

Abstract
We screened 29 strains of Neisseria gonorrhoeae and found 16/21 strains that resisted killing by normal human serum and 0/8 serum sensitive strains that bound the complement regulator, C4b-binding protein (C4bp). Microbial surface–bound C4bp demonstrated cofactor activity. We constructed gonococcal strains with hybrid porin (Por) molecules derived from each of the major serogroups (Por1A and Por1B) of N. gonorrhoeae, and showed that the loop 1 of Por1A is required for C4bp binding. Por1B loops 5 and 7 of serum-resistant gonococci together formed a negatively charged C4bp-binding domain. C4bp–Por1B interactions were ionic in nature (inhibited by high salt or by heparin), whereas the C4bp–Por1A bond was hydrophobic. Only recombinant C4bp mutant molecules containing the NH2-terminal α-chain short consensus repeat (SCR1) bound to both Por1A and Por1B gonococci, suggesting that SCR1 contained Por binding sites. C4bp α-chain monomers did not bind gonococci, indicating that the polymeric form of C4bp was required for binding. Using fAb fragments against C4bp SCR1, C4bp binding to Por1A and Por1B strains was inhibited in a complement-dependent serum bactericidal assay. This resulted in complete killing of these otherwise fully serum resistant strains in only 10% normal serum, underscoring the importance of C4bp in mediating gonococcal serum resistance.
Keywords: Neisseria gonorrhoeae, C4b-binding protein, porin, serum resistance, short consensus repeat
Introduction
Neisseria gonorrhoeae is one of two bacterial pathogens involved in the majority of cases of sexually transmitted genital infection and pelvic inflammatory disease (PID); the other is Chlamydia trachomatis. Gonococci that cause symptomatic local inflammation (PID) usually are sensitive to killing by nonimmune normal human serum (NHS) in vitro 1. Complement component C3 is present in functional amounts at the cervical level 2 3, is synthesized in the endometrial glandular epithelium 4 5, and binds to gonococci in vivo 6. Serum-sensitive (SS) gonococci can abrogate killing by NHS by adding sialic acid to their lipo- oligosaccharide (LOS 7 8 9 10 11), which permits direct binding of the complement regulatory protein factor H to sialylated LOS 12. This is termed unstable serum resistance (SR). In vivo, gonococci appear to be heterogeneously sialylated 6 13. Disseminated bloodstream infection with gonococci (DGI) often occurs in the absence of local genital inflammation and is caused predominantly by gonococci that resist killing by NHS independent of sialylation, termed stable serum resistance 14. These isolates sometimes lack the LOS acceptor site for sialic acid 15, or may not completely sialylate LOS for reasons that remain unclear. Commonly they also express the gonococcal major outer membrane protein serotype porin (Por)1A 16 17. Gonococcal Por comprises 60% of the outer membrane protein content 18. Por is a 34–35-kD protein comprising 8 transmembrane loops (16 membrane spanning segments) whose native configuration is a homogenous trimer that functions as a selective anion channel 18 19. Por is an allelic protein consisting of two main isoforms, Por1A and Por1B 17 20. There is moderate antigenic variation between the isoforms, which subdivides them into serotypes or serovars 21 22 23. Por1A gonococci frequently cause disseminated disease, whereas Por1B strains usually cause local urogenital disease and PID in women 16 17 20. We have recently shown that Por1A isolates resist killing by directly binding factor H, a regulator of the alternative pathway of complement, to the surface exposed loop 5 of Por1A 24.
Although the alternative pathway is important in C3 deposition on the gonococcal surface, the classical pathway of complement is required to initiate this deposition and resultant killing 25. A key soluble phase classical pathway regulatory protein is C4b-binding protein (C4bp), which is present in human plasma at concentrations of 200–250 μg/ml. C4bp is a cofactor in factor I–mediated cleavage of C4b to C4d, and accelerates dissociation of C2a from the classical pathway C3 convertase (C4b, 2a), thereby downregulating the classical pathway 26 27 28 29 30. C4bp (M r = 570 kD) is composed of seven identical 70-kD subunits (α-chains) linked by disulfide bridges 30 31. When visualized by electron microscopy, it appears as a spider-like structure with seven elongated tentacles (α-chains) extending from a central, ring-like core 32 33. The NH2-terminal 491 (of a total of 549) residues of each α-chain are organized into eight short consensus repeat (SCR) domains, and each SCR is comprised of ∼60 amino acids. A patch of positively charged residues at the interface between the first and second NH2-terminal SCRs plays an important role in the interaction between C4bp and C4b as well as heparin 34. C4bp also contains a 45-kD subunit (β-chain), organized into three SCRs, which is attached to the central core region of the molecule by a disulfide bond 35. Three isoforms with different subunit compositions have been identified in human plasma; the major isoform is comprised of seven α-chains and one β-chain (α7β1); the remaining isoforms are α7β0 and α6β1 36.
Many isolates of Streptococcus pyogenes have been reported to bind C4bp, which may contribute to pathogenicity 37. Binding of C4bp occurs through the NH2-terminal highly variable region of several members of the M protein family present in this species 38. In addition, C4bp binding has been demonstrated to all clinical isolates of Bordetella pertussis expressing the filamentous hemagglutinin 39. Because the classical pathway is crucial in initiating complement deposition on gonococci 25, regulation of this pathway could provide a very efficient means for the bacteria to evade the bactericidal action of serum at an early stage of complement activation. In this work, we detail the interactions between C4bp and gonococcal Por, and demonstrate the function of C4bp in mediating stable SR (SR not mediated by LOS sialylation).
Materials and Methods
Bacterial Strains and Plasmids.
29 strains of N. gonorrhoeae were screened for binding to C4bp; these are listed in Table . Strain FA6616, containing the MS11 Por molecule reintroduced into an MS11 background using plasmid pUNCH61 40, was used in this study, and for convenience will be referred to as MS11 hereafter. Bacteria grown on chocolate agar supplemented with Isovitalex equivalent in 5% CO2 for 10 to 11 h 41 were suspended in HBSS containing 0.15 mM CaCl2 and 1 mM MgCl2 (HBSS2+) in C4bp binding assays. Alternatively, bacteria were harvested from chocolate agar plates after overnight growth and grown in gonococcal liquid media 41. Results of C4bp binding were equivalent when strains were grown in either media. Plasmids pUNCH61 and pUNCH62 contained the por1B and por1A genes of strains MS11 and FA19, respectively, plus ∼1 kB of gonococcal DNA 3′ to the por gene, respectively, containing a chloramphenicol-resistance marker (CmR [40, 42]). Strains MS11 and FA19 are resistant to killing (SR) by 33% nonimmune NHS. pUNCH61 and pUNCH62 were each used to transform the SS strain F62 to replace F62 Por with either MS11 Por (F62PorMS11) or FA19 Por (F62PorFA19), respectively. In addition to replacing Por completely, transformation with pUNCH61 resulted in hybrids F62loop1MS11loop2–8, and F62loop1–4MS11loop5–8 (see Fig. 4), and transformation of F62 with pUNCH62 yielded hybrids F62loop1FA19loop2–8 and F62loop1–4FA19loop5–8 (see Fig. 3). Because C4bp binding studies with these MS11/F62 hybrids suggested that the C4bp binding region in MS11 Por was contained in the COOH-terminal half of the Por molecule (see Results), we then constructed hybrid MS11loop1F62loop2–7MS11loop8 by replacing the BbsI-BsgI fragment of MS11 Por encompassing loops 2 through 7 in pUNCH61 with the corresponding BbsI-BsgI fragment of F62 Por. Using this plasmid, designated pBUMC1, we constructed hybrids that were mutated at loops 5, 6, or 7 individually or in combination, using either overlap extension PCR for loops 5 and 6, or site-directed mutagenesis for loop 7 (see below: Por mutagenesis). The amino acid sequences of the putative exposed regions of Por loops 5, 6, and 7 of F62 and MS11 are indicated in Table ; variations in sequence are indicated in bold type. The net charge of the surface exposed loop regions was calculated by assigning +1 for arginine (r) and lysine (K), and +0.5 for histidine (H), and –1 for aspartic acid (D) and glutamic acid (E), as described previously 43.
Table 1.
C4bp Binding and Phenotype of 29 Strains of Neisseria gonorrhoeae
| Strain (reference) | Phenotype | C4bp binding |
|---|---|---|
| Por1A strains | ||
| 15253 (78, 79) | SR | + |
| 401082 (24) | SR | + |
| 339063 (24) | SR | + |
| DGI isolate 1 (14) | SR | + |
| 179008 (24) | SR | + |
| FA19 (80, 81) | SR | + |
| 442089 (24) | SR | + |
| 273043 (24) | SR | + |
| UU1 (46, 47) | SS | − |
| 374073 (24) | SR | − |
| DGI isolate 56 (this study) | SR | − |
| Por1B strains | ||
| FA1090 82 | SR | + |
| 156001 (24) | SR | + |
| 1291 83 | SR | + |
| BMC-1 84 | SR | + |
| FA6616 (40) | SR | + |
| WG (14) | SR | + |
| 422083 (24) | SR | − |
| 252 (24) | SR | − |
| 336062 (24) | SR | − |
| DGI isolate 40 (14) | SR | + |
| DGI isolate 17 (14) | SR | + |
| 398079 (24) | SS | − |
| 256036 (24) | SS | − |
| 255034 (24) | SS | − |
| F62 85 | SS | − |
| 24-1 (61) | SS | − |
| Pgh 3-2 (46, 47) | SS | − |
| 150002 (24) | SS | − |
Figure 4.

MS11 Por1B loops 5 and 7 together participate in forming a C4bp-binding region. Transformation of F62 with pUNCH61 (reference 40) resulted in strains with hybrid Por molecules F62loop1MS11loop2–8 and F62loop1–4MS11loop5–8. F62 sequence is indicated by black bars; MS11 sequence is represented by hatched bars. Both hybrids bound C4bp, suggesting that the C4bp binding region in MS11 lay in a region encompassed by loops 5–8. The BbsI-BsgI fragment of F62 was cloned into pUNCH61 to obtain pBUMC1 and this plasmid was used to transform F62. This yielded a gonococcal strain with hybrid Por molecule MS11loop1 F62loop2–7MS11loop1–8, which did not bind C4bp, thus eliminating MS11 loop 8 as the C4bp binding loop. Using overlap extension PCR or site-directed mutagenesis on pBUMC1, we mutated loops 5, 6, or 7 either individually or in combination and then transformed F62. We noted that only hybrid Por molecules that contained both MS11 loop 5 and loop 7 bound C4bp. To further demonstrate the absolute requirement of MS11 loops 5 and 7, we mutated MS11 loops 5, 6, and 7 individually in pUNCH61 to resemble the corresponding F62 loop, and then transformed F62. Mutations of loop 5 (MS11loop1–4F62loop5MS11loop6–8) and loop 7 (MS11loop1–6F62loop7MS11loop8) resulted in complete abrogation of C4bp binding, whereas mutating loop 6 (MS11loop1–5F62loop6MS11loop7–8) did not influence C4bp binding. Collectively, these data suggest that MS11 loops 5 and 7 together participate in forming a C4bp-binding domain.
Figure 3.

FA19 Por1A loop 1 is required for C4bp binding. Transformation of F62 with pUNCH62 (containing the FA19 por1A gene) resulted in two classes of hybrid Por molecules, F62loop1FA19loop2–8 and F62loop1–4FA19loop2–8. F62 (Por1B) sequence is shown by solid black bars, and FA19 (Por1A) sequence by white bars. Neither class of hybrids bound C4bp. Loop 1 of FA19 Por1A is indicated by the gray shaded box; only parent strain FA19 that contains Por1A loop 1 binds C4bp, suggesting that loop 1 of Por1A is required for C4bp binding.
Table 2.
Comparison of Putative Exposed Sequences of Por Loops 5, 6, and 7 of Strains MS11 and F62
| Por loop | Strain | Sequence of putative surface-exposed region | Net charge |
|---|---|---|---|
| Loop 5 | MS11 | RYGEGTKKIE-YEHQVYSIPSLFVEKL | +0.5 |
| F62 | RYGEGTKKMEGY---AYNIPSLFVEKL | +1 | |
| Loop 6 | MS11 | DAKLYQNQLVRDNSHNSQTE | −0.5 |
| F62 | DAKLYGTW--RANSHNSQTE | +0.5 | |
| Loop 7 | MS11 | VDSADHDNTYDQV | −3.5 |
| F62 | VHSADYDNTYDQV | −2.5 |
Por Mutagenesis.
We used overlap extension PCR to replace MS11 loop 5 or 6 with F62 loops 5 or 6, respectively, and vice versa 44. To construct each mutation, we performed two separate PCR reactions with the primer pairs listed in Table . Denaturation for this PCR was carried out at 94°C for 1 min, annealing temperatures varied between 58 and 61°C, and extension was performed at 72°C for 1 min. PCR products were gel purified using the GeneClean kit (Bio 101). The products of gel purification were then subjected to a second PCR reaction using primers PorI and PorII, followed by gel purification of the PCR product. This PCR reaction consisted of 10 cycles of overlap (94°C for 30 s followed by 72°C for 2 min) followed by 15 extension cycles of denaturation at 94°C for 1 min, annealing at 58°C for 1 min, and extension at 72°C for 1 min. The products obtained from this PCR reaction were then digested with BbsI and BsgI, and the resultant ∼800-bp fragment gel purified and cloned into the BbsI-BsgI site in pUNCH61 or pBUMC1. Loop 7 mutations were made using pUNCH61 (to replace MS11 loop 7 with F62 loop 7) or the reverse in pBUMC1 (to replace F62 loop 7 with MS11 loop 7), using the QuikChange™ site-directed mutagenesis kit (Stratagene) according to the manufacturer's instructions. In these reactions, primer 5′-AGG CAC TGT TGA TAG TGC AGA CCA CGA CAA TAC-3′ and its complimentary antisense counterpart were used to exchange MS11 loop 7 with F62 loop 7, and 5′-AGG CAC TGT TCA TAG TGC AGA CTA CGA CAA TAC-3′ and its antisense counterpart were used to exchange F62 loop 7 with MS11 loop 7. Gonococcal transformants were screened using mAb 1F5 (21; see below) that recognizes MS11 Por loop 1 42, but does not react with F62. To ensure that the desired mutation had been obtained in F62, entire por genes of mAb 1F5-positive clones were amplified using primers PorI and PorII, and DNA sequencing was performed at the DNA core facility at Boston University School of Medicine using an Applied Biosystems automated sequencer.
Table 3.
Primer Pairs Used for Constructing MS11-F62 Hybrid Por Molecules Using Overlap Extension PCR
| Mutation | Primer sequence (5′→3′) |
|---|---|
| F625 to MS115 | GGGATACTATAAACTTGATGTTCGTATTCGATTTTTTTAGTG/PorI |
| CGAATACGAACATCAAGTTTATAGTATCCCCAGCCTGTTTGT/PorII | |
| F626 to MS116 | TCACGCACTAATTGATTTTGATACAATTTGGCATCTTGTTG/PorI |
| TTGTATCAAAATCAATTAGTGCGTGATAATTCGCACAACTCTCAA/PorII | |
| MS115 to F625 | AAACTGGGGATATTATATGCATATCCTTCCATTTTTTTAGTGCCTTCGCCG/PorI |
| AAAAATGGAAGGATATGCATATAATATCCCCAGTTTGTTTGTTTGTTGAAAA/PorII | |
| MS116 to F626 | GCACGCCATGTTCCATACAATTTGGCATCTTGTTGTTGTG/PorI |
| CACAACAACAAGATGCCAAATTGTATGGAACATGGCGTGC/PorII |
In an effort to obtain a strain containing loop 1 of FA19 and loops 2–8 of F62, we cloned the chloramphenicol-resistance marker into the HincII site of gonococcal DNA 5′ to the FA19 por1A gene in pUNCH 30 45, and used this plasmid, designated pBUMC2, to transform F62. Attempts to create a hybrid Por molecule containing the NH2 terminus of the FA19 Por and the COOH terminus of F62 Por were not successful; therefore, we attempted to construct a hybrid Por molecule that contained loop 1 from strain FA19 and loops 2–8 from another gonococcal strain, UU1 46 47. UU1 is a Por1A strain that is SS and does not bind C4bp. We introduced a SpeI site 2 bp 3′ to the UU1 por gene by PCR using PorI as the forward primer and 5′-GCG ACT AGT ATT AGA ATT TGT GGC GCAG-3′ as the reverse primer. Then, using the QuikChange™ site-directed mutagenesis kit (Stratagene), a SpeI site was similarly introduced 3′ to the FA19 por1A gene in pUNCH62 using primer 5′-GAC AGA CTA GTT GTT GAT ACC GAT-3′ and its complementary primer. A new construct was made by ligating the BbsI-SpeI fragment of UU1 into pUNCH62, which had been predigested with the same enzymes. The resultant plasmid, pBUMC3, that contained hybrid Por molecule FA19loop1UU1loop2–8 was used to attempt transformation of strains F62 and UU1 (see Results: C4bp Binding Region in Por1A).
Sera and Complement Reagents.
Nonimmune NHS was obtained from 10 healthy volunteers with no prior history of neisserial infection. Sera were aliquoted and stored at −70°C until further use. In some experiments, NHS was incubated at 56°C for 30 min to yield heat-inactivated serum (HIS). NHS was adsorbed against glutaraldehyde-fixed bacteria (strain F62PorMS11) on ice to deplete serum of bacteria-specific Ig, as described previously 41, and used as an antibody-free complement source in certain experiments (see Results: Enhanced IgM Binding to F62PorMS11: Por influences IgM Binding to Gonococci). Purified C4b was obtained from Advanced Research Technologies. C4bp that was either complexed to or devoid of protein S (a component of the protein C anticoagulation system) was purified from human plasma using previously described methods 48.
Recombinant C4bp Mutant Proteins.
Recombinant human C4bp and seven polymeric rC4bp mutant molecules lacking individual SCRs were constructed by an overlapping PCR technique that used as a template full-length cDNA coding for the α-chain of human C4bp 49 cloned in vector pcDNA3 (Invitrogen). The amino acids deleted (represented as deletions of SCRs 1 to 7 deleted sequentially) in each construct and sequences of primers used are listed in Table . The final PCR product was cloned into HindII and NotI sites in vector pcDNA3 and used as the transfecting construct. Nucleotide sequences of all mutations were determined by automated DNA sequencing (PerkinElmer).
Table 4.
Primers Used for Construction of Recombinant Human C4bp Deletion Mutant Molecules
| rC4bp protein | Amino acids deleted | Sense primer (5′→3′) |
|---|---|---|
| rC4bp ΔSCR1 | N1 to Y62 | CCT GCT GTT CTT GGC AAA CGA TGC AGA CAC |
| rC4bp ΔSCR2 | K63 to I124 | ACC TTC TGT ATC TAC GTC AAG TGT AAG CCT |
| rC4bp ΔSCR3 | V125 to K188 | CCA CAA TGT GAA ATT ATC ACC TGT CGC AAG |
| rC4bp ΔSCR4 | K188 to N249 | CCT CCT ACC TGT GAA AAT AGT TGT ATT AAT |
| rC4bp ΔSCR5 | N 249 to A314 | CCT GCT TGT GAG CCC TTA TGT TGC CCT GAA |
| rC4bp ΔSCR6 | A314 to G375 | TAC CAA GGA TGT GAG GAC ATT TGC AAT TTT |
| rC4bp ΔSCR7 | D376 to K433 | ACA CCA TCA TGT GGA GCT CTG TGC CGG AAA |
Human kidney 293 cells (American Type Culture Collection catalog no. 1573-CRL) were transfected with individual C4bp constructs, and the expressed proteins, with the exception of rC4bpΔSCR1, were purified by affinity chromatography using mAb 104 (specific for C4bp SCR1) as described previously 50. rC4bpΔSCR1 was affinity purified using mAb 67 (specific for C4bp SCR 4; reference 51).
C4bp α-chain monomers containing all eight SCRs were constructed by introducing the STOP codon after SCR 8 using primer 5′-CCC AAG TGT GAG TGG TAG ACC CCC GAA GGC TGT-3′ (stop codon indicated in bold).
Ab.
Transformants were screened using serotyping mAbs against gonococcal Por 21. These included mAb 2F12 (specific for FA19 Por loop 1), 9D2 (central region of FA19 Por), 1F5 (MS11 Por loop 1), and 3C8 (loop 5 of MS11 Por). These mAbs were used in colony blotting experiments or in whole cell ELISA at a dilution of 1 μg/ml in PBS-0.05% Tween 20.
Anti-C4bp mAb (Quidel Corp.) was used in flow cytometry experiments (see below) at a concentration of 20 μg/ml in HBSS2+. In some experiments, a sheep polyclonal Ab against human C4bp (Biodesign International) was used in flow cytometry experiments at a dilution of 1:50 in HBSS2+. mAbs specific for C4c and C4d were purchased from Quidel Corp., and used in flow cytometry experiments at a dilution of 20 μg/ml in HBSS2+. C4c and C4d are the fragments of C4b generated when the enzyme factor I acts together with its cofactor C4bp to cleave C4b. C3 bound to bacteria was detected using an IgG fraction of goat anti–human C3 (Organon Teknika-Cappel) at a concentration of 100 μg/ml. C5b-9 on the bacterial surface was detected by flow cytometry using an IgG fraction of rabbit anti–human SC5b-9 neoantigen (Advanced Research Technologies) at a 1:50 dilution in HBSS2+. To identify the region in C4bp that bound gonococcal Por, we used five mAbs directed against C4bp SCR 1 to 2 (mAb 104, mAb 102, mAb 96, mAb 92, and mAb 70) and mAb 67 (specific for SCR 4 of the α-chain of C4bp; reference 51), in experiments to attempt to inhibit binding of C4bp to the bacterial surface. The functional effects of abrogating C4bp binding to the bacterial surface were studied by using fAb fragments of mAb 104 to avoid confounding by possible Fc related effects of the inhibiting Ab; fAb fragments of mAb 67 (specific for C4bp SCR 4), shown not to influence C4bp binding to bacteria, were used as controls. fAb fragments were generated from mAb 104 by digestion with immobilized papain using a kit (Pierce Chemical Co.) according to the manufacturer's instructions. We observed that papain digestion of mAb 67 did not yield the desired ∼50-kD fAb fragment, but instead a 110-kD (possibly F[ab′]2) fragment. However, digestion with immobilized pepsin yielded 50-kD fragments that retained the capacity to bind C4bp by ELISA (see below).
Flow Cytometry.
Flow cytometry was used to quantify IgM, C4bp, C4c, C4d, C3, and C5b-9 binding to the bacterial surface, using methods described previously 12. In brief, 108 bacteria suspended in HBSS2+ were incubated either with NHS, purified C4bp, or rC4bp deletion mutant molecules (quantity specified in each experiment, final volume of reaction mixture 100 μl), followed by detection of the indicated component using the specified primary and appropriate FITC-labeled secondary conjugate (Sigma-Aldrich). In some experiments, inhibition of C4bp binding was attempted using high salt conditions, heparin, pure C4b, anti-C4bp mAbs, or their fAb fragments (see above). Conditions are specified for each experiment.
Bactericidal Testing.
Serum bactericidal testing was performed as described previously 41. In some experiments, C4bp binding to the bacterial surface was abrogated using 25 μg fAb 104. This represents an ∼70-fold molar excess of fAb 104 over C4bp in a bactericidal assay using 10% NHS. In bactericidal experiments to demonstrate that enhanced Ab binding was responsible for the serum sensitive phenotype of F62PorMS11, organisms were incubated with either 15 μl HIS (antibody source) or buffer (control), and NHS adsorbed against glutaraldehyde-fixed F62PorMS11 was used as a source of complement. The final volume of the bactericidal reaction mixture in every experiment was maintained at 150 μl.
ELISA.
To determine whether fAb fragments of mAb 67 retained the ability to bind C4bp, we coated ELISA plates with either 10 μg/ml mAb 67 or the derived fAb fragment in bicarbonate buffer pH 9.6 for 2 h at 37°C, followed by blocking with 1% porcine gelatin (Sigma-Aldrich) for 30 min at 37°C. Plates were then washed twice with PBS-0.05% Tween 20, and cell culture supernatant containing rC4bp was applied to each well for 1 h at 37°C. After washing with PBS-Tween, bound C4bp was detected using a sheep polyclonal anti-C4bp (Biodesign) and alkaline phosphatase–conjugated anti–sheep IgG (Sigma-Aldrich). The OD410nm obtained with binding of rC4bp to the fAb fragment was compared with the OD410nm obtained with rC4bp binding to the intact mAb.
Results
C4bp Binding Correlates with Serum Resistance
We screened 29 clinical and laboratory gonococcal isolates (Table ) for C4bp binding using flow cytometry. Strains were incubated with 10% NHS for 30 min, followed by detection with an anti-C4bp mAb. The geometric mean fluorescence intensity of C4bp binding did not differ among strains that bound C4bp, when tested under similar assay conditions (representative positive and negative tracings are shown in Fig. 2 by F62PorFA19 and F62PorF62, respectively). Resistance of each of the 29 strains to killing by 10% NHS is indicated as phenotype in Table . 11 strains belonged to the Por1A serogroup; the remaining 18 were Por1B. 10 of 11 Por1A strains were resistant to killing by 10% NHS (SR). 8 of the 10 SR Por1A strains bound C4bp; the only SS Por1A strain that was tested did not bind C4bp. 11 of the 18 Por1B strains were SR, and 8 of these strains bound C4bp; no SS Por1B strains bound C4bp. A strong correlation was observed between C4bp binding and a SR phenotype (P < 0.0001 by Fisher's exact test).
Figure 2.

Por1A of FA19 and Por1B of MS11 are the acceptor molecules for C4bp. To demonstrate this for FA19 Por1A, we used pUNCH62 (reference 40) to replace F62 Por1B (C4bp nonbinder) with FA19 Por1A. The resultant strain, termed F62PorFA19, bound C4bp in a flow cytometry assay when incubated with 10% NHS (left), indicating that FA19 Por1A was an acceptor molecule for C4bp. Similarly, using plasmid pUNCH61 (reference 40) we replaced F62 Por1B with MS11 Por1B, and observed that the isogenic mutant F62PorMS11 bound C4bp (right), indicating that MS11 Por1B was an acceptor molecule for C4bp.
We noted that all Por1B strains that bound C4bp belonged to closely related serovars, and most bound the serotyping mAb 3C8 21 that is specific for loop 5 of certain Por1B strains 52. This suggested the possibility that the central region of Por1B might be important in C4bp binding.
C4bp Binds Directly to Gonococci; Protein S Does Not Influence C4bp Binding
The major isoform of C4bp found in blood is seven α-chains and one β-chain (α7β1), and virtually all C4bp (α7β1) is complexed with protein S, a component of the protein C anticoagulation system 48. To consider a possible effect of protein S in influencing C4bp binding to gonococci, we compared the binding of 2.5 μg of pure C4bp free of protein S and an equivalent amount of C4bp coupled to protein S to strains FA19 (Por1A) and MS11 (Por1B). The amount of C4bp bound to the two strains under each condition of incubation was similar (data not shown), thereby suggesting that C4bp binding is not influenced by protein S.
C4bp Bound to Gonococci Has Cofactor Function
C4bp regulates classical complement pathway activation by serving as a cofactor in the inactivation of C4b by factor I, and yields the C4 fragments C4d (which remains bound to the bacterial surface) and C4c (released into solution). Cofactor function of C4bp was assessed using mAbs directed against C4c and C4d. mAb against C4d is specific for the parent molecule C4b as well as the fragment C4d, whereas mAb against C4c recognizes C4b and C4c, but not C4d. Therefore, cofactor activity will not alter the amount of C4 measured on the bacterial surface by the mAb against C4d, but will decrease the amount of C4 bound to the organism measured by mAb against C4c, resulting in a higher C4d/C4c ratio 53. We measured C4c and C4d on strains incubated in 20% NHS for 30 min at 37°C. We used strain F62 (SS, Por1B, does not bind C4bp), FA19 (SR, Por1A, binds C4bp), and MS11 (relatively SR, Por1B, binds C4bp). We observed that both C4bp-binding strains showed a higher C4d/C4c ratio (4 for MS11 and 5.4 for FA19) than that observed with strain F62 (1.17; Fig. 1). Thus, C4bp that binds to the bacterial surface exhibits cofactor activity.
Figure 1.


Demonstration of cofactor activity on gonococcal strains that bind C4bp. Strains F62, MS11, and FA19 were incubated with 20% NHS for 30 min at 37°C, followed by detection of intact C4b fragments remaining on the bacterial surface by flow cytometry. C4bp cofactor activity would result in factor I–mediated cleavage of C4b to C4c and C4d. C4d remains bound to the bacterial surface, whereas the C4c fragment is released into solution. Anti-C4d mAb recognizes both C4b as well as C4d, whereas anti-C4c mAb recognizes only C4b bound to the organism. Therefore, cofactor activity would result in decreased intact C4b detected on the organism (measured with the anti-C4c mAb), with a resultant increase in the C4d/C4c ratio. Consistent with their ability to bind C4bp, both MS11 as well as FA19 show higher C4d/C4c ratios. geo mean flu, geometric mean fluorescence.
Defining Por as the Acceptor for C4bp
We next examined if Por1A was the acceptor molecule for C4bp by replacing the Por1B of F62 (does not bind C4bp) with the Por1A molecule of strain FA19 (binds C4bp) using plasmid pUNCH62 40. Colony lifts of transformants that resulted from homologous recombination were screened using serotyping mAbs 2F12 and 9D2 21 that are specific for the NH2-terminal and central region of FA19 Por, respectively 42 54. Clones that reacted with both mAbs were deemed to have acquired most of the FA19 Por1A molecule in their F62 background. The presence of the entire FA19 Por molecule was confirmed by DNA sequencing. One such isogenic mutant, designated F62PorFA19, bound C4bp in a flow cytometry assay (Fig. 2, left), and demonstrated 100% survival in 10% NHS (SR), akin to the parent strain FA19.
Similarly, we used plasmid pUNCH61 40 to replace the Por molecule of F62 with MS11 Por. Transformants were screened using mAbs 1F5 (specific for MS11 loop 1) and 3C8 (specific for the central region of MS11, but not F62, Por). Again, the presence of the entire MS11 Por molecule was confirmed by DNA sequencing. The resulting strain (F62PorMS11) also bound C4bp (Fig. 2, right), proving that the Por molecule functioned as a C4bp acceptor on strain MS11. Unlike F62PorFA19, F62PorMS11 remained fully SS to the bactericidal action of 10% NHS (SS). To understand this apparent discrepancy between C4bp binding and phenotype, we analyzed other properties of this strain (discussed below).
Defining Por Loops Required for C4bp Binding
C4bp Binding Region in Por1A.
Transformation of F62 with plasmid pUNCH62 40 produced clones with hybrid Por1A/1B molecules that were recognized by mAb 9D2 (recognizes a central region of FA19 Por) but not mAb 2F12 (specific for FA19 loop 1), indicating that a recombination had occurred between loop 1 and loop 5. Two classes of hybrids were identified, one with F62loop1 FA19loop2–8, and the other with F62loop1–4FA19loop5–8 (Fig. 3). Neither class of hybrid bound C4bp, suggesting that the loop 1 of FA19 Por1A was essential for C4bp binding.
We attempted to construct a hybrid Por in an F62 background that contained the NH2 terminus of FA19 Por (loop 1), and the COOH terminus of F62, to test if such a hybrid bound C4bp, thereby proving that loop 1 of FA19 contained the C4bp binding region exclusively. We were unsuccessful having used the following strategies. The first strategy we used was to transform F62 with pBUMC2 (see Materials and Methods). A recombinational event that occurred within the por gene would have resulted in a hybrid containing the NH2 terminus of FA19 and the COOH terminus of F62. Approximately 100 chloramphenicol-resistant colonies (the result of two separate transformation experiments) were screened using mAb 2F12 (specific for FA19 Por loop 1), but none of the clones reacted with this mAb. The second strategy was to construct a plasmid derived from pUNCH62 that contained loop 1 from FA19 and loops 2–8 of a C4bp-nonbinding Por1B molecule by cloning the BbsI-XbaI fragment of pBUMC1 into the corresponding region of pUNCH62. This plasmid coded for a Por hybrid FA19loop1F62loop2–7MS11loop8. Attempts to transform F62 with this plasmid also did not yield clones that contained the desired hybrid. These results strongly suggested that formation of this hybrid resulted in a lethal mutation. Finally, we attempted to construct a more “physiologic” hybrid Por molecule containing FA19 loop 1 and UU1 loop 2–8. Plasmid pBUMC3, containing the Por hybrid FA19loop1UU1loop2–8, was constructed and used to transform F62, as well as UU1. However, none of ∼50 colonies from each transformation attempt reacted with the FA19 loop 1 specific mAb 2F12, suggesting that the desired hybrid had not been obtained. We also observed that rates of transformation in all the above experiments were significantly lower than with control plasmids pUNCH61 and pUNCH62, again strongly suggesting that a hybrid containing loop 1 from FA19 and loops 2 through 8 from either F62 or UU1 were lethal to gonococci.
The results of the above experiments demonstrate that FA19 loop 1 is required for C4bp binding. However, we cannot conclude that the presence of this loop alone is sufficient for C4bp binding.
C4bp Binding Region in Por1B.
The C4bp binding region in MS11 Por1B was mapped using 12 strains containing F62/MS11 hybrid Por molecules, as shown in Fig. 4. Transformation of strain F62 with plasmid pUNCH61 produced colonies that bound mAbs 3C8 (recognizes central region of MS11 Por; reference 42), but not mAb 1F5 (specific for MS11 loop 1; reference 42), indicating again that recombination had occurred between loop 1 and loop 5. Two classes of hybrids were identified, one with F62loop1MS11loop2–8 and F62loop1–4MS11loop5–8. Both of these hybrids bound C4bp, suggesting that the C4bp binding domain in Por1B lay in the COOH-terminal half of the molecule (Fig. 4).
We replaced the BbsI-BsgI fragment of pUNCH61 with the corresponding BbsI-BsgI fragment from the F62 Por1B molecule to obtain plasmid pBUMC1 that contained a hybrid Por sequence MS11loop1F62loop2–7MS11loop8 (Fig. 4). Gonococcal colonies were screened using mAb 1F5 (specific for MS11 loop 1) to identify clones that were likely to contain the constructed hybrid molecule. DNA sequencing of the por gene confirmed that the gonococcal clones contained the desired hybrid molecule. This hybrid did not bind C4bp, thus narrowing down C4bp binding to a region encompassed by MS11 loops 5–7. Using overlap extension PCR or site-directed mutagenesis, we mutated the exposed regions of loops 5, 6, and 7 individually in pBUMC1 to reinsert MS11 sequence back into this region of the F62 Por. Individual mutations of loops 5, 6, and 7 did not restore C4bp binding. We next studied the effects of mutating the combinations of either loops 5 and 6, loops 6 and 7, or loops 5 and 7 together in pBUMC1 to simulate MS11 loops. Only the presence of MS11 loop 5 and 7 simultaneously resulted in C4bp binding (as demonstrated by the hybrid MS11loop1F62loop2–4MS11loop5F62loop6MS11loop7–8; Fig. 4), suggesting that these two loops together participated in formation of a C4bp-binding domain (indicated by the gray shaded boxes in Fig. 4).
To further demonstrate the necessity of MS11 Por loops 5 and 7, but not loop 6, for C4bp binding, we mutated each of these loops individually in pUNCH61 (contains the entire MS11 por sequence), to yield hybrid Por molecules that differed from MS11 only at loops 5, 6, or 7, which were mutated to duplicate these regions in F62. Substitution of loops 5 and 7 separately (hybrids MS11loop1–4F62loop5 MS11loop6–8 and MS11loop1–6F62loop7MS11loop8, respectively; Fig. 4), but not loop 6 (hybrid MS11loop1–5F62loop6 MS11loop7–8), completely abrogated C4bp binding (Fig. 4). Collectively, these data suggest that MS11 loops 5 and 7 participate in the formation of a C4bp binding domain.
Enhanced IgM Binding to F62PorMS11: Por Influences IgM Binding to Gonococci
Serum bactericidal testing revealed that F62PorMS11 was fully susceptible (0% survival) to the killing action of 10% NHS, despite its ability to bind C4bp. In contrast, hybrid F62loop1MS11loop2–8 that differed from F62PorMS11 only at Por loop 1 was fully resistant to 10% NHS. We compared the Ig and complement (C3 and C5b-9) binding properties of F62PorMS11 with F62loop1MS11loop2–8 and F62 to explain the apparent paradox between the ability of F62PorMS11 to bind C4bp and its SS phenotype. We observed that F62PorMS11 bound two- and fourfold more IgM than parent strain F62 and mutant strain F62loop1MS11loop2–8, respectively (Fig. 5). The amount of C3 and C5b-9 bound to F62PorMS11 was twofold more than that observed with F62loop1MS11loop2–8, and fivefold less than that detected on F62 (Fig. 5). IgG binding to the three strains was low (geometric mean fluorescence <10 for all strains), making meaningful interstrain comparisons difficult. To demonstrate that Ig was responsible for killing F62PorMS11, we adsorbed NHS against glutaraldehyde-fixed bacteria (F62PorMS11) at 4°C to deplete NHS of bacteria-specific antibody 41, which abrogated bactericidal activity against this strain (100% survival observed). The addition of heat-inactivated NHS to a final concentration of 10%, which acted as an antibody source, restored 99% killing of F62PorMS11. MS11 and F62loop1MS11loop2–8 showed 100% survival (remained SR) under both bactericidal assay conditions, and served as controls for this experiment. Similarly, hybrid F62loop1–4MS11loop5–8, which was also found to be SS despite binding C4bp, bound levels of IgM that were similar to those observed with F62PorMS11. These data provide evidence that the Por molecule, in addition to determining C4bp binding, may also dictate levels of IgM binding to the gonococcal surface, and that the two factors may interact to determine bacterial killing.
Figure 5.
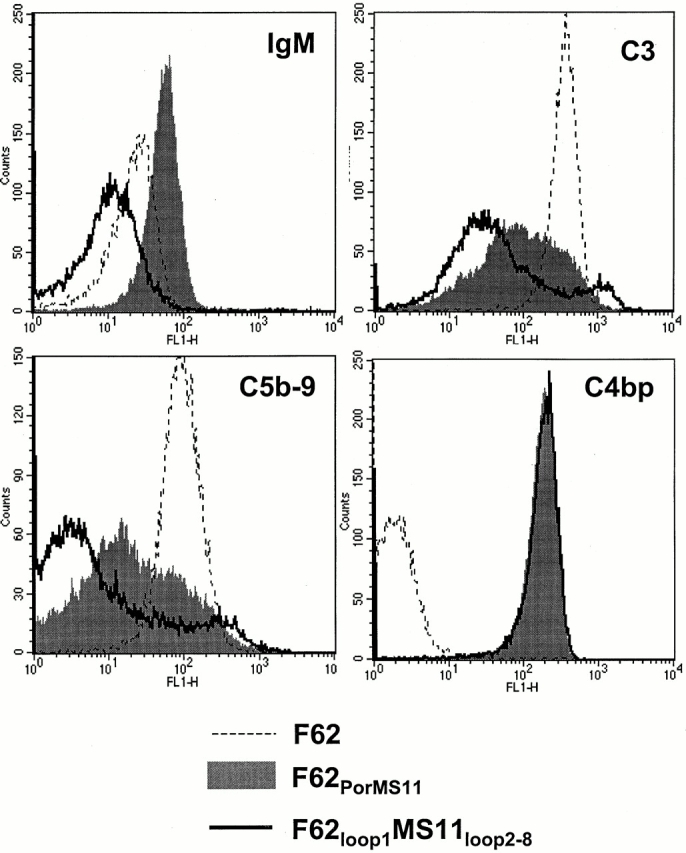
Enhanced IgM binding to F62PorMS11: Por influences IgM binding to gonococci. Despite the ability of F62PorMS11 to bind C4bp (see Fig. 2), the resulting transformant possessed the SS phenotype on bactericidal testing (100% killing in 10% NHS at 30 min). To determine the mechanism of serum sensitivity, we studied the binding of IgM, C3, C5b-9, and C4bp to F62PorMS11 (gray shaded areas) in comparison with F62 (dotted lines) and F62loop1MS11loop2–8 (solid lines). In contrast to F62PorMS11, F62loop1MS11loop2–8 was fully SR (100% survival in 10% NHS at 30 min), and differed from the former only at Por loop 1. F62PorMS11 bound two- and fourfold more IgM than F62 and F62loop1MS11loop2–8, respectively. Levels of C3 and C5b-9 binding to F62PorMS11 were intermediate compared with the two other strains. C4bp binding to F62PorMS11 and F62loop1MS11loop2–8 were identical.
Characterization of the bonds between C4bp and Por. The effects of ionic strength and the influence of two known ligands of C4bp (C4b and heparin) on C4bp–Por interactions were studied. C4b, when added in a 60-fold molar excess of C4bp, decreased binding of C4bp to MS11 by 50% (Fig. 6 A). In the presence of 125 units of heparin (Fig. 6 B) or 575 mM NaCl (Fig. 6 C), binding of C4bp to MS11 was decreased by >90% and ∼85%, respectively. None of these three conditions influenced the binding of C4bp to Por1A strain FA19. Based on these observations, it may be concluded that the C4bp–Por1B bond is ionic in nature, and that the binding site for Por1B in C4bp may reside at or very near binding sites in C4bp for heparin and C4b, which has been mapped to the interface between SCRs 1 and 2 34. This region also overlaps the binding site for streptococcal M proteins 55. In contrast, the Por1A–C4bp interaction could be hydrophobic, and is not influenced by ionic strength of buffers or heparin.
Figure 6.

Characterization of Por–C4bp bonds. The influence of C4b, heparin and high-ionic strength on Por-C4bp binding was studied by flow cytometry. C4b, when added to C4bp in a 60-fold molar excess, inhibited C4bp binding to MS11 (Por1B) by 40% (A). 125 units of heparin completely inhibited C4bp binding to MS11 (B), and 575 mM NaCl reduced C4bp binding to MS11 by >80% (C). None of the above conditions influenced C4bp binding to FA19 (Por1A). To minimize bacterial lysis in these experiments, organisms were first fixed with 1% paraformaldehyde. Fixing gonococci before incubation with C4bp did not alter C4bp binding properties.
C4bp SCR1 contains Por1A as well as Por1B binding sites. We determined that the binding site for gonococcal Por1A as well as Por1B resided in the α-chain of C4bp because recombinant polymeric human C4bp (rC4bp) that lacked the β-chain bound to FA19 as well as MS11 using flow cytometry. Native C4bp purified from human serum (containing the β-chain) bound to these two strains with similar fluorescent intensity as rC4bp (data not shown). Neither type of C4bp bound to strain F62. To determine the specific SCR(s) of C4bp that bound gonococci, we used rC4bp molecules expressed in human kidney cell line 293 that lacked individual α-chain SCRs and studied their binding to strains FA19 and MS11. rC4bp mutant molecule that lacked SCR1 bound neither FA19 or MS11, suggesting that SCR1 was required for binding to both gonococcal Por types (Fig. 7 A). Deletion of other domains individually had no significant impact on C4bp binding to gonococci. Further proof that SCR1 bound to both Por1A (strain FA19) and Por1B (strain MS11) was evidenced by showing that five mAbs directed against the NH2-terminal end of the α-chain of C4bp (mAb 70, mAb 92, mAb 96, mAb 102, and mAb 104; reference 51) each were able to completely inhibit C4bp binding to both strains (as an example, mAb 104 inhibition of C4bp binding shown in Fig. 7 B). mAb 67, directed against C4bp α-chain SCR4 51, did not influence C4bp binding to either strain and served as a control for this experiment (Fig. 7 B).
Figure 7.



C4bp SCR 1 contains both Por1A as well as Por1B binding regions. (A) Mapping of Por binding sites in C4bp. Binding of recombinant C4bp (rC4bp) and seven rC4bp mutant molecules lacking individual SCRs to FA19 (Por1A) and MS11 (Por1B) were studied using flow cytometry. 108 organisms were incubated with 2.5 μg of each rC4bp molecule, followed by detection with sheep polyclonal anti–human C4bp and disclosed with anti–sheep IgG-FITC. All rC4bp molecules that contained SCR 1 bound both FA19 as well as MS11; only the mutant that lacked SCR 1 (rC4bp ΔSCR1) did not bind either strain. Because artifactually lower fluorescence (threefold less binding compared with rC4bp) was observed when rC4bp ΔSCR6 was detected with the polyclonal anti-C4bp Ab, we used anti-C4bp mAb (Quidel Corp.) and mAb 67 (specific for SCR 4) to detect rC4bp ΔSCR6 bound to both strains. Compared with rC4bp, rC4bp ΔSCR6 bound with similar fluorescence intensities, when either mAb was used as a probe. Binding of rC4bp ΔSCR6 to both strains using the Quidel Corp. mAb is shown here (indicated by the asterisk). rC4bp ΔSCR1 did not bind to either strains when mAb 67 or the Quidel Corp. anti-C4bp mAb was used as the detection Ab. (B) Anti-C4bp SCR1 mAb blocks C4bp binding to gonococci. Further evidence that SCR 1 contains Por1A and Por1B binding regions was provided because mAb 104 that is specific for C4bp SCR 1 completely inhibits binding of C4bp to both FA19 (Por1A) as well as MS11 (Por1B; solid lines); control mAb 67 (specific for the irrelevant C4bp SCR 4) did not influence C4bp binding to either strain (gray shaded area) when compared with binding of pure C4bp alone (dashed lines).
To determine if the organization of the heptameric α-chain structure of the C4bp molecule was required for C4bp binding to Por, we studied binding of 5 μg of recombinant monomeric α-chain of C4bp that contained all eight α-chain SCRs, to strains FA19 and MS11 in a flow cytometry assay. The monomeric molecule did not bind to either Por, indicating a requirement for the polymeric form of C4bp α-chains to bind gonococcal Por (data not shown). Both mAb 67 as well as polyclonal anti-C4bp (Biodesign), which were used as detection Abs in flow cytometry, were capable of binding either monomeric or polymeric C4bp (data not shown).
Abrogating C4bp binding to gonococci results in complete bacterial killing. We have shown that C4bp that binds directly to the bacterial surface exhibits cofactor activity (see above). The ability to kill SR gonococci by selectively inhibiting C4bp binding would provide more definitive proof of the biological importance of this molecule. We prepared fAb fragments from mAb 104, and demonstrated that fAb 104, when added to NHS, completely inhibited C4bp binding to FA19 as well as to MS11 in a flow cytometry assay (Fig. 8 A). fAb 67 was used as a control, and did not influence C4bp binding to gonococci (not shown). We confirmed that fAb 67 retained 60% of the ability of mAb 67 to bind C4bp in ELISA (data not shown). Abrogating C4bp binding with fAb 104 resulted in a decreased C4d/C4c ratio, as well as enhanced C5b-9 insertion in the bacterial membrane (Table ), demonstrating that classical pathway regulatory activity was lost when C4bp binding to FA19 and MS11 was inhibited. We also performed a bactericidal assay including fAb 104 in the reaction mixture (molar ratio of fAb 104 to C4bp in the reaction mixture ∼70) to divert C4bp away from these bacterial surfaces. We observed complete killing of strain FA19, and >80% killing of MS11 at t = 30 min (Fig. 8 B). fAb 67, used as a control, showed no effect. Kinetics of bacterial killing showed an almost linear and steady decrease in bacterial viability over time, with almost complete killing observed at 30 min (Fig. 8 C) when C4bp binding was abrogated. The importance of C4bp in contributing to serum resistance of FA19 and MS11 can be gauged from the fact that both strains resist bactericidal activity of up to 33% NHS 24; FA19 resists killing of up to 66% NHS.
Figure 8.



Functional effects of blocking C4bp binding to gonococci. (A) fAb 104 inhibits C4bp binding to gonococci. 20 μg of fAb fragments generated from mAb 104, when added to 10% NHS, inhibited C4bp binding to FA19 and MS11 equally (gray shaded area). C4bp binding in the presence of NHS alone is shown by the solid line. (B) Diverting C4bp from the bacterial surface converts SR gonococci to an SS phenotype. A serum bactericidal assay was performed to assess the functional effects of inhibiting the binding of C4bp binding to the bacteria. FA19 and MS11 were incubated either with 10% NHS alone, 10% NHS plus 25 μg fAb 104, or 10% NHS plus 25 μg fAb 67 (negative control) for 30 min at 37°C. fAb 104 abrogated C4bp binding to the bacterial surface resulting in 100% killing of FA19 and 80% killing of MS11. NHS alone or NHS with (irrelevant) fAb 67 resulted in no significant killing of either strain. (C) Kinetics of bacterial killing by an unimpeded classical pathway. Abrogation of C4bp binding to the bacterial surface resulted in slow, sustained bacterial killing, with an almost linear decrease in bacterial viability over time of both FA19 (solid line) and MS11 (dotted line).
Table 5.
Inhibition of C4bp Binding to Gonococci Results in Complement Deposition on the Bacterial Surface
| Strain | Opsonization condition | C4c | C4d | C4d/C4c | C5b-9 |
|---|---|---|---|---|---|
| FA19 | NHS | 2.9 | 9.3 | 3.2 | 4.0 |
| NHS plus fAb 104 | 5.7 | 6.2 | 1.1 | 25.3 | |
| MS11 | NHS | 2.6 | 6.9 | 2.7 | 4.4 |
| NHS plus fAb 104 | 4.8 | 4.4 | 0.9 | 30.1 |
The effects of inhibiting C4bp binding in 20% NHS to FA19 and MS11 on the resultant binding of C4c, C4d, and C5b-9 to FA19 and MS11 were examined. Diminished binding of C4bp to the bacteria resulted in an increased C4d/C4c ratio, and increased C5b-9 binding to both strains.
Discussion
Complement is present in cervical secretions 3, and evasion of complement-mediated killing is vital for survival of gonococci. N. gonorrhoeae have evolved intricate mechanisms to evade complement. All gonococcal strains initially recovered from the human genital tract are resistant to the bactericidal action of NHS; strains that lose this property upon serial subculture 56 are termed unstably serum resistant, and this phenotype is facilitated by binding of factor H to sialic acid attached to LOS 12.
Strains that remain resistant after subculture are termed SR strains. Prior work in our laboratory has also defined factor H binding to the loop 5 of Por1A strains as a probable mechanism of stable serum resistance 24. In these studies, we have shown C4bp binding to Por1A as an additional mechanism whereby Por1A strains can evade complement. However, Por1B isolates form a significant proportion gonococcal isolates worldwide 57 58 59. Por1B strains are generally non- or weak binders of factor H, and must therefore evade complement by other mechanisms. This study defines C4bp binding to Por1B as an important mechanism of stable serum resistance in gonococci that belong to this serogroup.
We have shown that a strong correlation between C4bp binding and stable serum resistance in gonococci. None of the serum sensitive strains recovered from persons with gonococcal disease that we tested were C4bp binders (Table ). However, during the course of our studies, we observed that introduction of the entire MS11 Por1B-9 into an F62 background (F62PorMS11) resulted in C4bp binding as expected (Fig. 1), but unexpectedly did not convert the transformant to an SS phenotype. However, hybrid Por molecule F62loop1MS11loop2–8 moved into the same F62 background also bound the same amount of C4bp, but was fully SR. Almost fourfold higher levels of IgM bound to F62PorMS11 compared with F62loop1MS11loop2–8 (Fig. 5). These data provide evidence that, in addition to binding C4bp, the Por molecule may also dictate levels of IgM binding to surface targets, most likely LOS 15 60 61. In addition, levels of C3 and C5b-9 binding to F62PorMS11 were greater than for F62loop1MS11loop2–8, but less so than for F62 (Fig. 5). These data suggest that C4bp bound to F62PorMS11 was indeed capable of regulating complement downward, but this was still not sufficient to overcome the initial overwhelming complement activation by IgM. Thus, in the face of high levels of IgM and kinetically overwhelming complement activation, C4bp binding alone may not be sufficient for serum resistance.
We did not succeed in constructing hybrid Por molecules that contained loop 1 of FA19 loops 2 through 8 of F62 or UU1. The apparent transformation rates with plasmids that contained these Por hybrid sequences were significantly lower than observed with pUNCH61, suggesting that recombination events that might result in generating this Por hybrid were deleterious or possibly lethal. Therefore, we can conclude that the NH2-terminal Por1A loop is required for C4bp binding, but it may not necessarily contain the entire C4bp binding region.
Using rC4bp mutant molecules as well as anti-C4bp mAbs, we have shown that the NH2-terminal SCR of C4bp contains both Por1A as well as Por1B binding regions (Fig. 7). Por1A and Por1B binding regions in SCR 1 of C4bp may differ because Por1B–C4bp, but not Por1A–C4bp, interactions can be inhibited by C4b, heparin, as well as high ionic strength (Fig. 6). Akin to Por1B–C4bp interactions, the bond between streptococcal M protein and C4bp appears to occur in close proximity to the C4b binding region in C4bp. However, the M protein–C4bp bond, unlike the entirely electrostatic C4bp–C4b bond, is governed by additional noncovalent forces 55. We have shown that loops 5 and 7 of Por1B loop in strain MS11 are both required for C4bp binding (Fig. 4). Both of these loops together may participate in forming a negatively charged patch that is required for Por1B–C4bp interactions. The overall charge of the exposed regions of loops 5 and 7 together is −3, while F62 (a C4bp nonbinder) loops 5 and 7 together possess a net negative charge of −1.5 (Table ).
C4bp function was also shown to be important because C4bp binding to the bacterial surface was abrogated using fAb 104. This resulted in complete killing of strain FA19, and >80% killing of MS11, both in the presence of only 10% NHS (Fig. 8 B). The pivotal role of C4bp in mediating stable serum resistance of these two strains can be gauged by the fact that both strains are otherwise fully resistant to 33% NHS 24.
The relatively slow, but sustained killing by unimpeded activation of the classical pathway is evident from the kinetics of bacterial killing when C4bp binding to the bacterial surface was inhibited by fAb 104 (Fig. 8 C). In contrast, abrogating factor H binding to strain FA19 results in almost instantaneous, although incomplete, killing 24. These observations indicate that both pathways together are critical for efficient and complete bacterial killing. The relatively slow killing by the classical pathway (alone) can be explained because C1s, the enzyme that generates the classical pathway convertases, remains bound to the organism surface and is therefore not available to cycle and again act on its substrate. In contrast, factor D, the enzyme that generates the alternative pathway convertases, is liberated after it acts on its substrate, C3bB, thus enabling recycling and efficient amplification of C3b deposition. Notably, MS11, which did not bind factor H, was also killed slowly, perhaps unexpectedly if rapid alternative pathway recruitment and efficient killing of this strain were to be expected when C4bp binding to the surface of MS11 was blocked. In an effort to explain these findings, we analyzed and compared total C3, C3b, iC3b, and factor Bb on strains MS11 and 24-1 (the latter is an SS Por1B strain, and does not bind either factor H or C4bp; reference 12). After 10 min opsonization with 10% NHS, we observed that MS11 bound 45% less C3 than 24-1, and almost all the C3b on MS11 was converted to iC3b, whereas C3b on 24-1 remained unprocessed. MS11 bound fivefold less factor Bb than 24-1. These data suggest regulation of the alternative pathway on the surface of MS11 despite its inability to bind pure factor H. A possible explanation for this phenomenon could include the ability of MS11 to bind factor H–like protein 1 (FHL-1), which is an alternatively spliced form of full-length factor H found in human serum 62 63, and has been shown to bind M proteins of group A streptococcus and exert cofactor activity 64 65. Other possibilities include decreased affinity of factor B for C3b on MS11, or endogenous factor H-like activity of the organism; the latter has been reported with Epstein-Barr virus 66, and merits further investigation.
Por is antigenic during natural infection 67 68 69 and certain evidence suggests that women with gonococcal PID and female commercial sex workers with a history of multiple gonococcal infections may be protected against repeated infection with the same serovar 70. However, other studies, involving predominantly male populations, suggest that serovar-specific protection may not be a feature of gonococcal disease 71 72. Gonococcal Por undergoes antigenic variation in vivo over time as shown by diversity of serovars recognized in a sexual network 70 73. Subtle differences in Por sequence can occur even among isolates belonging to the same serovar 74. An interesting feature of nucleotide polymorphisms in the por gene is that maximum rates of nucleotide diversity occur in the putative exposed portion of Por1A loop 1 and Por1B loop 5 75; both of these regions are critical for C4bp binding. The ability of the Por molecule to undergo sequence alterations potentially could also abrogate binding of bactericidal antibodies 76 77, in the face of retention (and perhaps an increase) in the capacity to bind complement regulators such as factor H and C4bp. This is especially likely in the case of Por1B strains, where the C4bp binding domain is formed by two noncontiguous loops and is charge dependent.
In conclusion, gonococci have several mechanisms that act cooperatively to mediate serum resistance. Factor H binds to sialic acid on LOS and mediates unstable serum resistance. Several nonsialylated gonococci can use their Por molecule to bind regulatory molecules to evade complement-mediated killing. Por1A strains bind factor H through loop 5 and C4bp possibly through loop 1, and the greater negative charge of the exposed regions of loops 5 and 7 of certain Por1B strains enable them to bind C4bp.
Acknowledgments
This work was supported by National Institutes of Health grants AI32725 and DK35081, Swedish Medical Council, Swedish Natural Science Research Council, Swedish Foundation for Strategic Research, and a research grant from the University Hospitals in Malmö. Dr. Ram is the recipient of the 1998–2000 American Social Health Association/Burroughs Wellcome Fund Post Doctoral Research Fellowship in Sexually Transmitted Diseases.
Footnotes
Abbreviations used in this paper: C4bp, C4b-binding protein; DGI, disseminated gonococcal infection; LOS, lipooligosaccharide; NHS, nonimmune normal human serum; PID, pelvic inflammatory disease; Por, porin; SCR, short consensus repeat; SR, serum resistant; SS, serum-sensitive.
References
- Rice P.A., McCormack W.M., Kasper D.L. Natural serum bactericidal activity against Neisseria gonorrhoeae isolates from disseminated, locally invasive, and uncomplicated disease. J. Immunol. 1980;124:2105–2109. [PubMed] [Google Scholar]
- Schumacher G.F. Immunology of spermatozoa and cervical mucus. Hum. Reprod. 1988;3:289–300. doi: 10.1093/oxfordjournals.humrep.a136698. [DOI] [PubMed] [Google Scholar]
- Price R.J., Boettcher B. The presence of complement in human cervical mucus and its possible relevance to infertility in women with complement-dependent sperm-immobilizing antibodies. Fertil. Steril. 1979;32:61–66. doi: 10.1016/s0015-0282(16)44117-8. [DOI] [PubMed] [Google Scholar]
- Bischof P., Planas-Basset D., Meisser A., Campana A. Investigations on the cell type responsible for the endometrial secretion of complement component 3 (C3) Hum. Reprod. 1994;9:1652–1659. doi: 10.1093/oxfordjournals.humrep.a138768. [DOI] [PubMed] [Google Scholar]
- Sayegh R.A., Tao X.J., Awwad J.T., Isaacson K.B. Localization of the expression of complement component 3 in the human endometrium by in situ hybridization. J. Clin. Endocrinol. Metab. 1996;81:1641–1649. doi: 10.1210/jcem.81.4.8636381. [DOI] [PubMed] [Google Scholar]
- McQuillen D.P., Gulati S., Ram S., Turner A.K., Jani D.B., Heeren T.C., Rice P.A. Complement processing and immunoglobulin binding to Neisseria gonorrhoeae determined in vitro simulates in vivo effects. J. Infect. Dis. 1999;179:124–135. doi: 10.1086/314545. [DOI] [PubMed] [Google Scholar]
- Nairn C.A., Cole J.A., Patel P.V., Parsons N.J., Fox J.E., Smith H. Cytidine 5′-monophospho-N-acetylneuraminic acid or a related compound is the low Mr factor from human red blood cells which induces gonococcal resistance to killing by human serum. J. Gen. Microbiol. 1988;134:3295–3306. doi: 10.1099/00221287-134-12-3295. [DOI] [PubMed] [Google Scholar]
- Parsons N.J., Patel P.V., Tan E.L., Andrade J.R., Nairn C.A., Goldner M., Cole J.A., Smith H. Cytidine 5′-monophospho-N-acetyl neuraminic acid and a low molecular weight factor from human blood cells induce lipopolysaccharide alteration in gonococci when conferring resistance to killing by human serum. Microb. Pathog. 1988;5:303–309. doi: 10.1016/0882-4010(88)90103-9. [DOI] [PubMed] [Google Scholar]
- Goldner M., Penn C.W., Sanyal S.C., Veale D.R., Smith H. Phenotypically determined resistance of Neisseria gonorrhoeae to normal human serumenvironmental factors in subcutaneous chambers in guinea pigs. J. Gen. Microbiol. 1979;114:169–177. doi: 10.1099/00221287-114-1-169. [DOI] [PubMed] [Google Scholar]
- Martin P.M., Patel P.V., Parsons N.J., Smith H. Induction of phenotypically determined resistance of Neisseria gonorrhoeae to human serum by factors in human serum. J. Gen. Microbiol. 1981;127:213–217. doi: 10.1099/00221287-127-1-213. [DOI] [PubMed] [Google Scholar]
- Veale D.R., Penn C.W., Smith H. Factors affecting the induction of phenotypically determined serum resistance of Neisseria gonorrhoeae grown in media containing serum or its diffusible components. J. Gen. Microbiol. 1981;122:235–245. doi: 10.1099/00221287-122-2-235. [DOI] [PubMed] [Google Scholar]
- Ram S., Sharma A.K., Simpson S.D., Gulati S., McQuillen D.P., Pangburn M.K., Rice P.A. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae . J. Exp. Med. 1998;187:743–752. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella M.A., Mandrell R.E., Shero M., Wilson M.E., Griffiss J.M., Brooks G.F., Lammel C., Breen J.F., Rice P.A. Modification by sialic acid of Neisseria gonorrhoeae lipooligosaccharide epitope expression in human urethral exudatesan immunoelectron microscopic analysis. J. Infect. Dis. 1990;162:506–512. doi: 10.1093/infdis/162.2.506. [DOI] [PubMed] [Google Scholar]
- O'Brien J.P., Goldenberg D.L., Rice P.A. Disseminated gonococcal infectiona prospective analysis of 49 patients and a review of pathophysiology and immune mechanisms. Medicine. 1983;62:395–406. [PubMed] [Google Scholar]
- Schneider H., Griffiss J.M., Mandrell R.E., Jarvis G.A. Elaboration of a 3.6-kilodalton lipooligosaccharide, antibody against which is absent from human sera, is associated with serum resistance of Neisseria gonorrhoeae . Infect. Immun. 1985;50:672–677. doi: 10.1128/iai.50.3.672-677.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunham R.C., Plummer F., Slaney L., Rand F., DeWitt W. Correlation of auxotype and protein I type with expression of disease due to Neisseria gonorrhoeae . J. Infect. Dis. 1985;152:339–343. doi: 10.1093/infdis/152.2.339. [DOI] [PubMed] [Google Scholar]
- Cannon J.G., Buchanan T.M., Sparling P.F. Confirmation of association of protein I serotype of Neisseria gonorrhoeae with ability to cause disseminated infection. Infect. Immun. 1983;40:816–819. doi: 10.1128/iai.40.2.816-819.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake M.S., Gotschlich E.C. Functional and immunological properties of pathogenic neisserial surface proteins. In: Inouye M., editor. Bacterial Outer Membranes as Model Systems. John Wiley; New York: 1986. pp. 377–400. [Google Scholar]
- Douglas J.T., Lee M.D., Nikaido H. Protein I of Neisseria gonorrhoeae outer membrane is a porin. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1981;12:305–309. [Google Scholar]
- Sparling P.F. Biology of Neisseria gonorrhoeae. 2nd ed. In: Holmes K.K., Mardh P.A., Sparling P.F., Wiesner P.J., Cates J., Lemon W.S.M., Stamm W.E., editors. Sexually Transmitted Diseases. McGraw-Hill; New York: 1990. pp. 433–449. [Google Scholar]
- Knapp J.S., Tam M.R., Nowinski R.C., Holmes K.K., Sandstrom E.G. Serological classification of Neisseria gonorrhoeae with use of monoclonal antibodies to gonococcal outer membrane protein I. J. Infect. Dis. 1984;150:44–48. doi: 10.1093/infdis/150.1.44. [DOI] [PubMed] [Google Scholar]
- Smith N.H., Maynard Smith J., Spratt B.G. Sequence evolution of the porB gene of Neisseria gonorrhoeae and Neisseria meningitidisevidence of positive Darwinian selection. Mol. Biol. Evol. 1995;12:363–370. doi: 10.1093/oxfordjournals.molbev.a040212. [DOI] [PubMed] [Google Scholar]
- Ward M.J., Lambden P.R., Heckels J.E. Sequence analysis and relationships between meningococcal class 3 serotype proteins and other porins from pathogenic and non-pathogenic Neisseria species. FEMS (Fed. Eur. Microbiol. Soc.) Microbiol. Lett. 1992;73:283–289. doi: 10.1016/0378-1097(92)90644-4. [DOI] [PubMed] [Google Scholar]
- Ram S., McQuillen D.P., Gulati S., Elkins C., Pangburn M.K., Rice P.A. Binding of complement factor H to loop 5 of porin protein 1Aa molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae . J. Exp. Med. 1998;188:671–680. doi: 10.1084/jem.188.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingwer I., Petersen B.H., Brooks G. Serum bactericidal action and activation of the classic and alternate complement pathways by Neisseria gonorrhoeae . J. Lab. Clin. Med. 1978;92:211–220. [PubMed] [Google Scholar]
- Facinelli B., Giovanetti E., Varaldo P.E., Casolari P., Fabio U. Antibiotic resistance in foodborne listeria. Lancet. 1991;338:1272. doi: 10.1016/0140-6736(91)92138-r. [DOI] [PubMed] [Google Scholar]
- Fujita T., Gigli I., Nussenzweig V. Human C4-binding protein. II. Role in proteolysis of C4b by C3b-inactivator. J. Exp. Med. 1978;148:1044–1051. doi: 10.1084/jem.148.4.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T., Nussenzweig V. The role of C4-binding protein and beta 1H in proteolysis of C4b and C3b. J. Exp. Med. 1979;150:267–276. doi: 10.1084/jem.150.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigli I., Fujita T., Nussenzweig V. Modulation of the classical pathway C3 convertase by plasma proteins C4 binding protein and C3b inactivator. Proc. Natl. Acad. Sci. USA. 1979;76:6596–6600. doi: 10.1073/pnas.76.12.6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfstein J., Ferreira A., Gigli I., Nussenzweig V. Human C4-binding protein. I. Isolation and characterization. J. Exp. Med. 1978;148:207–222. doi: 10.1084/jem.148.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa S., Unno H., Ichihara C., Koyama J., Koide T. Human C4b-binding protein, C4bp. Chymotryptic cleavage and location of the 48 kDa active fragment within C4bp. FEBS Lett. 1983;164:135–138. doi: 10.1016/0014-5793(83)80036-2. [DOI] [PubMed] [Google Scholar]
- Dahlback B., Smith C.A., Muller-Eberhard H.J. Visualization of human C4b-binding protein and its complexes with vitamin K-dependent protein S and complement protein C4b. Proc. Natl. Acad. Sci. USA. 1983;80:3461–3465. doi: 10.1073/pnas.80.11.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlback B., Muller-Eberhard H.J. Ultrastructure of C4b-binding protein fragments formed by limited proteolysis using chymotrypsin. J. Biol. Chem. 1984;259:11631–11634. [PubMed] [Google Scholar]
- Villoutreix B.O., Hardig Y., Wallqvist A., Covell D.G., Garcia de Frutos P., Dahlback B. Structural investigation of C4b-binding protein by molecular modelinglocalization of putative binding sites. Proteins. 1998;31:391–405. doi: 10.1002/(sici)1097-0134(19980601)31:4<391::aid-prot6>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Hillarp A., Dahlback B. Novel subunit in C4b-binding protein required for protein S binding. J. Biol. Chem. 1988;263:12759–12764. [PubMed] [Google Scholar]
- Hessing M., Kanters D., Hackeng T.M., Bouma B.N. Identification of different forms of human C4b-binding protein lacking beta-chain and protein S binding ability. Thromb. Haemostasis. 1990;64:245–250. [PubMed] [Google Scholar]
- Thern A., Stenberg L., Dahlback B., Lindahl G. Ig-binding surface proteins of Streptococcus pyogenes also bind human C4b-binding protein (C4BP), a regulatory component of the complement system. J. Immunol. 1995;154:375–386. [PubMed] [Google Scholar]
- Johnsson E., Thern A., Dahlback B., Heden L.O., Wikstrom M., Lindahl G. A highly variable region in members of the streptococcal M protein family binds the human complement regulator C4BP. J. Immunol. 1996;157:3021–3029. [PubMed] [Google Scholar]
- Berggard K., Johnsson E., Mooi F.R., Lindahl G. Bordetella pertussis binds the human complement regulator C4BProle of filamentous hemagglutinin. Infect. Immun. 1997;65:3638–3643. doi: 10.1128/iai.65.9.3638-3643.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbonetti N., Simnad V., Elkins C., Sparling P.F. Construction of isogenic gonococci with variable porin structureeffects on susceptibility to human serum and antibiotics. Mol. Microbiol. 1990;4:1009–1018. doi: 10.1111/j.1365-2958.1990.tb00673.x. [DOI] [PubMed] [Google Scholar]
- McQuillen D.P., Gulati S., Rice P.A. Complement-mediated bacterial killing assays. Methods Enzymol. 1994;236:137–147. doi: 10.1016/0076-6879(94)36013-8. [DOI] [PubMed] [Google Scholar]
- Carbonetti N.H., Simnad V.I., Seifert H.S., So M., Sparling P.F. Genetics of protein I of Neisseria gonorrhoeaeconstruction of hybrid porins Proc. Natl. Acad. Sci. USA. 85 1988. 6841 6845[published erratum at 86:1317] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson J., Dorward D., Lubke L., Kao D. Porin polypeptide contributes to surface charge of gonococci. J. Bacteriol. 1997;179:3541–3548. doi: 10.1128/jb.179.11.3541-3548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- Elkins C., Carbonetti N.H., Coimbre A.J., Thomas C.E., Sparling P.F. Cloning and constitutive expression of structural genes encoding gonococcal porin protein in Escherichia coli and attenuated Salmonella typhimurium vaccine strains. Gene. 1994;138:43–50. doi: 10.1016/0378-1119(94)90781-1. [DOI] [PubMed] [Google Scholar]
- Wetzler L.M., Gotschlich E.C., Blake M.S., Koomey J.M. The construction and characterization of Neisseria gonorrhoeae lacking protein III in its outer membrane. J. Exp. Med. 1989;169:2199–2209. doi: 10.1084/jem.169.6.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzler L.M., Blake M.S., Gotschlich E.C. Characterization and specificity of antibodies to protein I of Neisseria gonorrhoeae produced by injection with various protein I–adjuvant preparations. J. Exp. Med. 1988;168:1883–1897. doi: 10.1084/jem.168.5.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlback B. Purification of human C4b-binding protein and formation of its complex with vitamin K-dependent protein S. Biochem. J. 1983;209:847–856. doi: 10.1042/bj2090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuguchi T., Okamura S., Aso T., Sata T., Niho Y. Molecular cloning of the cDNA coding for proline-rich protein (PRP)identity of PRP as C4b-binding protein. Biochem. Biophys. Res. Commun. 1989;165:138–144. doi: 10.1016/0006-291x(89)91045-0. [DOI] [PubMed] [Google Scholar]
- Blom A.M., Webb J., Villoutreix B.O., Dahlback B. A cluster of positively charged amino acids in the C4BP alpha-chain is crucial for C4b binding and factor I cofactor function. J. Biol. Chem. 1999;274:19237–19245. doi: 10.1074/jbc.274.27.19237. [DOI] [PubMed] [Google Scholar]
- Hardig Y., Hillarp A., Dahlback B. The amino-terminal module of the C4b-binding protein alpha-chain is crucial for C4b binding and factor I-cofactor function. Biochem. J. 1997;323:469–475. doi: 10.1042/bj3230469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkins, C., N.H. Carbonetti, V.A. Varela, D. Stirewalt, D.G. Klapper, and P.F. Sparling. Antibodies to N-terminal peptides of gonococcal porin are bactericidal when gonococcal lipopolysaccharide is not sialylated. Mol. Microbiol. 6:2617–2628. [DOI] [PubMed]
- Liszewski M.K., Leung M.K., Atkinson J.P. Membrane cofactor proteinimportance of N- and O-glycosylation for complement regulatory function. J. Immunol. 1998;161:3711–3718. [PubMed] [Google Scholar]
- Elkins C., Carbonetti N.H., Varela V.A., Stirewalt D., Klapper D.G., Sparling P.F. Antibodies to N-terminal peptides of gonococcal porin are bactericidal when gonococcal lipopolysaccharide is not sialylated. Mol. Microbiol. 1992;6:2617–2628. doi: 10.1111/j.1365-2958.1992.tb01439.x. [DOI] [PubMed] [Google Scholar]
- Blom A.M., Berggard K., Webb J.H., Lindahl G., Villoutreix B.O., Dahlback B. Human C4b-binding protein has overlapping, but not identical, binding sites for C4b and streptococcal M proteins. J. Immunol. 2000;164:5328–5336. doi: 10.4049/jimmunol.164.10.5328. [DOI] [PubMed] [Google Scholar]
- Ward M.E., Watt P.J., Glynn A.A. Gonococci in urethral exudates possess a virulence factor lost on subculture. Nature. 1970;227:382–384. doi: 10.1038/227382a0. [DOI] [PubMed] [Google Scholar]
- Ross J.D., Weir M., Horn C.K., Moyes A., Young H. Gonococcal serovar patterns in Glasgow1990-1992. Br. J. Biomed. Sci. 1995;52:87–92. [PubMed] [Google Scholar]
- Moyes A., Young H. Epidemiological typing of Neisseria gonorrhoeaea comparative analysis of three monoclonal antibody serotyping panels. Eur. J. Epidemiol. 1991;7:311–319. doi: 10.1007/BF00144994. [DOI] [PubMed] [Google Scholar]
- Kohl P.K., Knapp J.S., Hofmann H., Gruender K., Petzoldt D., Tams M.R., Holmes K.K. Epidemiological analysis of Neisseria gonorrhoeae in the Federal Republic of Germany by auxotyping and serological classification using monoclonal antibodies. Genitourin. Med. 1986;62:145–150. doi: 10.1136/sti.62.3.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apicella M.A., Westerink M.A.J., Morse S.A., Schneider H., Rice P.A., Griffiss J.M. Bactericidal antibody response of normal human serum to the lipooligosaccharide of Neisseria gonorrhoeae . J. Infect. Dis. 1986;153:520–526. doi: 10.1093/infdis/153.3.520. [DOI] [PubMed] [Google Scholar]
- Densen P., Gulati S., Rice P.A. Specificity of antibodies against Neisseria gonorrhoeae that stimulate neutrophil chemotaxis. J. Clin. Invest. 1987;80:78–87. doi: 10.1172/JCI113067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel P.F., Skerka C. Complement factor H and related proteinsan expanding family of complement-regulatory proteins? Immunol. Today. 1994;15:121–126. doi: 10.1016/0167-5699(94)90155-4. [DOI] [PubMed] [Google Scholar]
- Zipfel P.F., Jokiranta T.S., Hellwage J., Koistinen V., Meri S. The factor H protein family. Immunopharmacology. 1999;42:53–60. doi: 10.1016/s0162-3109(99)00015-6. [DOI] [PubMed] [Google Scholar]
- Johnsson E., Berggard K., Kotarsky H., Hellwage J., Zipfel P.F., Sjobring U., Lindahl G. Role of the hypervariable region in streptococcal M proteinsbinding of a human complement inhibitor. J. Immunol. 1998;161:4894–4901. [PubMed] [Google Scholar]
- Perez-Caballero D., Alberti S., Vivanco F., Sanchez-Corral P., Rodriguez de Cordoba S. Assessment of the interaction of human complement regulatory proteins with group A Streptococcus. Identification of a high-affinity group A Streptococcus binding site in FHL-1. Eur. J. Immunol. 2000;30:1243–1253. doi: 10.1002/(SICI)1521-4141(200004)30:4<1243::AID-IMMU1243>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Mold C., Bradt B.M., Nemerow G.R., Cooper N.R. Epstein-Barr virus regulates activation and processing of the third component of complement. J. Exp. Med. 1988;168:949–969. doi: 10.1084/jem.168.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan A., McNeillage G., Young H., Bain S.S. Secretory antibody response of the cervix to infection with Neisseria gonorrhoeae . Br. J. Vener. Dis. 1979;55:265–270. doi: 10.1136/sti.55.4.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly R.J., Lee L., Welch B.G. Secretory IgA antibody responses to Neisseria gonorrhoeae in the genital secretions of infected females. J. Infect. Dis. 1976;133:113–125. doi: 10.1093/infdis/133.2.113. [DOI] [PubMed] [Google Scholar]
- Lammel C.J., Sweet R.L., Rice P.A., Knapp J.S., Schoolnik G.K., Heilbron D.C., Brooks G.F. Antibody-antigen specificity in the immune response to infection with Neisseria gonorrhoeae . J. Infect. Dis. 1985;152:990–1001. doi: 10.1093/infdis/152.5.990. [DOI] [PubMed] [Google Scholar]
- Plummer F.A., Simonsen J.N., Chubb H., Slaney L., Kimata J., Bosire M., Ndinya-Achola J.O., Ngugi E.N. Epidemiologic evidence for the development of serovar-specific immunity after gonococcal infection. J. Clin. Invest. 1989;83:1472–1476. doi: 10.1172/JCI114040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young H., Moyes A., Ross J., McMillan A. A serovar analysis of heterosexual gonorrhoea in Edinburgh 1986-90. Genitourin. Med. 1992;68:16–19. doi: 10.1136/sti.68.1.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox K.K., Thomas J.C., Weiner D.H., Davis R.H., Sparling P.F., Cohen M.S. Longitudinal evaluation of serovar-specific immunity to Neisseria gonorrhoeae . Am. J. Epidemiol. 1999;149:353–358. doi: 10.1093/oxfordjournals.aje.a009820. [DOI] [PubMed] [Google Scholar]
- Brunham R.C., Plummer F.A., Stephens R.S. Bacterial antigenic variation, host immune response, and pathogen-host coevolution. Infect. Immun. 1993;61:2273–2276. doi: 10.1128/iai.61.6.2273-2276.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C.R., Koeuth T., Estabrook M.M., Lupski J.R. Rapid determination of outbreak-related strains of Neisseria meningitidis by repetitive element-based polymerase chain reaction genotyping. J. Infect. Dis. 1996;174:760–767. doi: 10.1093/infdis/174.4.760. [DOI] [PubMed] [Google Scholar]
- Fudyk T.C., Maclean I.W., Simonsen J.N., Njagi E.N., Kimani J., Brunham R.C., Plummer F.A. Genetic diversity and mosaicism at the por locus of Neisseria gonorrhoeae . J. Bacteriol. 1999;181:5591–5599. doi: 10.1128/jb.181.18.5591-5599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy T.F., Yi K. Mechanisms of recurrent otitis mediaimportance of the immune response to bacterial surface antigens. Ann. NY Acad. Sci. 1997;830:353–360. doi: 10.1111/j.1749-6632.1997.tb51907.x. [DOI] [PubMed] [Google Scholar]
- Haase E.M., Campagnari A.A., Sarwar J., Shero M., Wirth M., Cumming C.U., Murphy T.F. Strain-specific and immunodominant surface epitopes of the P2 porin protein of nontypeable Haemophilus influenzae . Infect. Immun. 1991;59:1278–1284. doi: 10.1128/iai.59.4.1278-1284.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrell R., Apicella M.A., Boslego J., Chung R., Rice P.A., Griffiss J.M. Human immune response to monoclonal antibody-defined epitopes of Neisseria gonorrhoeae lipooligosaccharides. In: Poolman J.T., Zanen H., Mayer T., Heckels J., Makela P.H., Smith H., Beuvery C., editors. Gonococci and Meningococci. Kluwer Academic Publishers; Dordrecht, Netherlands: 1988. pp. 569–574. [Google Scholar]
- Yamasaki R., Kerwood D.E., Schneider H., Quinn K.P., Griffiss J.M., Mandrell R.E. The structure of lipooligosaccharide produced by Neisseria gonorrhoeae, strain 15253, isolated from a patient with disseminated infectionevidence for a new glycosylation pathway of gonococcal lipooligosaccharide. J. Biol. Chem. 1994;269:30345–30351. [PubMed] [Google Scholar]
- Meyer T.F., Mlawer N., So M. Pilus expression in Neisseria gonorrhoeae involves chromosomal rearrangement. Cell. 1982;30:45–52. doi: 10.1016/0092-8674(82)90010-1. [DOI] [PubMed] [Google Scholar]
- Reyn A. Sensitivity of Neisseria gonorrhoeae to antibiotics. Br. J. Vener. Dis. 1961;27:145–157. doi: 10.1136/sti.37.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S.E., Clark V.L. Genetic loci and linkage associations in Neisseria gonorrhoeae and Neisseria meningitidis Clin. Microbiol. Rev. 2Suppl1989. S92 S103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas K.C., Apicella M.A. Selection and immunochemical analysis of lipooligosaccharide mutants of Neisseria gonorrhoeae . Infect. Immun. 1988;56:499–504. doi: 10.1128/iai.56.2.499-504.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babl F.E., Ram S., Barnett E.D., Rhein L., Carr E., Cooper E.R. Neonatal gonococcal arthritis after negative prenatal screening and despite conjunctival prophylaxis. Pediatr. Infect. Dis. J. 2000;19:346–349. doi: 10.1097/00006454-200004000-00017. [DOI] [PubMed] [Google Scholar]
- Schneider H., Griffiss J.M., Williams G.D., Pier G.B. Immunological basis of serum resistance of Neisseria gonorrhoeae . J. Gen. Microbiol. 1982;128:13–22. doi: 10.1099/00221287-128-1-13. [DOI] [PubMed] [Google Scholar]
- Hobbs M.M., Alcorn T.M., Davis R.H., Fischer W., Thomas J.C., Martin I., Ison C., Sparling P.F., Cohen M.S. Molecular typing of Neisseria gonorrhoeae causing repeated infectionsevolution of porin during passage within a community. J. Infect. Dis. 1999;179:371–381. doi: 10.1086/314608. [DOI] [PubMed] [Google Scholar]