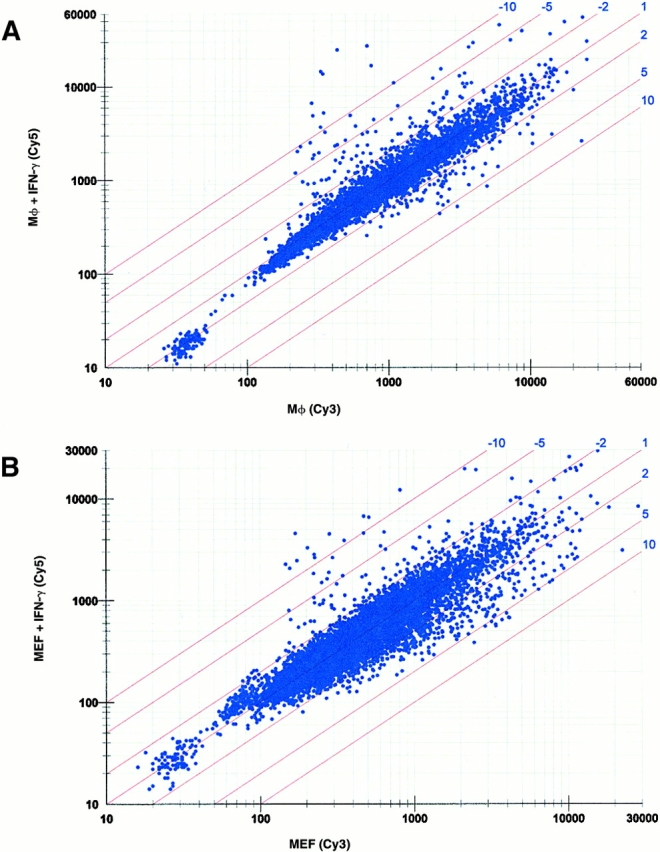
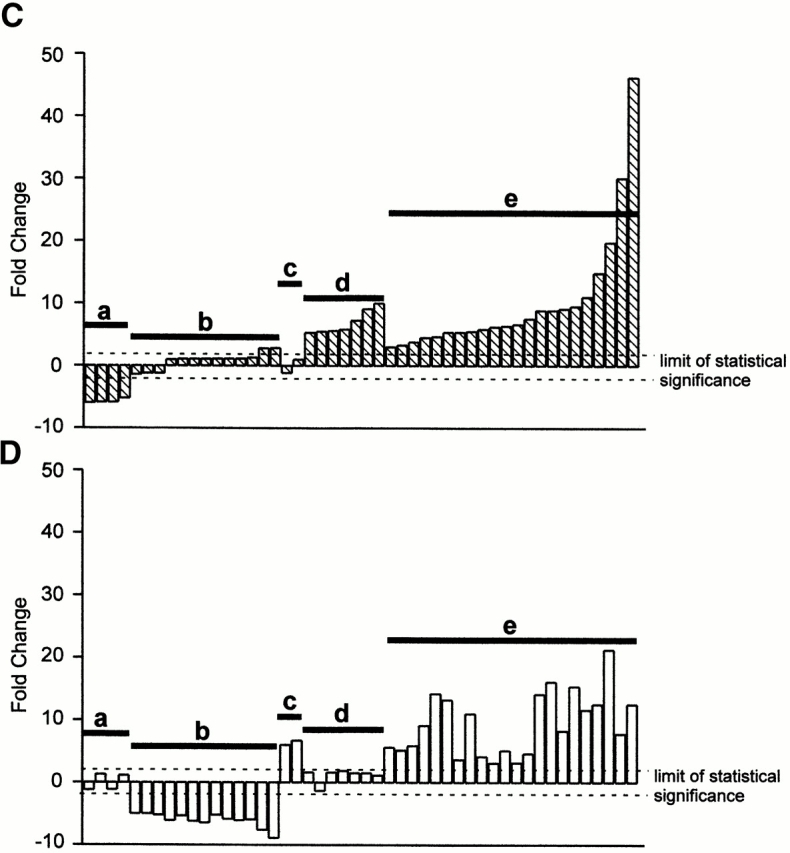
Comparative expression of transcripts in BMMϕ and MEFs after IFN-γ treatment. (A) Scatter plot of fluorescence intensity comparing gene expression in BMMϕ with or without IFN-γ treatment for 8,717 genes. The Cy5 and Cy3 signal values indicate the amount of transcript measured in BMMϕ plus IFN-γ and BMMϕ plus media treatment only, respectively. (B) The data is plotted for MEFs as in A with Cy5 and Cy3 signal values indicating the amount levels of transcripts in MEF plus IFN-γ and MEF plus media treatment only, respectively. (C and D) Bar graphs showing the expression of 49 genes which were changed more than fivefold by IFN-γ treatment in either (C) BMMϕ or (D) MEF samples. These genes are ordered identically in C and D. The lowercase letters represent groupings of transcripts with different expression patterns and are described in the text and Table . The genes are listed in Table . The limit of statistical significance is set at twofold based on data from Incyte Genomics analyzing the variance in the method across multiple experiments.
Table 1.
| Category | GenBank accession no. | Gene name or product | Fold change in BMMϕ versus BMMϕ plus IFN-γ | Fold change in MEF versus MEF plus IFN-γ |
|---|---|---|---|---|
| a: Specifically | AA163727 | ZW1- homolog (Drosophila), centromere/kinetochore protein | −5.9 | −1.2 |
| down in BMMϕ | AA276440 | Selenoprotein P, plasma, 1 | −5.8 | 1.3 |
| W59165 | Folate binding protein 2 | −5.8 | −1.1 | |
| AA138966 | EST | −5.2 | 1.1 | |
| b: Specifically | AA058055 | EST | −1.4 | −5 |
| down in MEF | AA030821 | EST | −1.2 | −5 |
| AA008052 | EST | −1.2 | −5.2 | |
| W14275 | EST | 1 | −6.1 | |
| W85641 | EST | 1.1 | −5.3 | |
| AA175695 | EST | 1.1 | −6.2 | |
| W63822 | Homeo box A2 | 1.1 | −6.4 | |
| W14332 | EST | 1.2 | −5.2 | |
| AA390032 | EST | 1.2 | −5.8 | |
| W97155 | EST | 1.2 | −6.1 | |
| AA216849 | EST | 1.3 | −5.9 | |
| AA538511 | Histocompatibility 2, D region locus 1 | 2.8 | −7.6 | |
| AA240404 | Putative purine nucleotide binding protein mRNA | 2.9 | −8.9 | |
| c: Specifically | AA245029 | EST | −1.1 | 6 |
| up in MEF | AA209006 | EST | 1 | 6.7 |
| d: Specifically | AA217290 | EST | 5.3 | 1.6 |
| up in BMMϕ | AA177481 | EST | 5.5 | −1.3 |
| AA458171 | EST | 5.6 | 1.6 | |
| AA230649 | Histocompatibility 2, class II, locus DMa | 5.9 | 1.9 | |
| AA269724, AA277329 | H-2 class II histocompatibility antigen, I-A β chain precursor | 7.3 | 1.5 | |
| AA462202 | BP-3 alloantigen | 9.1 | 1.5 | |
| W34612 | Transglutaminase 2, C polypeptide | 10 | 1.1 | |
| e: Upregulated | AA186012 | Histocompatibility 2, Q region locus 7 | 3 | 5.6 |
| in BMMϕ | AA177731 | EST | 3.3 | 5.2 |
| and MEF | AA143986 | EST | 3.8 | 5.9 |
| AA109951 | β2 microglobulin | 4.5 | 9.1 | |
| AA140542 | EST | 4.7 | 14.2 | |
| AA138757 | EST | 5.4 | 13.2 | |
| AA451385 | EST | 5.4 | 3.7 | |
| AA174721 | EST | 5.5 | 11 | |
| AA286393, AA172456 | Small inducible cytokine A12 | 5.9 | 4.2 | |
| AA123837 | Transporter 1, ABC (ATP binding cassette) | 6.2 | 3.1 | |
| AA388678 | STAT 1 | 6.4 | 5.1 | |
| AA475774 | Cathepsin C | 6.6 | 3.2 | |
| AA272807 | Histocompatibility 2, class II antigen A, alpha | 7.5 | 4.6 | |
| AA172624 | EST | 8.9 | 14.1 | |
| AA145136 | EST | 8.9 | 16.1 | |
| AA260490 | EST | 9.1 | 8.3 | |
| AA204588 | EST | 9.5 | 15.4 | |
| AA122443 | EST | 11 | 11.6 | |
| AA472492 | Monokine induced by IFN-γ | 14.9 | 12.5 | |
| AA153021, AA277451 | IFN-induced guanylate binding protein | 19.8 | 21.3 | |
| W83447, AA210495 | Spi2 proteinase inhibitor (spi2/eb1) | 30.1 | 7.8 | |
| AA000712, AA472994AA145865 | Lymphocyte antigen 6 complex | 46.2 | 12.5 |
GenBank accession numbers, gene names, and fold changes in gene expression are listed from top to bottom in the same order as left to right in Fig. 5C and Fig. D, for all genes changed more than fivefold in either MEFs or BMMϕ by IFN-γ treatment. The lower case letters in the left column correspond to those in Fig. 5C and Fig. D.
IFN-γ–induced changes in gene expression differed between MEFs and BMMϕ. These differences included genes whose expression was either increased or decreased in BMMϕ but not altered in MEFs (groups a and d, Fig. 5C and Fig. D, and Table ), and genes whose expression was either increased or decreased in MEFs but not altered in BMMϕ (groups b and c, Fig. 5C and Fig. D, and Table ). There was a group of genes whose expression was increased in both MEFs and BMMϕ, and these include several genes well known to be IFN inducible (group e, Fig. 5C and Fig. D, and Table ). This data showed that despite similarities in STAT-1α activation, there are significant differences in IFN-γ effects in MEFs versus BMMϕ, providing a rationale for the differences in IFN-γ effectiveness at controlling MCMV growth in these two cell types.
IFN-γ Decreases Viral Yield per Infected BMMϕ.
To determine why IFN-γ is more effective at controlling MCMV growth in BMMϕ than MEFs, we analyzed the effect of IFN-γ on sequential stages in the MCMV replication cycle in MEFs and BMMϕ. We first determined the frequency of cells productively infected with MCMV using limiting dilution analysis and a MEF indicator monolayer previously shown to detect 1–10 PFU of MCMV even in the presence of IFN-γ 7 35. IFN-γ did not alter the frequency of productively infected MEFs (Fig. 6 A). However, 100 U/ml of IFN-γ decreased the frequency of productively infected BMMϕ two- to fourfold (Fig. 6 A). In the absence of an IFN-γ–induced decrease in MCMV yield per infected cell, this difference would not explain the 100-fold decline in MCMV titer caused by the same dose of IFN-γ in BMMϕ (Fig. 1). We next quantitated IFN-γ effects on the frequency of BMMϕ expressing the IE1 protein by immunofluorescence. IFN-γ did not significantly change the frequency of BMMϕ expressing the IE1 protein (Fig. 6). This ruled out significant effects of IFN-γ on viral binding, internalization, and the frequency of cells expressing IE1 protein as an explanation for the antiviral effects of IFN-γ in BMMϕ. However, we did notice that immunofluorescent staining for IE1 was qualitatively fainter in IFN-γ–treated than control BMMϕ (see below). Results from limiting dilution analysis and staining for IE1 protein demonstrated that IFN-γ effects on MCMV titer were due to changes in the frequency of productively infected cells and viral yield per infected cell.
Figure 6.


IFN-γ inhibits MCMV replication in both BMMϕ and MEFs by decreasing viral yield per cell rather than decreasing the number of infected cells. (A) BMMϕ or MEFs were treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1 for 2 h on ice, and then incubated for 2 h at 37°C to allow internalization of the virus. Cells were washed, serially diluted, and the frequency of productively infected cells determined. Data shown is the percentage of wells with cytopathic effect (24 wells/dilution/experiment) from one of two experiments with similar results. (B–D) BMMϕ were treated with medium (B) or 100 U/ml IFN-γ (C), infected at an MOI of 1 for 1 h at 4°C, or mock infected. Cells were stained with an IE1-specific mAb (B and C) or an isotype-matched control mAb (D), a FITC-conjugated secondary Ab, and counterstained with bisbenzimide. Data shown are double exposures of bisbenzimide staining (converted from the blue to red plane) and FITC staining. IE1-positive cells can be visualized as light yellow nuclei. Shown is a representative one of four experiments. For this experiment shown, two independent, randomly selected regions of each of two slides for each condition were photographed and counted. 21% (252 of 1,206 cells counted) of media-treated BMMϕ (B) expressed nuclear IE1 protein, whereas 17% (136 of 792 cells counted) of IFN-γ–treated BMMϕ (C) expressed nuclear IE1 protein. Infected cells stained with the isotype-matched mAb, HI-gamma-1-109.3 specific for DNP, were uniformly red (shown are IFN-γ–treated infected BMMϕ (D). Mock-infected cells stained with either mAb specific for IE1 or control mAB were indistinguishable from D.
IFN-γ Inhibits Expression of the MCMV IE1 mRNA during the First 3 h of Infection in BMMϕ but Not MEF.
We next examined whether steady-state levels of message from representative IE, early, or late genes were influenced by IFN-γ treatment. IFN-γ treatment of BMMϕ decreased expression of IE1 mRNA ∼10-fold at 1–2 h after infection (Fig. 7 A). By 4 h and thereafter, levels of IE1 mRNA differed by at most twofold between BMMϕ treated with IFN-γ or medium alone (Fig. 7 A, quantitation shown in Fig. 7 B). Early and late transcript levels were also decreased by IFN-γ treatment in Mϕ (Fig. 7 A). DNA polymerase was decreased 3–13-fold (across five experiments) and glycoprotein B levels were decreased 5–10-fold (across five experiments). In contrast to results in BMMϕ, levels of IE1 mRNA were unchanged by IFN-γ treatment in MEFs (Fig. 7 A). In MEFs there was a slight effect of IFN-γ on mRNA levels for one early gene, DNA polymerase, but not for another early gene, E1. Consistent with a previous report 11, the most dramatic effect in MEFs was seen on the level of late gene transcripts as assessed by glycoprotein B (Fig. 7 A).
Figure 7.
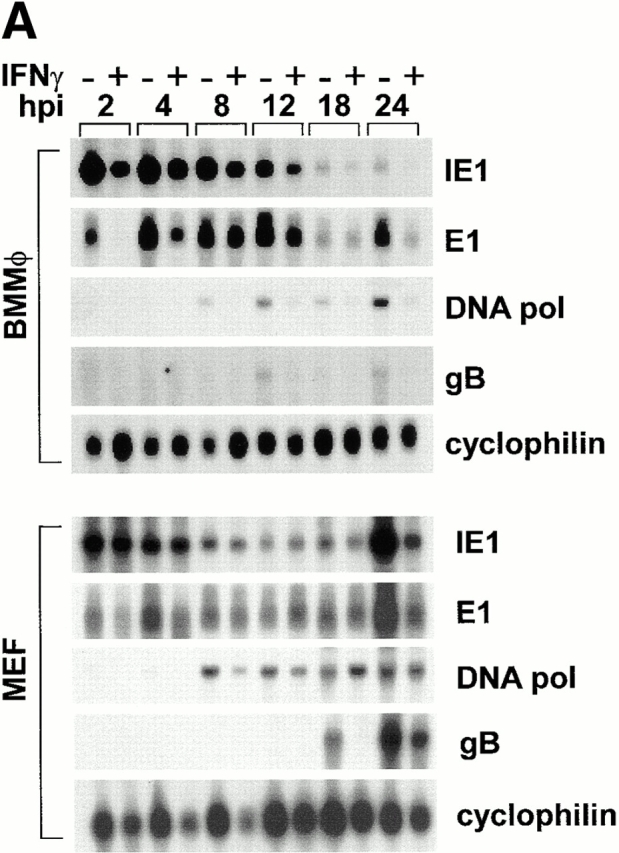

IFN-γ alters steady state levels of different viral transcripts in MEFs compared with BMMϕ. (A) BMMϕ or MEF were treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1, and at the times indicated Northern analysis was performed using probes directed to the viral IE gene, IE1; the viral early genes, E1 and DNA pol; or the viral late gene, gB. Cyclophilin expression is shown as a loading control. Shown are representative results from one of five (BMMϕ) or one of three (MEF) experiments. (B) Phosphorimage quantitation of mRNA levels of IE1 message levels in media-treated or IFN-γ–treated BMMϕ was used to determine the fold decrease in message levels in IFN-γ–treated cells. Fold decrease is expressed as the amount of IE1 message in media-treated BMMϕ divided by the amount in IFN-γ treated BMMϕ (mean ± SEM of two to six independent experiments).
IFN-γ Inhibits Expression of IE1 Protein for Prolonged Periods in BMMϕ but Not MEFs.
As IFN-γ significantly inhibited early and late gene expression in BMMϕ, but had relatively subtle effects on IE1 mRNA expression after 4 h (Fig. 6 B), we considered the possibility that IFN-γ inhibited the expression of IE proteins (Fig. 8 A), which are critical regulators of early and late gene expression 30 31 32.
Figure 8.


IFN-γ decreases expression of IE1 protein in BMMϕ but not MEFs. (A) BMMϕ or MEFs were treated with or without 100 U/ml IFN-γ for 48 h, infected at an MOI of 1, and at the times indicated Western analysis was performed with mAb directed against the viral IE protein, IE1, or the viral early protein, E1. β-actin is shown as a loading control. Shown are representative results from one of three experiments. (B) Net intensity measurements on ECL-developed Western blots were used to quantitate the fold decrease of IE1 protein levels in IFN-γ–treated BMMϕ compared with media-treated BMMϕ (mean ± SEM from three independent experiments).
IE1 (the pp89 isoform [43, 44], see Materials and Methods, referred to as IE1 here) and E1 protein expression was not altered by IFN-γ in MEFs. The decrease in IE1 expression seen at 8 and 12 h seen in both IFN-γ–treated and control MEFs samples has been described previously and is part of the normal viral replication cycle 44. There was no effect of IFN-γ or virus infection on actin levels, demonstrating that the effect of IFN-γ on IE1 protein expression was not due to a general degradation of protein in IFN-γ–treated cells.
Surprisingly, although there was no detectable effect of IFN-γ on the protein level of IE1 or E1 in MEFs, there was a significant decrease (∼6–24-fold in four experiments at all times from 3–24 h after infection) in IE1 and E1 protein levels in IFN-γ–treated BMMϕ (Fig. 8 A, quantitation shown in Fig. 8 B). There was also a decrease (about sixfold across six experiments) in the pp72 isoform of IE1 in BMMϕ and a smaller decrease of about two- to threefold in MEFs. The decrease in IE1 protein expression in BMMϕ persisted for 96 h after infection (data not shown). It was notable that the fold decrease in IE1 protein caused by IFN-γ was significantly greater than the fold decrease in IE1 mRNA at multiple time points after 3 h (compare Fig. 7 B and 8 B). Differences in IE1 expression between BMMϕ and MEFs were not MOI dependent. IE1 protein expression at 8 and 24 h after infection was not significantly inhibited in MEFs infected at MOIs ranging from 1 to 0.001 (data not shown, three independent experiments).
We considered the possibility that effects of IFN-γ on IE1 protein or RNA expression were due to known mediators of IFN action RNaseL and PKR, despite the fact that these molecules were not required for IFN-γ–mediated inhibition of MCMV replication (Fig. 2). It was possible that IFN-γ–mediated inhibition of IE1 protein expression was dependent on PKR, as PKR phosphorylates EIF2α, thereby inhibiting protein synthesis 64. It was also possible that activation of RNaseL might contribute to alterations in IE1 mRNA expression induced by IFN-γ treatment. Therefore, we examined mechanisms of IFN-γ action in more detail in PKR−/− and RNaseL−/− BMMϕ. By Northern and Western analysis performed on virally infected BMMϕ derived from PKR−/− or RNaseL−/− strains, we found that neither PKR nor RNaseL were required for IFN-γ–induced decreases in viral IE1 mRNA or protein levels (data not shown).
Discussion
In this paper we demonstrate the following important points: (a) IFN-γ is much more effective at blocking MCMV replication in primary BMMϕ than MEFs, (b) IFN-γ action in BMMϕ does not require known mediators of antiviral effects of IFN, (c) the mechanisms of action of IFN-γ differ significantly in BMMϕ and MEFs, and (d) IFN-γ acts by a novel mechanism, independent of RNaseL and PKR, to decrease IE1 protein expression in BMMϕ. Although IFN-γ–induced activation of STAT-1α is similar in BMMϕ and MEFs early after IFN-γ treatment, microarray analysis showed that IFN-γ regulated gene expression differently in BMMϕ and MEFs. Thus, IFN-γ may act in different primary cells types by inducing or decreasing expression of different sets of genes. The fact that IFN-γ action is cell type specific and utilizes a novel mechanism in BMMϕ, a cell type key for both acute and chronic MCMV infection, has important implications for understanding how IFNs regulate viral infection.
The Role of IFN-γ in Cytomegalovirus Infection and the Importance of Defining Antiviral Mechanisms of IFN-γ against CMV.
The role of IFN-γ in cytomegalovirus infection is controversial, with published studies arguing for a role of IFN-γ in either inhibiting or promoting infection. Some evidence has been presented for a role for IFN-γ enhancing infection with rat CMV in vivo 65, and for a role of high dose exogenously administered IFN-γ enhancing MCMV disease 3. However, multiple groups have convincingly demonstrated a protective effect of IFN-γ at multiple stages of cytomegalovirus (both rat and mouse CMV) infection, including acute infection 1 3 6 7 65, prevention of chronic persistent infection 4 5, induction of effective antigen presentation 5 66, and inhibition of reactivation from latency 7 10. We have additionally shown that IFN-γ plays an important role in protection against vascular disease induced by either MCMV or the γ-herpes virus γHV68 7 67.
Although these studies all argue for a protective role for IFN-γ against MCMV infection, in humans IFN-γ is involved in differentiation of peripheral blood mononuclear cells into Mϕ that are permissive for human CMV replication (HCMV 68). In these studies, IFN-γ was added to freshly isolated adherent human PBMCs and 9 d later cultures were infected with HCMV. Under these conditions, IFN-γ resulted in Mϕ that replicated HCMV very efficiently. This is in striking contrast to the results we present here in which pretreatment of already differentiated cells with IFN-γ inhibited replication 100-fold. We feel it likely that these differences reflect the fact that we add IFN-γ before infection of already differentiated cells, whereas in the studies by Soderberg-Naucler et al. 68, IFN-γ is added during the differentiation process. An argument was made by Soderberg-Naucler et al. that IFN-γ does not have antiviral activity in differentiated human Mϕ, as treatment of cultures with IFN-γ after HCMV infection did not inhibit replication of the virus 68. However, this conclusion is unlikely, as IFN-γ typically requires induction of gene expression to exert its effects, and thus pretreatment with IFNs is often required to observe maximal antiviral effects. Moreover, both HCMV and MCMV specifically inhibit IFN signaling 34 69 70 71. Thus, one would not expect IFN-γ to effectively inhibit CMV infection when added after viral infection. Notwithstanding this caveat, it is clear that under some conditions IFN-γ can generate cells that efficiently replicate HCMV. In this regard, it is notable that MCMV replicates well in cells derived from IFN-γR−/− mice (Fig. 3 A) and can reactivate from BMMϕ derived from latently infected IFN-γR−/− mice 7, demonstrating that IFN-γ is not required for differentiation of permissive Mϕ in vivo.
Although IFN-γ can influence Mϕ differentiation, resulting in HCMV permissive cells, other investigators have shown that IFN-γ has antiviral effects against HCMV. Human CD4 T cells, which play an important role in controlling HCMV infection 72, secrete IFN-γ 15. When the astrocytoma line, U373MG, 15 or human fibroblasts 73 are preincubated with IFN-γ, significant inhibition of HCMV replication and expression of early and late gene products is observed, a result consistent with our findings as well as previous studies showing that pretreatment with IFN-γ inhibits MCMV replication in fibroblasts 11 13. Interestingly, and similar to studies presented here, evidence was presented in one study supporting an effect of IFN-γ on expression of HCMV IE proteins 15.
Despite this, the mechanism of IFN-γ action against MCMV has been controversial. Conclusions based on work in infected primary 11 or transformed fibroblasts 13 have been contradictory with the proposal that IFN-γ targets either IE mRNA expression 13 or late mRNA expression 11. Experiments presented here help to clarify this conflict, as we found that the effects of IFN-γ are cell type specific with actions on the IE stage of MCMV replication in BMMϕ compared with actions on the late stage of infection in MEFs. Effects of IFN on the IE stage of MCMV infection are strikingly similar to mechanisms of action against HSV-1 74.
Potential Consequences of Cell Type Specificity.
We show here that IFN-γ has cell type–specific effects against MCMV infection in vitro. This may be a generally applicable finding. IFN-γ has cell type–specific effects in vivo against LCMV infection. IFN-γ treatment clears hepatocyte infection, but not intrahepatic nonparenchymal cells or splenocytes 75. Fibroblasts and Mϕ likely play distinct roles in the replication and pathogenesis of MCMV in vivo. Mϕ have been shown to harbor latent virus 27 28 29. They have also been implicated in spread of the virus 20 21 22 23, and play an important role in determining the outcome of MCMV disease 25 26. Fibroblasts are more likely to play a role in the initial acute infection. Thus, the cell type specificity of IFN-γ action in BMMϕ versus MEFs is likely to play a role in its function in vivo. Indeed, IFN-γ has been shown to play a key role in terminating persistent viral infection of both MCMV and other viruses 4 5 7 76 77. As Mϕ are a likely latent reservoir for MCMV, blockade to viral growth in these cells may explain why IFN-γ plays a strong role in protecting the host against reactivation both in vitro 7 and in vivo 10. Its protective effect in Mϕ is likely to aid in maintaining antigen presentation in light of multiple viral mechanisms which inhibit both the class I and class II pathways 34 78 79 80. IFN-γ has been shown to restore antigen presentation in the setting of viral infection 5 66. Thus, the effectiveness of IFN-γ against MCMV replication in Mϕ may have important consequences for the course of pathogenesis and disease in infected mice.
Potential Mediators of IFN-γ Effects on MCMV Growth and IE1 Protein Expression.
We found that the well-characterized potential effector mechanisms for IFN-γ including IFN-α/β receptors, PKR, iNOS, TNF-α receptors, Mx1, and RNaseL are not required for IFN-γ–mediated inhibition of MCMV growth in BMMϕ. However, IFN-γ action in BMMϕ does require the IFN-γ receptor and STAT-1, acts in cis in the infected cell rather than by inducing expression of additional soluble mediators, and targets a specific stage in MCMV replication. The existence of as yet undiscovered antiviral mechanisms of IFN is also suggested by residual IFN-α/β effects against encephalomyocarditis virus in mice triply deficient for Mx1, PKR, and RNaseL 81. These alternative mechanisms may be required by the host because viruses have evolved strategies for inhibiting certain pathways of IFN action. By using multiple antiviral mechanisms specifically targeted to vulnerable stages in the replication cycle of specific viruses, the host may be able to overcome immune evasion by viruses. Many viruses, particularly those which persist in the host, have developed mechanisms to block the effectiveness of interferons, including inhibition of PKR 82 83 and disruption of the Janus kinase (JAK)/STAT pathway 69 70 84. Considering the lack of a requirement for PKR or RNaseL in the antiviral effects of IFN-γ against MCMV, it is tempting to postulate that MCMV may also inhibit these pathways. It has already been reported that one downstream function of IFN-γ, induction of MHC class II, is inhibited by CMV infection 34 71.
Although we have identified one target of IFN-γ action against MCMV in BMMϕ (expression of the IE1 protein), the molecular pathways involved are not defined. The different transcriptional responses to IFN-γ in BMMϕ versus MEFs could provide a starting point for identification of molecules responsible for the effects of IFN-γ. If the stages of MCMV infection are the same in BMMϕ and MEFs, it seems unlikely that genes which are similarly regulated in BMMϕ and MEFs will explain cell type–specific effects of IFN-γ. However, it is possible that a difference in the stages of MCMV infection in BMMϕ as opposed to MEFs could provide a target for an IFN-γ–induced gene in one cell type and not the other.
There are several possibilities for the mechanism of IFN-γ action worth noting. It has been reported that IFN-γ inhibits IE gene transcription by downregulating nuclear factor (NF)κB activity 85. The interferon inducible protein, p202, can modulate transcription by binding NFκB sites 86, and may therefore be involved in this mechanism. In support of this hypothesis, a family member of p202, the human IFN-inducible protein IFI16, has been shown to repress transcription of the HCMV UL54 promoter 87. Although we only observed decreased IE1 mRNA during the first few hours of infection, it will be interesting to evaluate whether this mechanism may be playing a role in the effect of IFN-γ seen in Mϕ infection. A second alternative mechanism involves the indoleamine 2,3 dioxygenase (IDO) pathway. In retinal pigment epithelial cells, IFN-γ inhibits HCMV replication, and the effects of IFN-γ are reversed by addition of l-tryptophan, an inhibitor of IDO 88. However, preliminary studies in our lab suggest that the IDO pathway is not involved in BMMϕ-specific IFN-γ–mediated protection, as neither the addition of l-tryptophan nor a specific inhibitor of IDO, 1-methyl tryptophan, reversed IFN-γ–mediated inhibition of MCMV growth. A third potential mediator of IFN-γ effects in BMMϕ is IFN-γ regulation of cell cycle. Cell cycle regulation by herpes viruses is a critical part of the viral life cycle (for reviews, see references 89 and 90 91 92 93 94 95 96). IFN-γ treatment of BMMϕ with IFN-γ results in expression of p21waf-1, leading to arrest at the G1/S boundary 97. As the outcome of MCMV infection is dependent on cell cycle phase 94, IFN-γ's effects on the cell cycle may influence CMV infection. It is possible that differences in the effectiveness of IFN-γ in BMMϕ and MEFs reflect differences in proliferative capacity or cell cycle regulation between these two primary cell types. An interesting additional potential mechanism of IFN-γ action has been recently identified in hepatitis B virus (HBV) transgenic mice. HBV gene expression in these mice is down regulated by a posttranscriptional mechanism induced by IFN-γ and TNF-α 98. Recent data suggests that this is mediated by the La protein, an RNA binding protein which is associated with clearance of HBV viral RNA 99 100. These mechanisms will need to be evaluated in the MCMV system and compared between primary MEF and BMMϕ. Further analysis of the specific molecules involved in the novel Mϕ-specific mechanism of IFN-γ action demonstrated here may well lead to definition of novel pathways of IFN action.
Acknowledgments
We thank Martin Messerle and Ulrich Koszinowski for kindly providing mAbs specific for MCMV IE1 and E1, and Carl Nathan, Bryan Williams, and Robert Silverman for kindly providing iNOS, PKR, and RNaseL-deficient mice. We thank Sam Speck, David Leib, Margaret MacDonald, Lynda Morrison, as well as members of their laboratories, for helpful comments. We thank Robert Schreiber for comments during the development of this project and for assistance with analysis of STAT-1α activation.
H.W. Virgin was supported by grant AI39616 from the National Institute of Allergy and Infectious Diseases and the Monsanto-Searle Biomedical Agreement. R.M. Presti and D.L. Popkin were supported by National Institutes of Health grant GM07200. M. Connick was supported by a Fellowship from the Pediatric Infectious Disease Society.
Footnotes
Abbreviations used in this paper: BM, bone marrow; EMSA, electrophoretic mobility shift assay; HBV, hepatitis B virus; HCMV, human CMV; IE, immediate early; iNOS, inducible nitric oxide synthase; MCMV, murine CMV; MEF, murine embryonic fibroblast; Mϕ, macrophage; MOI, multiplicity of infection; PKR, protein kinase RNA activated; STAT, signal transducer and activator of transcription.
References
- Fennie E.H., Lie Y.S., Low M.A., Gribling P., Anderson K.P. Reduced mortality in murine cytomegalovirus infected mice following prophylactic murine interferon-gamma treatment. Antivir. Res. 1988;10:27–39. doi: 10.1016/0166-3542(88)90012-5. [DOI] [PubMed] [Google Scholar]
- Osborn J.E., Medearis D.N.J. Studies of relationship between mouse cytomegalovirus and interferon. Proc. Soc. Exp. Biol. Med. 1966;121:819–824. doi: 10.3181/00379727-121-30897. [DOI] [PubMed] [Google Scholar]
- Pomeroy C., Delong D., Clabots C., Riciputi P., Filice G.A. Role of interferon-gamma in murine cytomegalovirus infection. J. Lab. Clin. Med. 1998;132:124–133. doi: 10.1016/s0022-2143(98)90007-5. [DOI] [PubMed] [Google Scholar]
- Lucin P., Pavic I., Polic B., Jonjic S., Koszinowski U.H. Gamma interferon-dependent clearance of cytomegalovirus infection in salivary glands. J. Virol. 1992;66:1977–1984. doi: 10.1128/jvi.66.4.1977-1984.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hengel H., Lucin P., Jonjic S., Ruppert T., Koszinowski U.H. Restoration of cytomegalovirus antigen presentation by gamma interferon combats viral escape. J. Virol. 1994;68:289–297. doi: 10.1128/jvi.68.1.289-297.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orange J.S., Wang B., Terhorst C., Biron C.A. Requirement for natural killer cell-produced interferon-gamma in defense against murine cytomegalovirus infection and enhancement of this defense pathway by interleukin 12 administration. J. Exp. Med. 1995;182:1045–1056. doi: 10.1084/jem.182.4.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presti R.M., Pollock J.L., Dal Canto A.J., O'Guin A.K., Virgin H.W. Interferon-gamma regulates acute and latent murine cytomegalovirus infection and chronic disease of the great vessels. J. Exp. Med. 1998;188:577–588. doi: 10.1084/jem.188.3.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay C.H., Yu L.Y., Kumar V., Mason L., Ortaldo J.R., Welsh R.M. The role of LY49 NK cell subsets in the regulation of murine cytomegalovirus infections. J. Immunol. 1999;162:718–726. [PubMed] [Google Scholar]
- Heise M.T., Virgin H.W. The T cell independent role of IFN-gamma and TNF-alpha in macrophage activation during murine cytomegalovirus and herpes simplex virus infection. J. Virol. 1995;69:904–909. doi: 10.1128/jvi.69.2.904-909.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polic B., Hengel H., Krmpotic A., Trgovcich J., Pavic I., Lucin P., Jonjic S., Koszinowski U.H. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J. Exp. Med. 1998;188:1047–1054. doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucin P., Jonjic S., Messerle M., Polic B., Hengel H., Koszinowski U.H. Late phase inhibition of murine cytomegalovirus replication by synergistic action of interferon-gamma and tumor necrosis factor. J. Gen. Virol. 1994;75:101–110. doi: 10.1099/0022-1317-75-1-101. [DOI] [PubMed] [Google Scholar]
- Dyugovskaya L., Ginsburg H. Cure of fibroblast monolayers from murine cytomegalovirus infectionphenotypic assessment of rat lymphoid cell population developed on fibroblast monolayers. Cell. Immunol. 1996;169:30–39. doi: 10.1006/cimm.1996.0087. [DOI] [PubMed] [Google Scholar]
- Gribaudo G., Ravaglia S., Caliendo A., Cavallo R., Gariglio M., Martinotti M.G., Landolfo S. Interferons inhibit onset of murine cytomegalovirus immediate-early gene transcription. Virology. 1993;197:303–311. doi: 10.1006/viro.1993.1591. [DOI] [PubMed] [Google Scholar]
- Schut R.L., Gekker G., Hu S., Chao C.C., Pomeroy C., Jordan M.C., Peterson P.K. Cytomegalovirus replication in murine microglial cell culturessuppression of permissive infection by interferon-gamma. J. Infect. Dis. 1994;169:1092–1096. doi: 10.1093/infdis/169.5.1092. [DOI] [PubMed] [Google Scholar]
- Davignon J.-L., Castanie P., Yorke J.A., Gautier N., Clement D., Davrinche C. Anti-human cytomegalovirus activity of cytokines produced by CD4+ T-cell clones specifically activated by IE1 peptides in vitro. J. Virol. 1996;70:2162–2169. doi: 10.1128/jvi.70.4.2162-2169.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landolfo S., Gribaudo G., Angeretti A., Gariglio M. Mechanisms of viral inhibition by interferons. Pharmacol. Ther. 1995;65:415–442. doi: 10.1016/0163-7258(95)98599-l. [DOI] [PubMed] [Google Scholar]
- Samuel C.E. Antiviral actions of interferon, interferon-regulated cellular proteins and their surprisingly selective antiviral activities. Virology. 1991;183:1–11. doi: 10.1016/0042-6822(91)90112-o. [DOI] [PubMed] [Google Scholar]
- Sen G.C., Lengyel P. The interferon systema bird's eye view of its biochemistry. J. Biol. Chem. 1992;267:5017–5020. [PubMed] [Google Scholar]
- Smith M.G. Propagation of salivary gland virus of the mouse in tissue culture. Proc. Soc. Exp. Biol. Med. 1954;86:435–440. doi: 10.3181/00379727-86-21123. [DOI] [PubMed] [Google Scholar]
- Stoddart C.A., Cardin R.D., Boname J.M., Manning W.C., Abenes G.B., Mocarski E.S. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol. 1994;68:6243–6253. doi: 10.1128/jvi.68.10.6243-6253.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale J.F., Jr., O'Neil M.E. Detection of murine cytomegalovirus DNA in circulating leukocytes harvested during acute infection of mice. J. Virol. 1989;63:2667–2673. doi: 10.1128/jvi.63.6.2667-2673.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saltzman R.L., Quirk M.R., Jordan M.C. Disseminated cytomegalovirus infection. Molecular analysis of virus and leukocyte interactions in viremia. J. Clin. Invest. 1988;81:75–81. doi: 10.1172/JCI113313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins T.M., Quirk M.R., Jordan M.C. Biphasic viremia and viral gene expression in leukocytes during acute cytomegalovirus infection of mice. J. Virol. 1994;68:6305–6311. doi: 10.1128/jvi.68.10.6305-6311.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise M.T., Pollock J.L., Bromley S.K., Barkon M.L., Virgin H.W. Murine cytomegalovirus infection suppresses interferon-gamma-mediated MHC class II expression on macrophagesthe role of type I interferon. Virology. 1998;241:331–344. doi: 10.1006/viro.1997.8969. [DOI] [PubMed] [Google Scholar]
- Cavanaugh V.J., Stenberg R.M., Staley T.L., Virgin H.W., MacDonald M.R., Paetzold S., Farrell H.E., Rawlinson W.D., Campbell A.E. Murine cytomegalovirus with a deletion spanning HindIII-J and -I displays altered cell and tissue tropism. J. Virol. 1996;70:1365–1374. doi: 10.1128/jvi.70.3.1365-1374.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson L.K., Slater J.S., Karabekian Z., Virgin H.W., Biron C.A., Ruzek M.C., van Rooijen N., Ciavarra R.P., Stenberg R.M., Campbell A.E. Replication of murine cytomegalovirus in differentiated macrophages as a determinant of viral pathogenesis. J. Virol. 1999;73:5970–5980. doi: 10.1128/jvi.73.7.5970-5980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J.L., Presti R.M., Paetzold S., Virgin H.W. Latent murine cytomegalovirus infection in macrophages. Virology. 1997;227:168–179. doi: 10.1006/viro.1996.8303. [DOI] [PubMed] [Google Scholar]
- Brautigam A.R., Dutko F.J., Olding L.B., Oldstone M.B. Pathogenesis of murine cytomegalovirus infectionthe macrophage as a permissive cell for cytomegalovirus infection, replication, and latency. J. Gen. Virol. 1979;44:349–359. doi: 10.1099/0022-1317-44-2-349. [DOI] [PubMed] [Google Scholar]
- Mitchell B.M., Leung A., Stevens J.G. Murine cytomegalovirus DNA in peripheral blood of latently infected mice is detectable only in monocytes and polymorphonuclear leukocytes. Virology. 1996;223:198–207. doi: 10.1006/viro.1996.0468. [DOI] [PubMed] [Google Scholar]
- Koszinowski U.H., Keil G.M., Volkmer H., Fibi M.R., Ebeling Keil A., Munch K. The 89,000-Mr murine cytomegalovirus immediate-early protein activates gene transcription. J. Virol. 1986;58:59–66. doi: 10.1128/jvi.58.1.59-66.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler B., Keil G.M., Weiland F., Koszinowski U.H. Characterization of the murine cytomegalovirus early transcription unit e1 that is induced by immediate-early proteins. J. Virol. 1990;64:1907–1919. doi: 10.1128/jvi.64.5.1907-1919.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves R.F., Mocarski E.S. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus ie1 mutant. J. Virol. 1998;72:366–379. doi: 10.1128/jvi.72.1.366-379.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft G.J., Collins H.L., Sigola L.B., Cross C.E. Modulation of murine macrophage behavior in vivo and in vitro. Methods Cell Biol. 1994;45:129–146. doi: 10.1016/s0091-679x(08)61849-x. [DOI] [PubMed] [Google Scholar]
- Heise M.T., Connick M., Virgin H.W. Murine cytomegalovirus inhibits interferon-gamma induced antigen presentation to CD4 T cells by macrophages via regulation of MHC class II associated genes. J. Exp. Med. 1998;187:1037–1046. doi: 10.1084/jem.187.7.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock J.L., Virgin H.W. Latency, without persistence, of murine cytomegalovirus in spleen and kidney. J. Virol. 1995;69:1762–1768. doi: 10.1128/jvi.69.3.1762-1768.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virgin H.W., Wittenberg G.F., Unanue E.R. Immune complex effects on murine macrophages. I. Immune complexes suppress interferon-gamma induction of Ia expression. J. Immunol. 1985;135:3735–3743. [PubMed] [Google Scholar]
- Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Puglielli M.T., Woisetschlaeger M., Speck S.H. oriP is essential for EBNA gene promoter activity in Epstein-Barr virus-immortalized lymphoblastoid cell lines. J. Virol. 1996;70:5758–5768. doi: 10.1128/jvi.70.9.5758-5768.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil G.M., Ebeling-Keil A., Koszinowski U.H. Temporal regulation of murine cytomegalovirus transcription and mapping of viral RNA synthesized at immediate early times after infection. J. Virol. 1984;50:784–795. doi: 10.1128/jvi.50.3.784-795.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer J.A., Marks J.R., Spector D.H. Molecular cloning and restriction endonuclease mapping of the murine cytomegalovirus genome (Smith strain) Virology. 1983;129:94–106. doi: 10.1016/0042-6822(83)90398-7. [DOI] [PubMed] [Google Scholar]
- Danielson P.E., Forss-Petter S., Brow M.A., Calavetta L., Douglass J., Milner R.J., Sutcliffe J.G. p1B15a cDNA clone of the rat mRNA encoding cyclophilin. DNA. 1988;7:261–267. doi: 10.1089/dna.1988.7.261. [DOI] [PubMed] [Google Scholar]
- MacDonald M.R., Burney M.W., Resnick S.B., Virgin H.W. Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J. Virol. 1999;73:3682–3691. doi: 10.1128/jvi.73.5.3682-3691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil G.M., Fibi M.R., Koszinowski U.H. Characterization of the major immediate-early polypeptides encoded by murine cytomegalovirus. J. Virol. 1985;54:422–428. doi: 10.1128/jvi.54.2.422-428.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keil G.M., Ebeling-Keil A., Koszinowski U.H. Immediate-early genes of murine cytomegaloviruslocation, transcripts, and translation products. J. Virol. 1987;61:526–533. doi: 10.1128/jvi.61.2.526-533.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz M.A., White J.M., Sheehan K.C.F., Bach E.A., Rodig S.J., Dighe A.S., Kaplan D.H., Riley J.K., Greenlund A.C., Campbell D. Targeted disruption of the Stat 1 gene in mice reveals unexpected physiologic specificity of the JAK-STAT signalling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Rothe J., Lesslauer W., Lotscher H., Lang Y., Koebel P., Kontgen F., Althage A., Zinkernagel R., Steinmetz M., Bluethmann H. Mice lacking the tumour necrosis factor receptor 1 are resistant to TNF-mediated toxicity but highly susceptible to infection by Listeria monocytogenes . Nature. 1993;364:798–802. doi: 10.1038/364798a0. [DOI] [PubMed] [Google Scholar]
- Erickson S.L., de Sauvage F.J., Kikly K., Carver-Moore K., Pitts-Meek S., Gillett N., Sheehan K.C., Schreiber R.D., Goeddel D.V., Moore M.W. Decreased sensitivity to tumour-necrosis factor but normal T-cell development in TNF receptor-2-deficient mice. Nature. 1994;372:560–563. doi: 10.1038/372560a0. [DOI] [PubMed] [Google Scholar]
- Muller U., Steinhoff S., Reis L.F.L., Hemmi S., Pavlovic J., Zinkernagel R.M., Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- MacMicking J.D., Nathan C., Hom G., Chartrain N., Fletcher D.S., Trumbauer M., Stevens K., Xie Q.-W., Sokol K., Hutchinson N. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:1–10. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- Zhou A., Paranjape J., Brown T.L., Nie H., Naik S., Dong B., Chang A., Trapp B., Fairchild R., Colmenares C., Silverman R.H. Interferon action and apoptosis are defective in mice devoid of 2′,5′- oligoadenylate-dependent RNase L. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.L., Reis L.F., Pavlovic J., Aguzzi A., Schafer R., Kumar A., Williams B.R., Aguet M., Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO (Eur. Mol. Biol. Organ.) J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenlund A.C., Farrar M.A., Viviano B.L., Schreiber R.D. Ligand-induced IFN-gamma receptor tyrosine phosphorylation couples the receptor to its signal transduction system (p91) EMBO (Eur. Mol. Biol. Organ.) J. 1994;13:1591–1600. doi: 10.1002/j.1460-2075.1994.tb06422.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y.E., Laimins L.A. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 2000;74:4174–4182. doi: 10.1128/jvi.74.9.4174-4182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S.-H., Oakes J.E., Lausch R.N. Synergistic anti-HSV effect of tumor necrosis factor alpha and interferon gamma in human corneal fibroblasts is associated with interferon beta induction. Antivir. Res. 1993;22:15–29. doi: 10.1016/0166-3542(93)90083-u. [DOI] [PubMed] [Google Scholar]
- Feduchi E., Alonso M.A., Carrasco L. Human gamma interferon and tumor necrosis factor exert a synergistic blockade on the replication of herpes simplex virus. J. Virol. 1989;63:1354–1359. doi: 10.1128/jvi.63.3.1354-1359.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreil T.R., Eibl M.M. Viral infection of macrophages profoundly alters requirements for induction of nitric oxide synthesis. Virology. 1995;212:174–178. doi: 10.1006/viro.1995.1465. [DOI] [PubMed] [Google Scholar]
- Melkova Z., Esteban M. Interferon-gamma severely inhibits DNA synthesis of vaccinia virus in a macrophage cell line. Virology. 1994;198:731–735. doi: 10.1006/viro.1994.1087. [DOI] [PubMed] [Google Scholar]
- Harris N., Buller R.M.L., Karupiah G. Gamma interferon-induced, nitric oxide-mediated inhibition of vaccinia virus replication. J. Virol. 1995;69:910–915. doi: 10.1128/jvi.69.2.910-915.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karupiah G., Xie Q., Buller R.M.L., Nathan C., Duarte C., MacMicking J.D. Inhibition of viral replication by interferon-gamma induced nitric oxide synthase. Science. 1993;261:1445–1448. doi: 10.1126/science.7690156. [DOI] [PubMed] [Google Scholar]
- Karupiah G., Harris N. Inhibition of viral replication by nitric oxide and its reversal by ferrous sulfate and tricarboxylic acid cycle metabolites. J. Exp. Med. 1995;181:2171–2179. doi: 10.1084/jem.181.6.2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staeheli P., Horisberger M.A., Haller O. Mx-dependent resistance to influenza viruses is induced by mouse interferons alpha and beta but not gamma. Virology. 1984;132:456–461. doi: 10.1016/0042-6822(84)90050-3. [DOI] [PubMed] [Google Scholar]
- Horisberger M.A., Staeheli P., Haller O. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc. Natl. Acad. Sci. USA. 1983;80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin J.E., Hackenmiller R., Simon M.C., Levy D.E. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Barber G.N., Wambach M., Wong M.-L., Dever T.E., Hinnebusch A.G., Katze M.G. Translational regulation by the interferon-induced double-stranded-RNA-activated 68kDa protein kinase. Proc. Natl. Acad. Sci. USA. 1993;90:4621–4625. doi: 10.1073/pnas.90.10.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans B.L., Van Der Meide P.H., Stals F.S., Van Den Eertwegh A.J.M., Claassen E., Bruggeman C.A., Horzinek M.C., Schijns V.E.C.J. Suppression of rat cytomegalovirus replication by antibodies against gamma interferon. J. Virol. 1994;68:2305–2312. doi: 10.1128/jvi.68.4.2305-2312.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geginat G., Ruppert T., Hengel H., Holtappels R., Koszinowski U.H. IFN-gamma is a prerequisite for optimal antigen processing of viral peptides in vivo. J. Immunol. 1997;158:3303–3310. [PubMed] [Google Scholar]
- Weck K.E., Dal Canto A.J., Gould J.D., O'Guin A.K., Roth K.A., Saffitz J.E., Speck S.H., Virgin H.W. Murine gammaherpesvirus 68 causes large vessel arteritis in mice lacking interferon-gamma responsivenessa new model for virus induced vascular disease. Nat. Med. 1997;3:1346–1353. doi: 10.1038/nm1297-1346. [DOI] [PubMed] [Google Scholar]
- Soderberg-Naucler C., Fish K.N., Nelson J.A. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J. Clin. Invest. 1997;100:3154–3163. doi: 10.1172/JCI119871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.M., Zhang Y., Rahill B.M., Waldman W.J., Sedmak D.D. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J. Immunol. 1999;162:6107–6113. [PubMed] [Google Scholar]
- Miller D.M., Rahill B.M., Boss J.M., Lairmore M.D., Durbin J.E., Waldman J.W., Sedmak D.D. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med. 1998;187:675–683. doi: 10.1084/jem.187.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy E., Muhlethaler-Mottet A., Davrinche C., Mach B., Davignon J.L. Escape of human cytomegalovirus from HLA-DR-restricted CD4+ T-cell response is mediated by repression of gamma interferon-induced class II transactivator expression. J. Virol. 1999;73:6582–6589. doi: 10.1128/jvi.73.8.6582-6589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell S.R., Greenberg P.D. T cell therapy of human CMV and EBV infection in immunocompromised hosts. Rev. Med. Virol. 1997;7:181–192. doi: 10.1002/(sici)1099-1654(199709)7:3<181::aid-rmv200>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Chaudhuri A.R., St. Jeor S., Maciejewski J.P. Apoptosis induced by human cytomegalovirus infection can be enhanced by cytokines to limit the spread of virus. Exp. Hematol. 1999;27:1194–1203. doi: 10.1016/s0301-472x(99)00044-2. [DOI] [PubMed] [Google Scholar]
- Klotzbucher A., Mittnacht S., Kirchner H., Jacobsen H. Different effects of IFNgamma and IFNalpha/beta on “immediate early” gene expression of HSV-1. Virology. 1990;179:487–491. doi: 10.1016/0042-6822(90)90322-i. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G., Borrow P., Brown A., McClary H., Koch R., Chisari F.V. Noncytopathic clearance of lymphocytic choriomeningitis virus from the hepatocyte. J. Exp. Med. 1999;189:1555–1564. doi: 10.1084/jem.189.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tishon A., Lewicki H., Rall G., Von Herath M., Oldstone M.B.A. An essential role for type 1 interferon-gamma in terminating persistent viral infection. Virology. 1995;212:244–250. doi: 10.1006/viro.1995.1477. [DOI] [PubMed] [Google Scholar]
- Ruby J., Ramshaw I. The antiviral activity of immune CD8+ T cells is dependent on interferon-gamma. Lymphokine Cytokine Res. 1991;10:353–358. [PubMed] [Google Scholar]
- Kleijnen M.F., Huppa J.B., Lucin P., Mukherjee S., Farrell H., Campbell A.E., Koszinowski U.H., Hill A.B., Ploegh H.L. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO (Eur. Mol. Biol. Organ.) J. 1997;16:685–694. doi: 10.1093/emboj/16.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H., Thale R., Lucin P., Muranyi W., Flohr T., Hengel H., Farrell H., Rawlinson W., Koszinowski U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- DelVal M., Munch K., Reddehase M.J., Koszinowski U.H. Presentation of CMV immediate-early antigen to cytolytic T lymphocytes is selectively prevented by viral genes expressed in the early phase. Cell. 1989;58:305–315. doi: 10.1016/0092-8674(89)90845-3. [DOI] [PubMed] [Google Scholar]
- Zhou A., Paranjape J.M., Der S.D., Williams B.R., Silverman R.H. Interferon action in triply deficient mice reveals the existence of alternative antiviral pathways. Virology. 1999;258:435–440. doi: 10.1006/viro.1999.9738. [DOI] [PubMed] [Google Scholar]
- He B., Chou J., Brandimarti R., Mohr I., Gluzman Y., Roizman B. Suppression of the phenotype of gamma(1)34.5- herpes simplex virus 1failure of activated RNA-dependent protein kinase to shut off protein synthesis is associated with a deletion in the domain of the alpha47 gene. J. Virol. 1997;71:6049–6054. doi: 10.1128/jvi.71.8.6049-6054.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor D.R., Shi S.T., Romano P.R., Barber G.N., Lai M.M. Inhibition of the interferon-inducible protein kinase PKR by HCV E2 protein. Science. 1999;285:107–110. doi: 10.1126/science.285.5424.107. [DOI] [PubMed] [Google Scholar]
- Gutch M.J., Reich N.C. Repression of the interferon signal transduction pathway by the adenovirus E1A oncogene. Proc. Natl. Acad. Sci. USA. 1991;88:7913–7917. doi: 10.1073/pnas.88.18.7913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribaudo G., Ravaglia S., Gaboli M., Gariglio M., Cavallo R., Landolfo S. Interferon-alpha inhibits the murine cytomegalovirus immediate-early gene expression by down-regulaing NF-kappaB activity. Virology. 1995;211:251–260. doi: 10.1006/viro.1995.1398. [DOI] [PubMed] [Google Scholar]
- Min W., Ghosh S., Lengyel P. The interferon-inducible p202 protein as a modulator of transcriptioninhibition of NF-kappa B, c-Fos, and c-Jun activities. Mol. Cell. Biol. 1996;16:359–368. doi: 10.1128/mcb.16.1.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone R.W., Kerry J.A., Trapani J.A. The human interferon-inducible protein, IFI 16, is a repressor of transcription. J. Biol. Chem. 1998;273:17172–17177. doi: 10.1074/jbc.273.27.17172. [DOI] [PubMed] [Google Scholar]
- Bodaghi B., Goureau O., Zipeto D., Laurent L., Virelizier J.L., Michelson S. Role of IFN-gamma-induced indoleamine 2,3 dioxygenase and inducible nitric oxide synthase in the replication of human cytomegalovirus in retinal pigment epithelial cells. J. Immunol. 1999;162:957–964. [PubMed] [Google Scholar]
- Van Dyk L.F., Hess J.L., Katz J.D., Jacoby M., Speck S.H., Virgin H.W. The murine gammaherpesvirus 68 v-cyclin is an oncogene that promotes cell cycle progression in primary lymphocytes. J. Virol. 1999;73:5110–5122. doi: 10.1128/jvi.73.6.5110-5122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol C., Flemington E.K. The Epstein-Barr virus bZIP transcription factor Zta causes G0/G1 cell cycle arrest through induction of cyclin-dependent kinase inhibitors. EMBO (Eur. Mol. Biol. Organ.) J. 1996;15:2748–2759. [PMC free article] [PubMed] [Google Scholar]
- Schang L.M., Phillips J., Schaffer P.A. Requirement for cellular cyclin-dependent kinases in herpes simplex virus replication and transcription. J. Virol. 1998;72:5626–5637. doi: 10.1128/jvi.72.7.5626-5637.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M., Shenk T. Human cytomegalovirus infection inhibits cell cycle progression at multiple points, including the transition from G1 to S. J. Virol. 1996;70:8850–8857. doi: 10.1128/jvi.70.12.8850-8857.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnahan W.A., Boldogh I., Thompson E.A., Albrecht T. Human cytomegalovirus inhibits cellular DNA synthesis and arrests productively infected cells in late G1. Virology. 1996;224:150–160. doi: 10.1006/viro.1996.0516. [DOI] [PubMed] [Google Scholar]
- Salvant B.S., Fortunato E.A., Spector D.H. Cell cycle dysregulation by human cytomegalovirusinfluence of the cell cycle phase at the time of infection and effects on cyclin transcription. J. Virol. 1998;72:3729–3741. doi: 10.1128/jvi.72.5.3729-3741.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer D., Mocarski E.S. Human cytomegalovirus infection inhibits G1/S transition. J. Virol. 1997;71:1629–1634. doi: 10.1128/jvi.71.2.1629-1634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jault F.M., Jault J.-M., Ruchti F., Fortunato E.A., Clark C., Corbell J., Richman D.D., Spector D.H. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 1995;69:6697–6704. doi: 10.1128/jvi.69.11.6697-6704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xaus J., Cardo M., Valledor A.F., Soler C., Lloberas J., Celada A. Interferon gamma induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity. 1999;11:103–113. doi: 10.1016/s1074-7613(00)80085-0. [DOI] [PubMed] [Google Scholar]
- Guidotti L.G., Ishikawa T., Hobbs M.V., Matzke B., Schreiber R., Chisari F.V. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- Heise T., Guidotti L.G., Chisari F.V. La autoantigen specifically recognizes a predicted stem-loop in hepatitis B virus RNA. J. Virol. 1999;73:5767–5776. doi: 10.1128/jvi.73.7.5767-5776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise T., Guidotti L.G., Cavanaugh V.J., Chisari F.V. Hepatitis B virus RNA-binding proteins associated with cytokine-induced clearance of viral RNA from the liver of transgenic mice. J. Virol. 1999;73:474–481. doi: 10.1128/jvi.73.1.474-481.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]