

Abstract
Human airway epithelial cells appear specially programmed for expression of immune response genes implicated in immunity and inflammation. To better determine how this epithelial system operates in vivo, we analyzed its behavior in mouse models that allow for in vitro versus in vivo comparison and genetic modification. Initial comparisons indicated that tumor necrosis factor α induction of epithelial intercellular adhesion molecule 1 required sequential induction of interleukin (IL)-12 (p70) and interferon γ, and unexpectedly localized IL-12 production to airway epithelial cells. Epithelial IL-12 was also inducible during paramyxoviral bronchitis, but in this case, initial IL-12 p70 expression was followed by 75-fold greater expression of IL-12 p40 (as monomer and homodimer). Induction of IL-12 p40 was even further increased in IL-12 p35-deficient mice, and in this case, was associated with increased mortality and epithelial macrophage accumulation. The results placed epithelial cell overgeneration of IL-12 p40 as a key intermediate for virus-inducible inflammation and a candidate for epithelial immune response genes that are abnormally programmed in inflammatory disease. This possibility was further supported when we observed IL-12 p40 overexpression selectively in airway epithelial cells in subjects with asthma and concomitant increases in airway levels of IL-12 p40 (as homodimer) and airway macrophages. Taken together, these results suggest a novel role for epithelial-derived IL-12 p40 in modifying the level of airway inflammation during mucosal defense and disease.
Keywords: asthma, cell adhesion molecule, mucosal immunity, paramyxoviral bronchitis, macrophage
Introduction
The adaptive immune system (manifested by the diverse repertoire of T cells and B cells) requires signals about the origin of the antigen and the type of response to be induced 1. Furthermore, these signals appear to be provided by the innate immune system 1. In this general context, and in the particular context of the response to inhaled materials, we have proposed that the epithelial cells lining the airway surface (i.e., the airway epithelial cell) represents an ideal candidate to act as a primary sentinel site in innate immunity. This possibility derived from the observation that the immune cell response to inhaled agents was directed to the airway epithelium and the subsequent evidence that local factors generated by the airway epithelial cells themselves provided critical biochemical signals for immune cell influx and activation 2. Our cellular and molecular concepts of this paradigm have subsequently evolved to the point where we recognize that subsets of immune response genes in epithelial cells are specially programmed for normal host defense and furthermore, that the same gene network may be abnormally programmed for immune signaling in inflammatory diseases such as asthma 3. For example, it appears that epithelial expression of a cell adhesion molecule (i.e., intercellular adhesion molecule [ICAM]1-1) and a chemokine (i.e., regulated upon activation, normal T cell expressed and secreted [RANTES]) are critically coordinated to direct mucosal immune cell traffic 4 5 6 7 8 and that overexpression of ICAM-1 and RANTES are invariant features of asthmatic airway inflammation 7 9.
The present studies were initiated to better define epithelial immune signaling using a mouse model that could be subjected to genetic modification. Initial experiments using wild-type mice and same-strain mice with disruption of IL-12 or IFN-γ genes indicated that ICAM-1 expression was inducible either directly by IFN-γ or via a cytokine cascade initiated by TNF-α and proceeding to sequential induction of IL-12 and then IFN-γ. These findings fit with previous evidence for selective IFN-γ responsiveness of epithelial ICAM-1 gene expression 10 11 12, but also unexpectedly localized the site of IL-12 production to airway epithelial cells. As IL-12 is generally produced by antigen-presenting cells (i.e., macrophages, dendritic cells, and B cells) in other settings 13 14 15 16, we next defined the site of IL-12 induction by a more natural stimulus that is also relevant to asthmatic airway inflammation 17. In particular, we found that initial IL-12 expression was inducible by mouse parainfluenza type I (Sendai) virus and was confined to airway epithelial cells (the host cells in this setting). Initial IL-12 induction was followed by excessive expression of IL-12 p40 that could be further enhanced in IL-12 p35–deficient mice. Others have provided evidence that IL-12 p40 may function as an antagonist of IL-12 action 18 19 20 21, but in this case, its production resulted in airway inflammation and increased mortality. Although toxicity has been observed for overproduction of IL-12 22 23, inflammation due to IL-12 p40 had not been observed. Thus, the results placed excessive generation of IL-12 p40 as a key intermediate for virus-inducible inflammation and a candidate for epithelial immune response genes that are abnormally programmed in inflammatory disease. We next confirmed this possibility by finding IL-12 p40 overexpression in airway epithelial cells in subjects with asthma.
Taken together, the results provide for a new cellular source of IL-12 and IL-12 p40 that is inducible by viral infection, a new functional consequence of IL-12 p40 production in vivo that is not dependent on actions of IL-12 p70 or IFN-γ, and the first evidence that epithelial IL-12 p40 expression is abnormally programmed in asthma. In conjunction with previous observations of constitutive activation of signal transducer and activator of transcription (Stat)1 and Stat1-dependent genes (typified by ICAM-1) and inducible expression of RANTES in asthma 7 9, the findings further support the possibility that pathways normally responsive to T helper type 1 cytokines are also dysregulated in allergic disease.
Materials and Methods
Materials.
Recombinant murine TNF-α was from PeproTech; recombinant murine IFN-γ and biotinylated rat anti–mouse IL-12 mAb (clone C17.15 recognizing IL-12 p40 and IL-12) were from Genzyme; recombinant mouse IL-12 p40 monomer, IL-12, recombinant human IL-12 p40 monomer and IL-12, hamster anti–mouse ICAM-1 IgG mAb (clone 3E2), and rat anti–mouse Mac-3 mAb (clone M3/84) were from BD PharMingen; recombinant murine IL-12 p40 homodimer and goat anti–human IL-12 pAb were from R&D Systems; rabbit anti–human IL-12 p35 Ab and goat anti–mouse IL-12 p40 Ab were from Santa Cruz Biotechnology, Inc.; rat anti–IL-12 p40 IgG2a hybridoma C17.8 was a gift of G. Trinchieri (Wistar Institute, Philadelphia, PA); mouse anti-cytokeratin IgG1 mAb (clone C-11) and rat nonimmune IgG were from Sigma-Aldrich; goat anti–mouse IgG F(ab′)2 conjugated with Cy3, rabbit anti-Sp1 Ab, goat anti–armenian hamster IgG horseradish peroxidase conjugate and sheep anti–rabbit IgG horseradish peroxidase conjugate were from Jackson ImmunoResearch Laboratories; rat anti-Sendai Ab was from BioReliance; sheep anti–rabbit IgG and streptavidin horseradish peroxidase conjugates were obtained from Boehringer; and biotinylated rabbit anti–goat IgG, biotinylated goat anti–rabbit IgG, biotinylated rabbit anti–rat IgG, streptavidin horseradish peroxidase conjugate, and streptavidin alkaline phosphatase conjugate were from Vector Laboratories.
Procurement and Housing of Mice.
Wild-type C57BL/6J and same-strain IFN-γ–, IL-12 p40–, and IL-12 p35–deficient mice 24 25 26 were obtained from The Jackson Laboratory and were maintained under pathogen-free conditions in the University biohazard barrier facility in microisolator cages for study at 7–9 wk of age. Sentinel mice (specific pathogen-free ICN strain) and experimental control mice were handled identically to inoculated mice and exhibited no serologic or histologic evidence of exposure to 11 rodent pathogens, including Sendai virus (SdV).
Isolation and Characterization of Mouse Tracheal Epithelial Cells.
To isolate mouse tracheal epithelial (mTE) cells, mice were anesthetized with intraperitoneal injection of ketamine (80 mg/kg) and xylazine (16 mg/kg), and subjected to anterior neck dissection, insertion of a 22-gauge angiocatheter (Baxter Healthcare Corporation) into the trachea below the cricoid cartilage, and instillation of 1 ml of Laboratory of Human Carcinogenesis (LHC)-8E medium containing 0.1% Pronase E (Sigma-Aldrich). Proximal and distal trachea were ligated, and the trachea was removed and then incubated in PBS containing penicillin/streptomycin at 4°C for 12 h. The distal ligature was removed, and dissociated cells were collected, subjected to hypotonic lysis, washed in PBS, and transferred to LHC-8E medium. Isolated cells were >98% positive for cytokeratin immunostaining and exhibited characteristic respiratory epithelial features by transmission electron microscopy 27.
Analysis of Epithelial Cell Levels of ICAM-1.
Isolated mTE cells were treated without or with murine IFN-γ (0–1,000 μ/ml) or TNF-α (100 μ/ml) for 0–24 h and lysed in 50 mM Tris, pH 8.0, 150 mM sodium chloride, 0.5% Nonidet P-40, 1 mM EDTA, 1 mM PMSF, 25 mM iodoacetamide, 1 mM sodium orthovanadate, 10 mM sodium fluoride, 2 mM sodium pyrophosphate, 10 mg/ml leupeptin, and 10 mg/ml aprotinin. Whole cell protein was boiled for 5 min and subjected to SDS-PAGE with 7% polyacrylamide. Protein was electrophoretically transferred to a polyvinylidine difluoride (PVDF) membrane that was then immunoblotted with hamster anti–mouse ICAM-1 IgG mAb (1.0 μg/ml for 1 h at 37°C) followed by incubation with goat anti–armenian hamster IgG horseradish peroxidase conjugate (0.16 μg/ml for 1 h at 25°C) and detection with enhanced chemiluminescence (Amersham Pharmacia Biotech). To document ICAM-1 specificity and equal protein loading, the membrane was stripped and then reblotted using rabbit anti-Sp1 pAb (1 mg/ml for 1 h at 25°C) and sheep anti–rabbit IgG horseradish peroxidase conjugate (0.01 μg/ml for 1 h at 25°C). Image acquisition and densitometry were performed using a UMAX Power Look II scanner (UMAX Data Systems, Inc.) and Gel-Doc 1000 image analyzer (Bio-Rad Laboratories).
Intratracheal Cytokine Injection and Characterization of Response.
Mice were anesthetized and then injected intratracheally using a 29-gauge needle and microinjection syringe (Hamilton) containing PBS (30 μl) without or with murine IFN-γ or TNF-α (75 μg/kg) for a total of four injections at 3-h intervals. The response to cytokines was assessed at 12 h after the last injection using immunohistochemistry and analysis of bronchoalveolar lavage (BAL) fluid.
For immunohistochemistry, trachea or lung (at 25-cm water pressure) was fixed in 10% formalin, dehydrated in ethanol, embedded in paraffin, and cut into 5-μM thick sections. For ICAM-1 immunostaining, tissue sections were blocked with nonimmune goat serum and then incubated sequentially with hamster anti–mouse ICAM-1 IgG mAb 3E2 (2.0 μg/ml for 18 h at 4°C), biotinylated goat anti–hamster IgG Ab (7.5 μg/ml for 30 min at 25°C), streptavidin-conjugated horseradish peroxidase, and 3,3′-diaminobenzidine (Vector Laboratories). For IL-12 p40, tissue sections were blocked with nonimmune rabbit serum and then incubated with goat anti–mouse IL-12 p40 IgG Ab (2.0 μg/ml for 18 h at 4°C), biotinylated rabbit anti–goat IgG, streptavidin-conjugated alkaline phosphatase complex, and red chromogen. For IL-12 p35, tissue sections underwent antigen retrieval as described previously 9 and then were blocked with nonimmune goat serum followed by incubation with rabbit anti–human IL-12 p35 IgG Ab (1 μg/ml for 18 h at 4°C), biotinylated goat anti–rabbit IgG Ab, streptavidin-conjugated alkaline phosphatase complex, and red chromogen. The same protocol was followed for IL-12 p35 Ab peptide competition experiments, except that rabbit anti–human IL-12 p35 IgG Ab (1 μg/ml) was first incubated with 50 M excess recombinant human IL-12 or IL-12 p40 (for 1 h at 25°C). Tissue sections were counterstained with hematoxylin, dehydrated in graded ethanol, and mounted for viewing in a photomicrography system (model D-7082; Carl Zeiss, Inc.). To quantify ICAM-1 and IL-12 p40 immunostaining, tissue section images were transferred to an image analysis program (Optimas) that was set to calculate brown or red intensity relative to the same-sized reference area (in cartilage or subepithelium) as described previously 9. Intensity was calculated for three different areas of epithelium and averaged to obtain a value for each tissue section.
For analysis of BAL fluid, mice underwent intraperitoneal anesthesia followed by anterior neck dissection and cannulation of the trachea with a 22-gauge angiocatheter as described above. BAL was performed with three aliquots of 0.8 ml of sterile PBS, and the BAL fluid was subjected to centrifugation, and the supernatant and cell pellet used for determinations of cytokine levels and cell differentials, respectively. Murine IL-12, IFN-γ, and IL-12 p40 were measured in duplicate using ELISA kits (R&D Systems) with sensitivities of 2.5 pg/ml for IL-12, 2 pg/ml for IFN-γ, and 4 pg/ml for IL-12 p40 with 20% cross-reactivity for IL-12. Accordingly, the BAL fluid was concentrated 10-fold using a Centricon-10 filter (Amicon) before analysis.
Viral Infection of Mice and Characterization of Response.
SdV, strain 52, was obtained from American Type Culture Collection and stored at −70°C. After anesthesia, mice were inoculated intranasally with the indicated dose (50% egg infectious dose [EID50]) of SdV or with UV-inactivated SdV diluted in 30 μl PBS. After inoculation, mice underwent daily inspection and body weight measurement, and at indicated intervals were used to perform immunohistochemistry and BAL fluid analysis as described above. However, in these experiments we also monitored SdV protein in tissue sections that were blocked with nonimmune rabbit serum and then incubated with rat anti-SdV Ab (1:750 vol/vol for 18 h at 4°C), biotinylated rabbit anti–rat IgG Ab, streptavidin-conjugated alkaline phosphatase complex, and red chromogen. In these experiments, the BAL fluid (without concentration) was subjected to additional analyses of TNF-α levels by ELISA (with a sensitivity of 5 pg/ml) and IL-12 by Western blotting. For immunoblotting, cell-free BAL fluid was subjected to 10% PAGE under nonreducing or reducing (10 mM dithiothreitol [DTT]) conditions, and protein was transferred to PVDF membrane for blotting against biotinylated rat anti–mouse IL-12 p40 mAb or control anti-IgG Ab (10 μg/ml for 1 h at 25°C) followed by incubation with streptavidin-conjugated horseradish peroxidase (0.5 U/ml for 1 h at 25°C) and detection with enhanced chemiluminescence. In these experiments, the BAL fluid cell pellet was also subjected to cytocentrifugation, methanol fixation, and Wright-Giemsa staining to obtain differential cell counts as the mean of values from two to three blinded observers, and tissue sections were also used to assess macrophage accumulation using rat anti–mouse Mac-3 mAb (1 μg/ml for 18 h at 4°C), biotinylated rabbit anti–rat IgG, streptavidin-conjugated horseradish peroxidase, and 3′3′-diaminobenzidine. To quantitate epithelial macrophages, two blinded observers counted Mac-3–positive cells per 1 mm of epithelial basement membrane in 10 randomly chosen airways from 3 animals in each cohort. For IL-12 p40 blocking experiments, wild-type or IL-12 p35 (−/−) cohorts underwent treatment with control rat nonimmune IgG or rat anti–IL-12 p40 IgG mAb that was purified from athymic nude mouse ascites fluid using protein G affinity chromatography. Preliminary experiments indicated that neutralization of IL-12 p40 levels in BAL fluid was accomplished by treatment with anti–IL-12 p40 mAb given by intraperitoneal injection on postinfection days 2 and 6 (1 mg of mAb in 0.66 ml PBS per injection).
Human Tissue Procurement and Analysis.
Healthy control, asthma, and chronic bronchitis subjects were recruited, characterized, and subjected to endobronchial biopsy and BAL as described previously 7 9. In brief, subjects were recruited using informed consent for a protocol approved by the University Committee for Human Studies and were characterized by spirometry, airway reactivity to inhaled methacholine, and skin test reactivity as summarized below in Table . Control subjects had no clinical history of lung disease and normal spirometry and airway reactivity, whereas asthma and chronic bronchitis subjects met clinical diagnostic criteria for those diseases 28, and asthmatic subjects had hyperreactivity to inhaled methacholine. To further determine the effect of glucocorticoid treatment, a group of asthmatic subjects were treated with inhaled fluticasone (1,760 μg/d) for 30 d before an initial bronchoscopy/BAL; fluticasone was then discontinued and subjects were monitored for an additional 6 wk or until peak expiratory flow had decreased by 25% or forced expiratory volume in 1 s by 15% at which time a second bronchoscopy/BAL was performed. For all subjects, there was no history of respiratory infection for the previous 3 mo. Endobronchial biopsies were obtained using fiberoptic bronchoscopy and then were washed with PBS and incubated with 10% neutral buffered formalin for 18 h at 25°C, followed by graded ethanol dehydration, paraffin embedding, and cut into 5-μm thick sections for immunostaining of IL-12 p40 and p35 using anti–human IL-12 p40 and p35 Abs as described above. BAL fluid was concentrated 35-fold using a Centriprep YM-3 concentrator (Millipore) and then used to determine levels of IL-12 p40 and p70 by ELISA kits from R&D Systems with sensitivities of 15 pg/ml for IL-12 p40 and 0.5 pg/ml for IL-12 p70 and cross-reactivities of 2.5 and <0.1%, respectively. Concentrated BAL fluid also underwent immunoprecipitation with goat anti–human IL-12 p40 IgG or nonimmune goat IgG (2 μg/ml for 4 h at 4°C), incubation with protein G Sepharose (Amersham Pharmacia Biotech) for 16 h at 4°C, and Western immunoblotting under reducing or nonreducing conditions with biotinylated anti–human IL-12 mAb (5.0 μg/ml for 1 h at 25°C) followed by incubation with streptavidin horseradish peroxidase conjugate (0.16 μg/ml for 1 h at 25°C) and detection with enhanced chemiluminescence (Amersham Pharmacia Biotech). Total BAL cell count was determined using a hemocytometer chamber and the BAL fluid cell pellet was subjected to cytocentrifugation, methanol fixation, and Wright-Giemsa staining to obtain differential cell counts.
Table 1.
Characteristics of Subjects
| Characteristic | Normal control subjects (n = 17) | Asthma subjects (n = 18) | Chronic bronchitis subjects (n = 8) |
|---|---|---|---|
| Age (yr) | |||
| Mean | 32 | 35 | 62 |
| Range | 23–54 | 19–65 | 21–76 |
| Sex (male/female) | 11/6 | 7/11 | 4/4 |
| Atopy | 3 | 13 | ND |
| Glucocorticoid treatment | 0 | 13 | 1 |
| FEV1 (liter/min) ± SD | 4.01 ± 0.66 | 2.82 ± 0.85 | 1.27 ± 0.81 |
| Percent predicted | 104 ± 15 | 86 ± 22 | 47 ± 15 |
| Range of percent predicted | 81–129 | 42–114 | 41–69 |
| FEV1 PC20 (mg/ml) ± SD | >16 | 1.38 ± 1.74 | ND |
| Range | >16 | 0.047–5.71 | ND |
FEV1, forced expiratory volume in 1 s; FEV1 PC20, provocative concentration of methacholine required to cause a 20% decrease in baseline FEV1; ND not determined.
Statistical Analysis.
Values for cytokine- or virus-dependent changes were analyzed for statistical significance using a one-way analysis of variance (ANOVA) for a factorial experimental design. The multicomparison significance level for the one-factor ANOVA was 0.05. If significance was achieved by one-way analysis, post-ANOVA comparison of means were performed using Scheffe's F test. Mouse survival rates were analyzed using the Wilcoxon rank-sum test with a significance level of 0.05. Levels of IL-12 in BAL fluid from normals and asthmatics were analyzed using the two-sample, independent groups t test with a significance level of 0.05 (two-tailed) and a paired t test with a significance level of 0.05 (two-tailed) when comparing treatment with and without glucocorticoids. Correlation between BAL levels of IL-12 p40 and macrophages was analyzed using the t test for correlation with a significance level of 0.05 (two-tailed).
Results
TNF-α Drives IL-12 that in turn Drives IFN-γ Production to Achieve Epithelial ICAM-1 Induction.
In studies of primary culture human airway epithelial cells and endobronchial explants, we found that apical and basolateral ICAM-1 expression is required for leukocyte adhesion and transmigration and is highly sensitive to IFN-γ but poorly responsive to TNF-α (references 4, 5, 7, 9, 10, and 12 and unpublished observations). For these experiments, we first verified that this profile of cytokine responsiveness for ICAM-1 gene expression was similar in isolated mTE cells (Fig. 1 A).
Figure 1.


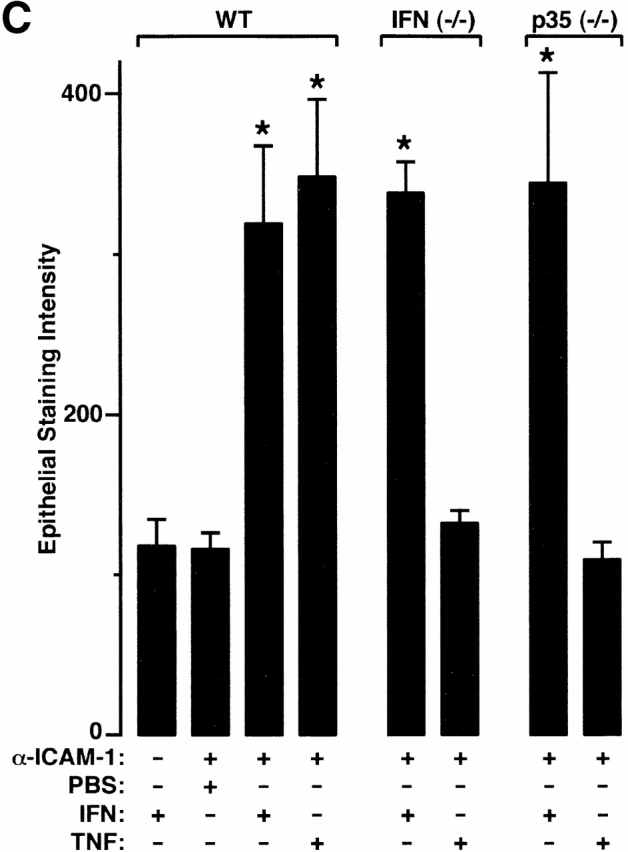
TNF-α responsiveness of epithelial ICAM-1 gene expression is not found in vitro (A) but is found in vivo (B and C), where it depends on IL-12 and IFN-γ production. In A, airway epithelial (mTE) cells were treated with IFN-γ (0–1,000 U/ml for 24 h and 100 U/ml for 0–24 h) or with IFN-γ vs. TNF-α (100 U/ml for 24 h). For each condition, cell lysates were subjected to Western blotting with anti–ICAM-1 mAb detected by enhanced chemiluminescence. Equality of protein loading and specificity was demonstrated by reblotting against anti-Sp1 mAb (data not shown). For each condition, fold increase above control was determined using densitometry, and values represent mean ± SEM for three experiments. A significant increase from untreated control value (by ANOVA) is indicated by (*). In B, wild-type (WT), IFN-γ–deficient (−/−), and IL-12 p35 (−/−) mice (all in C57BL/6J background) underwent intratracheal injection of IFN-γ or TNF-α. At 12 h after treatment, tracheal sections were immunostained with anti–ICAM-1 mAb, biotinylated goat anti–hamster IgG, streptavidin-conjugated horseradish peroxidase, and 3,3′-diaminobenzidine, and then counterstained with hematoxylin. Wild-type, IFN-γ (−/−), and IL-12 p35 (−/−) mice injected with PBS vehicle exhibited levels of epithelial ICAM-1 similar to background (data not shown). Immunostaining with control nonimmune IgG also gave no signal above background (data not shown). Bar, 20 μm. In C, tracheal sections from conditions in B underwent quantification of epithelial ICAM-1 immunostaining relative to a cartilage reference set at a value of 100. For each condition, values represent mean ± SEM for three experiments, and a significant increase from PBS-treated wild-type cohort is indicated by (*).
We then extended these observations to an in vivo system by intratracheal injection of cytokines in mice using a schedule that depended on the minimal doses of IFN-γ and TNF-α required to induce epithelial ICAM-1 expression by tissue immunostaining. In this system, treatment with both IFN-γ and TNF-α resulted in equivalent ICAM-1 induction by immunostaining (Fig. 1b and Fig. c) or immunoblot of tracheal tissue (data not shown). The pattern of ICAM-1 expression on both apical and basolateral surfaces was similar among groups and to one that we have described previously using laser scanning confocal microscopy 7 9. Because TNF-α did not induce epithelial ICAM-1 in vitro, we reasoned that its effectiveness in vivo might depend on direct or indirect stimulation of IFN-γ production. This possibility was confirmed when we found that TNF-α induction of epithelial ICAM-1 was blocked in mice deficient in IFN-γ (Fig. 1b and Fig. c). In this case, TNF-α was still capable of stimulating ICAM-1 expression on capillary endothelium consistent with TNF-α responsiveness of isolated endothelial cells 4 5.
In other systems, especially Th1 cell development, it appears that IFN-γ production may depend on stimulation by macrophage-derived IL-12 29 30. Although TNF-α–driven IL-12 production leading to IFN-γ generation has not been clearly ordered in vivo, we reasoned that this sequence might lead to epithelial ICAM-1 expression in the present model. This possibility was first supported by the finding that TNF-α induction of epithelial ICAM-1 was blocked in mice rendered deficient in IL-12 production by disruption of the IL-12 p35 gene (Fig. 1b and Fig. c). To next define whether TNF-α induction of IL-12 release was upstream of IFN-γ production, we determined levels of IL-12 and IFN-γ in wild-type versus IL-12 p35–deficient mice. We found that TNF-α–driven induction of IL-12 release proceeded as expected in the IFN-γ–deficient mouse, whereas induction of IFN-γ was blocked in the IL-12 p35–deficient mouse (Fig. 2). Taken together, these findings support the possibility that TNF-α initiates a cytokine cascade that includes initial IL-12 followed by IFN-γ release to achieve target gene (in this case, ICAM-1) expression on airway epithelial cells. In these experiments, it appears that induction of IL-12, at least as assessed in BAL fluid, is induced to a level that overcomes any antagonistic action of endogenous IL-12 p40 (Fig. 2).
Figure 2.

TNF-α induction of IL-12 expression drives downstream production of IFN-γ. Wild-type (WT), IL-12 p35 (−/−), and IFN-γ (−/−) C57BL/6J mice were treated with vehicle alone (PBS) or TNF-α as described in the legend to Fig. 1, followed 12 h later by BAL. The BAL fluid was concentrated 10-fold and used for duplicate measurements of IL-12 and IFN-γ levels by ELISA. Values represent mean ± SEM (n = 4). Levels of IL-12 were undetectable in IL-12 p35 (−/−) mice. A significant increase from PBS-treated wild-type cohort (by ANOVA) is indicated by (*).
TNF-α and Viral Tracheobronchitis Selectively Induce IL-12 p40 Expression in Airway Epithelial Cells.
In other systems, IL-12 is selectively produced by immune cells, especially antigen-presenting cells, and production depends on induction of the IL-12 p40 subunit in the context of constitutive p35 expression 16. However, when we submitted tracheal and lung tissue from TNF-α–treated mice to immunohistochemistry, we found that airway epithelial cells were the predominant site of induction of IL-12 p40 expression (Fig. 3). We also found constitutive IL-12 p35 expression in airway epithelial as well as other parenchymal and immune cells (Fig. 3). To next determine whether a more natural stimulus of airway inflammation might cause similar upregulation of IL-12 expression, we examined the response to an inoculum of Sdv (5,000 EID50) that causes reversible tracheobronchitis and bronchiolitis with transient epithelial expression of SdV protein (that is maximal at day 5 and undetectable by day 8 after viral inoculation) and mononuclear cell influx that is limited to the adjacent bronchovascular tissue compartment (Fig. 4, and data not shown). In this setting, we found that induction of IL-12 p40 was again localized to airway epithelial cells, rather than adjacent mononuclear cells, and was colocalized (temporally and spatially) with SdV protein expression, whereas constitutive expression of IL-12 p35 remained unchanged in all cell populations (Fig. 4). Thus, airway epithelial cells (rather than immune cells) appear to be the major cellular source for IL-12 p40 production during airway inflammatory conditions initiated by TNF-α administration or respiratory viral infection.
Figure 3.

TNF-α induction of IL-12 p40 without a change in constitutive p35 expression in airway epithelial cells. In A, wild-type (WT) and same-strain IFN-γ (−/−) C57BL/6J mice were treated with PBS vehicle or TNF-α as described in the legend to Fig. 1. At 12 h after treatment, tracheal (rows 1 and 3) or bronchial (rows 2 and 4) tissue was fixed in formalin, blocked with nonimmune goat or rabbit serum, and then incubated with anti–IL-12 p40 or p35 Ab. For p35 immunostaining, tissues were also subjected to antigen retrieval (using 10 mM Citra solution for 10 min at 98°C). Primary Ab binding was detected by incubation with biotinylated rabbit anti–goat or goat anti–rabbit IgG, streptavidin-conjugated alkaline phosphatase complex, and a red chromogenic substrate, and tissues were counterstained with hematoxylin. Control goat nonimmune IgG gave no signal above background (data not shown). In B, wild-type C57BL/6J mice were treated with PBS as described in the legend Fig. 1. Tracheal tissue sections were incubated with control rabbit nonimmune IgG, anti–IL-12 p35 pAb, or anti–IL-12 p35 pAb in the presence of recombinant murine IL-12 p40 or IL-12 followed by detection of primary Ab binding and hematoxylin counterstaining as described in A. Bars, 20 μm.
Figure 4.

Viral induction of IL-12 p40 without a change in constitutive p35 expression in airway epithelial cells. Wild-type C57BL/6J mice underwent intranasal inoculation with SdV (5,000 EID50 in 30 μl of PBS), and lungs were removed and fixed in formalin on day 1, 3, 5, and 8 after inoculation. In each case, tissue was immunostained for IL-12 P-40 and p35 as described in the legend to Fig. 3 and similarly for viral protein (labeled SdV) using rat anti-SdV pAb. In mice inoculated with UV-inactivated Sdv, IL-12 p40 immunostaining was not detected and IL-12 p35 constitutive immunostaining was unchanged from untreated control mice (not shown). Control rat, goat, or rabbit nonimmune IgG gave no signal above background (data not shown). Bar, 20 μm.
Early Induction of Epithelial IL-12 Followed by IL-12 p40 Homodimer Expression during Viral Bronchitis.
As noted above, IL-12 production is often (but not always) limited by production of IL-12 p40 16 31, so we expected these measurements to track together (as was the case for TNF-α stimulation experiments noted above). Indeed, this appeared to be the case at early times after viral inoculation (e.g., day 1 and 3; Fig. 5 A) when concentrations of IL-12 p40 were relatively low (<50 pg/ml) and similar to the range that we observed for TNF-α stimulation experiments. In addition, however, at later times after SdV inoculation (e.g., day 5 and 8; Fig. 5 A), we observed a marked increase in IL-12 p40 relative to IL-12 levels, so that the ratio of IL-12 p40/p70 was in excess of 75:1 (Fig. 5 A). Western blots of BAL fluid indicated that a significant proportion of IL-12 p40 existed as the IL-12 p40 homodimer (designated IL-12 p80; Fig. 5 B).
Figure 5.
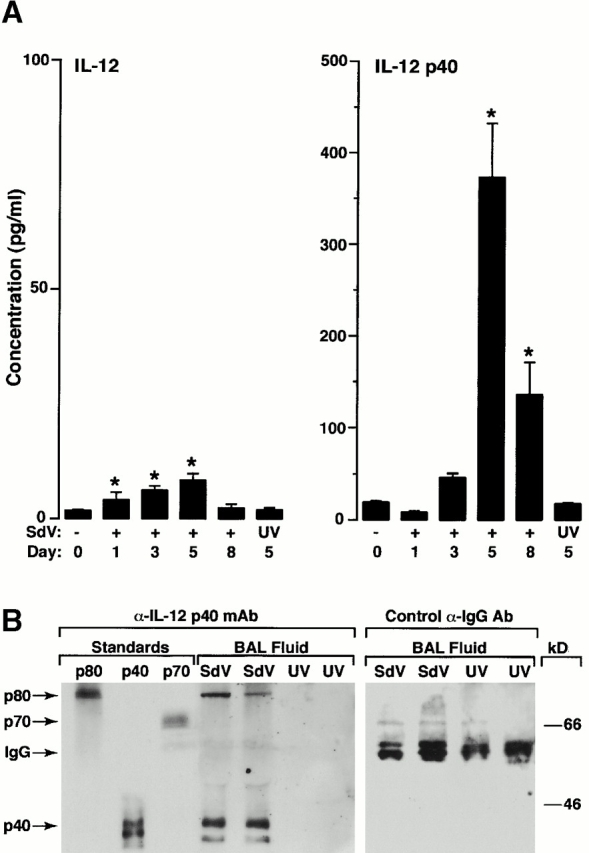
Early induction of IL-12 p70 followed by predominant release of IL-12 p40 into the airway during viral bronchitis. Wild-type mice were inoculated with SdV or UV-inactivated SdV as described in the legend to Fig. 4. In A, at the indicated times after inoculation, BAL fluid was obtained for duplicate measurements of IL-12 and IL-12 p40 levels by ELISA. All values represent mean ± SEM (n = 4). A significant increase from PBS-treated cohort or UV-activated SdV cohort (by ANOVA) is indicated by (*). In B, BAL fluid from day 5 after inoculation was subjected to Western blotting against anti–IL-12 p40 mAb under nonreducing conditions or control anti–mouse IgG Ab under reducing conditions with detection by enhanced chemiluminescence. Bands corresponding to IL-12 p40 homodimer (p80), IL-12 (p70), IL-12 p40 monomer (p40), and mouse IgG are indicated by arrows.
IL-12 p40 Overproduction and Macrophage Accumulation Linked to Viral Bronchitis.
To define the roles for IL-12 and IL-12 p40 homodimer production during viral bronchitis, we compared wild-type mice to mice rendered deficient in IL-12 p35 (but still capable of IL-12 p40 and p80 generation) versus mice deficient in IL-12 p40 (and so incapable of generating IL-12 or functional IL-12 p40). After SdV inoculation at 5,000 EID50 we found a trend towards greater weight loss in the p35-deficient animals (data not shown), and at 50,000 EID50 we observed an increased mortality rate and a persistent viral pneumonia in IL-12 p35 (−/−) mice compared with wild-type and IL-12 p40 (−/−) mice (Fig. 6). We found no difference in susceptibility to infection between wild-type and IL-12 p40 (−/−) mice, and no differences in viral persistence or histologic features of viral pneumonia between these two groups of mice (Fig. 6, and data not shown). Similarly, we found no difference in IFN-γ levels in BAL fluid in the presence or absence of IL-12 or IL-12 p40 (Fig. 7 A), indicating that SdV infection triggers IFN-γ production pathways that do not depend on IL-12 but may instead depend on IFN-α/β 32. However, even if there were differences in IFN-γ production, we have also found that IFN-γ (−/−) mice exhibit no increase in susceptibility in this model 33. In these same groups of mice, we found concomitant virus-inducible release of TNF-α (Fig. 7 A), suggesting (as noted above) that TNF-α may help drive IL-12 p40 gene expression in this setting.
Figure 6.

Decreased survival from viral bronchopneumonia in IL-12 p35–deficient mice. In A, wild-type (WT), and IL-12 p35 (−/−) and p40 (−/−) C57BL/6J mice were inoculated with SdV (50,000 EID50) and monitored for survival by Kaplan-Meier analysis (n = 29, 19, and 27 in each group, respectively). A significant decrease in survival (by Wilcoxon rank-sum test) is indicated by (*). In B, the same cohorts were inoculated with SdV (50,000 EID50), and lungs were removed at days 7, 9, and 10 after inoculation for hematoxylin/eosin staining and photomicrography. Bar, 50 μm.
Figure 7.

Decreased survival from viral bronchopneumonia driven by overexpression of IL-12 p40 homodimer and macrophage accumulation. In A and B, wild-type (WT) and IL-12 p35 (−/−) and p40 (−/−) C57BL/6J mice were inoculated with PBS vehicle, UV-inactivated SdV, or SdV (50,000 EID50), and IFN-γ, TNF-α, and IL-12 p40 levels were determined in BAL fluid obtained at day 7 after inoculation. In C, wild-type and IL-12 p35 (−/−) and p40 (−/−) mice were inoculated with SdV (50,000 EID50) and differential cell counts were determined in BAL fluid obtained at day 7 after inoculation. In A–C, values represent mean ± SEM (n = 4), and a significant difference from the wild-type cohort is indicated by (*). In D, wild-type, IL-12 p35 (−/−), and IL-12 p40 (−/−) mice were inoculated with SdV (50,000 EID50) and lungs at day 7 were removed and immunostained with anti-Mac3 mAb and counterstained with hematoxylin. Control nonimmune IgG gave no signal above background (not shown). Bar, 20 μm. Quantitation of macrophages per mm in length of basement membrane are provided as mean ± SEM (n = 5) for each condition.
The selective increase in mortality rate for the IL-12 p35 (−/−) mice indicated that IL-12 p40 exerts a biologic function in the absence of IL-12. Furthermore, postmortem histopathology indicated that organ abnormalities were confined to the lung (data not shown). To better define the mechanism for how IL-12 p40 expression (in the absence of IL-12) may lead to increased morbidity from SdV infection, we next determined the lung levels of IL-12 p40 under these conditions. We found that the increased mortality was associated with increased levels of IL-12 p40 in BAL fluid (Fig. 7 B) and serum (data not shown) relative to wild-type mice. These findings indicated that IL-12 and/or IL-12 p35 may prevent overexpression of IL-12 p40 (i.e., negative feedback). We note that the persistence of low levels of p40 in p40 null mice is likely due to the generation of a nonfunctional p40 fragment as described previously 25, as we observed SdV induction of this fragment at least at the mRNA level by reverse transcription PCR (data not shown). By contrast, IL-12 p80 exhibits selective macrophage chemoattractant activity in vitro and in vivo 34. Consistent with this observation, we detected a selective enrichment in macrophages in BAL fluid (Fig. 7 C) and accumulation of macrophages in bronchial and bronchiolar epithelium (Fig. 7 D) in IL-12 p35–deficient mice that overproduce IL-12 p80. Quantitation of tissue macrophages more precisely supported these findings (Fig. 7 D). Moreover, neutralization of IL-12 p40 and p80 (by treatment with anti–IL-12 p40 mAb) prevented the enhanced macrophage accumulation in the BAL fluid (Fig. 8) and reversed the increased mortality in IL-12 p35–deficient mice (from 12% survival after treatment with control IgG to 22% with anti–IL-12 p40 mAb compared with 25% in wild-type mice treated with control IgG; n = 8–16 mice per group). However, we note that all cohorts exhibit higher mortality rates when injected with mAb, IgG, or PBS compared with uninjected mice, likely reflecting the added stress of intraperitoneal injection procedures during viral bronchopneumonia. Taken together, it appears that epithelial overexpression of IL-12 p80 may cause macrophage accumulation and so contribute to airway inflammation and consequent morbidity during viral bronchitis.
Figure 8.
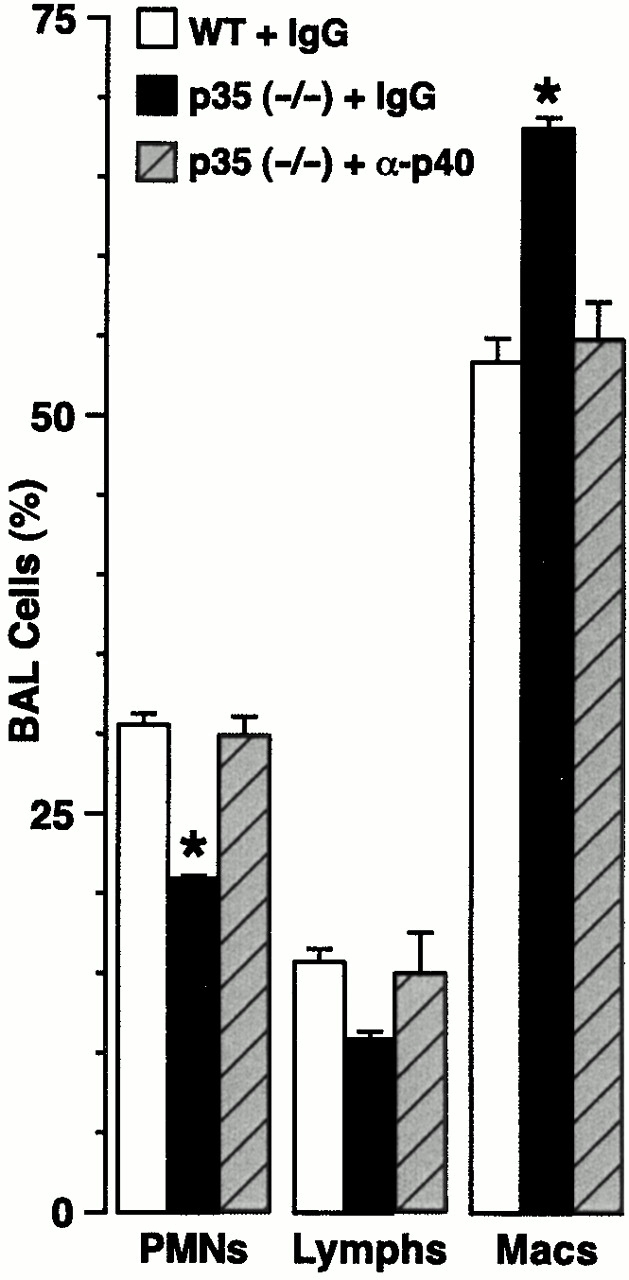
Inhibition of macrophage accumulation by anti–IL-12 p40 Ab treatment. Wild-type (WT) and IL-12 p35 (−/−) mice were inoculated with SdV (50,000 EID50) and treated with control rat IgG or rat anti–IL-12 p40 IgG (1 mg given intraperitoneally on postinoculation days 2 and 6), and differential cell counts were determined in BAL fluid obtained at day 7 after inoculation. Values represent mean ± SEM (n = 4), and a significant difference from the wild-type cohort treated with control IgG is indicated by (*).
Selective Induction of IL-12 p40 Expression in Airway Epithelial Cells in Asthmatic Subjects.
The results in mice suggest that overexpression of IL-12 p40 by the airway epithelium may lead to airway inflammation. As noted above, we have suggested that abnormal programming of epithelial immune response genes may serve as a basis for airway inflammation due to asthma 7 9. Accordingly, we next determined the level of IL-12 p40 and p35 expression in endobronchial biopsies and IL-12 and IL-12 p40 levels in BAL fluid obtained from normal versus asthma or chronic bronchitis subjects (characterized in Table ). In endobronchial biopsies, we found that airway epithelial cell IL-12 p40 expression was present in each of seven asthmatic but none of seven normal or eight chronic bronchitis subjects, whereas IL-12 p35 expression was constitutively expressed in airway epithelial, parenchymal, and inflammatory cells from all groups of subjects (Fig. 9). In addition, we found that increased epithelial IL-12 p40 expression in asthmatic subjects resulted in increased BAL fluid IL-12 p40 (but not IL-12 p70) concentrations that was unaltered by the concomitant administration of inhaled or systemic glucocorticoids (Fig. 10A and Fig. B). Additional experiments using a glucocorticoid withdrawal protocol to compare six asthmatic subjects to themselves with and without glucocorticoid treatment confirmed the lack of correlation between treatment status and IL-12 p40 levels in BAL fluid (20.5 ± 8.5 pg/ml and 31.4 ± 8.0 pg/ml with and without treatment, respectively; P > 0.05). Further analysis of BAL fluid samples indicated that IL-12 p40 appeared to be expressed predominantly as the homodimer (although background in concentrated BAL fluid is necessarily increased) and to correlate with macrophage accumulation (Fig. 10C and Fig. D). The asthmatic BAL fluid IL-12 p40/p70 ratio was elevated relative to normal subjects at a level (mean ratio of 221 ± 86) similar to the one found in viral bronchitis. Taken together, this data indicated that airway epithelial IL-12 p40 overexpression (particularly as the homodimer) may similarly contribute to airway inflammation in asthmatic subjects as it does during viral infection.
Figure 9.

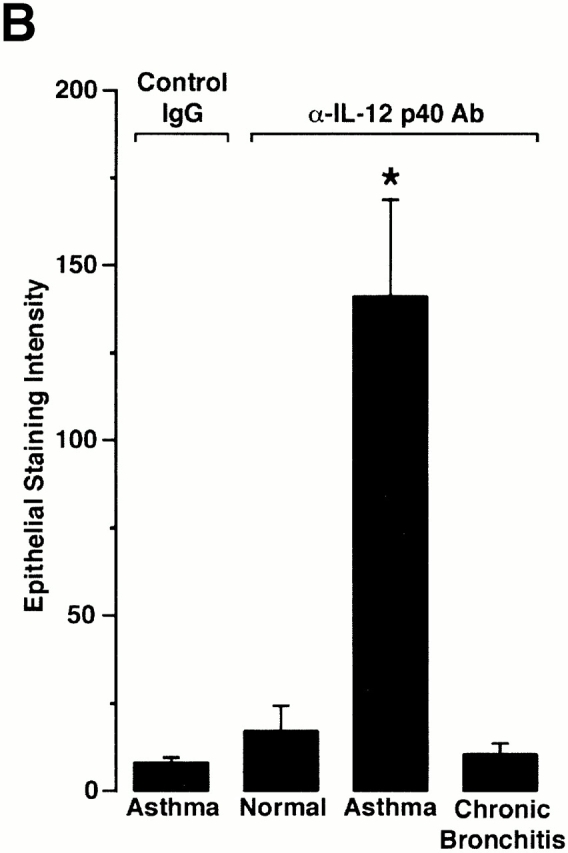
Selective induction of epithelial IL-12 p40 expression in asthmatic subjects. In A, endobronchial biopsies from normal control, asthmatic, and chronic bronchitis subjects were immunostained with control nonimmune IgG or with anti–IL-12 p40 or p35 Ab as described in the legend to Fig. 3. Bar, 20 μm. Representative photomicrographs of biopsies from seven control, seven asthmatic, and eight chronic bronchitis subjects are shown. In B, endobronchial biopsy sections from conditions in A underwent quantification of epithelial IL-12 p40 immunostaining relative to a subepithelial reference set at a value of 100. For each condition, values represent mean ± SEM for each cohort, and a significant increase from immunostaining with control nonimmune IgG is indicated by (*).
Figure 10.

Selective IL-12 p40 release in asthmatic subjects is independent of glucocorticoid (GC) treatment and associated with macrophage accumulation. In A and B, BAL fluid from 10 normal, 4 asthmatic (without glucocorticoid treatment,) and 7 asthmatic subjects (with glucocorticoid treatment) was concentrated 35-fold and used for duplicate measurements of IL-12 p70 and p40 levels by ELISA. Mean value for IL-12 p40 is represented by bold line and P values are indicated. In C, concentrated BAL fluid from normal and asthmatic subjects was subjected to immunoprecipitation and Western blotting against anti–IL-12 p40 Ab using nonreducing (top blot) or reducing (bottom blot) conditions and enhanced chemiluminescence detection. Bands corresponding to IL-12 (p70) and IL-12 P-40 monomer (p40) were identified using recombinant protein standards (Std) and are indicated by arrows. Immunoprecipitation with control nonimmune IgG gave no signal above background (data not shown). In D, BAL fluid levels of macrophages and IL-12 p40 were determined for each sample obtained from each asthmatic subject and then subjected to correlation analysis (r, correlation coefficient).
Discussion
IL-12 (p70) is produced by antigen-presenting cells (i.e., macrophages, dendritic cells, and B cells) in other settings, presumably because of restricted expression of the IL-12 p40 gene. Thus, original identification of IL-12 p40 in mice was limited to lymphoid tissue 35 and induction requires a signaling pathway that appropriately activates nuclear factor (NF)-κB 14 36 37 38 39 40. In turn, IL-12 action depends at least in part on its capacity to drive IFN-γ production and Th1 cell responses 41 42 43 44 45 46 47 48. Thus, mice deficient in IL-12 are unable to produce IFN-γ and exhibit a Th2 response and increased susceptibility to infection with certain bacteria, parasites, and viruses 25 26 29 32 49. Similarly, human subjects with inactivating mutations of the IL-12 receptor exhibit enhanced susceptibility to Mycobacterial and Salmonella infections, a phenotype also observed in subjects deficient in the IFN-γ receptor 50 51 52. These previous studies implied a critical role for IL-12 in Th1-dependent immunity, but did not yet define a distinct role for IL-12 p40. In fact, most evidence indicated that IL-12 p40 served only as a physiologic antagonist to downregulate excessive and potentially harmful IL-12 action 18 19 20 21 53 54 55 56. Two recent reports indicated that p40 may also act as an agonist to enhance alloantigen-specific Th1 development or to recruit macrophages in a tumor model system 34 57. However, these recent reports did not use genetically modified mice to define endogenous IL-12 p40 action in these settings, and none of the reports examined the behavior of IL-12 p40 in the setting of infection.
Here we describe induction of IL-12 p40 gene expression in response to TNF-α or respiratory paramyxoviral infection in mice and localize the predominant site of expression to the airway epithelial cell. We further demonstrate that overexpression of IL-12 p40 in this setting leads to virus-inducible airway inflammation in mice, and we define the same pattern of expression in association with airway inflammation in asthmatic subjects. These findings therefore have implications for IL-12 p40 gene expression in relation to antiviral immunity and inflammation, barrier epithelial cell function, and pathogenesis of asthma, and we discuss each separately.
In relation to antiviral immunity, we note that IL-12–dependent events have a variable role depending on the type of viral infection. The role of IL-12 in this setting depends on the relative dependence on CD8+ T cells, CD4+ T cells that provide variable help for B cells and CTL responses, and the direct effects of antiviral cytokines such as TNF-α and type I and II interferons. Depending on the type of virus, IL-12 treatment may or may not be protective, and conversely, IL-12–deficient mice may or may not show an increase in susceptibility to infection 32 49. In the case of Sdv, it appears that mucosal immunity may develop normally in the absence of IL-12 or IFN-γ production. These findings are consistent with the dominant role of class I MHC–restricted CD8+ T cells in clearing Sendai viral infection 58, but it is still possible that IL-12 has a protective role. Thus, IL-12 p40 homodimer has high affinity for the IL-12 receptor 14 54 59 and so functions as an efficient IL-12 antagonist. As protective effects of IL-12 may be distinct from those that allow for IFN-γ production 49, it is possible that these beneficial effects were lost in the presence of high levels of IL-12 p40 homodimer. In essence, wild-type mouse with high levels of p40 homodimer and consequent IL-12 antagonism would be rendered similar in IL-12 function to the IL-12 p40–deficient mouse. Thus, neither present nor previous studies adequately exclude a role for IL-12–dependent IFN-γ–independent events in mucosal immunity to viruses. As endogenous IL-12 may be derived from the viral host cell, in this case the airway epithelial cell, it appears that this cell population may still have an IL-12–dependent role in mediating innate immunity.
In addition to antagonist effects of IL-12 p40, especially as a homodimer, our report provides evidence of its action as an agonist in the setting of viral infection. Thus, excessive IL-12 p40 levels found in IL-12 p35–deficient mice led to macrophage accumulation in tissue and airspace compartments. Whether this abnormality is a direct effect of IL-12 p40 is not yet certain, but is consistent with IL-12 p40 homodimer (and IL-12) action as a selective chemoattractant for macrophages, especially because this action is additive rather than antagonistic with IL-12 34. It is uncertain whether IL-12 p40 also influences other macrophage functions for host defense and/or allergic disease, such as antigen presentation 60. However, this data indicate that the role of IL-12 p40 extends beyond one of IL-12 antagonism and in that capacity beyond one for triggering IFN-γ production.
The unusual action of IL-12 p40 in the setting of paramyxoviral bronchitis is coupled with the distinct cellular location. Thus, others have noted that IL-12 was expressed mainly by macrophages in viral infection 61, but in this study and others, the analysis was often restricted to recruited or circulating immune cells. For viruses trophic for hematopoietic or lymphoid cells, this approach may be appropriate, but in this case, direct examination of the viral host cell appears critical. In fact, the site of IL-12 gene expression may reflect direct activation by the virus. In other cell systems, it appears that expression of the murine and human IL-12 p40 genes depends in part on transcriptional activation via a conserved NF-κB half-site that binds NF-κB complexes 39 40 62. TNF-α blocking strategies (anti–TNF-α Ab and TNF-α receptor [−/−] mice) have demonstrated that TNF-α is necessary for efficient IL-12 p40 expression 36 37. TNF-α can also act to inhibit induction of IL-12, especially in the setting of IFN-γ priming 63 64 65. Taken together, it appears that TNF-α may participate in initiation as well as subsequent limitation of IL-12 production. Our report adds that TNF-α is sufficient for acute induction of IL-12 expression and subsequent action on IFN-γ production, but insufficient (in the case of intratracheal TNF-α administration or IL-12 p35 deficiency) to induce IL-12 p40 overexpression. Similarly, TNF-α generated during paramyxoviral infection may drive induction of epithelial IL-12 gene expression, but this pathway may not yet fully explain the selective expression in infected epithelial cells. Thus, whether TNF-α synergizes with other viral actions on the host cells or whether virus may directly activate the IL-12 p40 gene via innate signaling pathways as is the case for other epithelial immune-response genes 66 is still under study.
The possibility that epithelial cells are a source for IL-12 is not completely without precedent. Others have shown IL-12 p40 mRNA expression and IL-12 release in epidermal cell preparations from skin treated with trinitrochlorobenzene, and IL-12 appeared to be generated by keratinocytes, as it persisted when immune cells were depleted from the preparation 67. Similarly, IL-12 and IL-12 p40 mRNA were detectable in cultured keratinocytes after treatment with phorbol-12,13-dibutyrate or ultraviolet light as well as after herpes simplex infection 68 69 70. However, none of these reports provided in situ evidence of IL-12 production in epithelial cells, any analysis of relative levels of IL-12 or IL-12 p40, or any definition of function in vitro or in vivo. Nonetheless, it is possible that regulated production of IL-12 p40 may be a general property of epithelial barrier cells and mucosal immunity. In that regard, one group has noted that Abs to IL-12 abrogate experimental colitis in a mouse model of inflammatory bowel disease 71, but the cellular source and molecular characteristics of IL-12 production in this case or other cases of mucosal inflammation was not defined.
However, in this study we examined the further possibility that the pattern of IL-12 expression in murine viral bronchitis might be informative for mechanisms of airway inflammation in human subjects. In particular, we provide the first evidence that asthma, often characterized as a condition that depends on overexpression of Th2 and underexpression of Th1 cytokines by immune cells, does in fact also exhibit overexpression of IL-12 p40 that appears to be chiefly derived from airway epithelial cells. This finding offers two new possibilities for a role of IL-12 p40 in asthma: (a) antagonism of endogenous IL-12, and so skewing the local cytokine environment towards a Th2 immune response; or (b) function as an agonist, e.g., as a macrophage chemotactic factor, and so precipitating inappropriate airway inflammation. In fact, some (but not all) previous studies find significant macrophage accumulation in the submucosal and intraepithelial airway tissue of asthmatic compared with normal subjects 72 73, whereas others provide evidence of macrophage activation in asthma 74 as well as an increase in number and activation state of airway macrophages during allergen challenge in asthma 75.
Although the precise action and characteristics of IL-12 p40 in asthma requires further definition in experimental models, its expression further establishes a pattern of epithelial cell behavior in asthma. Thus, our previous work indicated that airway epithelial cells express at least two subsets of immune response genes (typified by ICAM-1 and RANTES) that appear critical for mucosal immunity (Walter, M.J., and M.J. Holtzman, unpublished observations; and references 4–7, 9, and 12). Each set of genes also appears to be overexpressed in asthma even in the absence of signs of a viral infection 7 9. The present results therefore further support an altered paradigm in which epithelial immune response genes are specially programmed for innate immunity and abnormally expressed in asthma. The results further raise the possibility that this component of the innate immune response is abnormally programmed for an antiviral response in this disease.
Acknowledgments
The authors gratefully acknowledge Guangshun Fan, Jill Roby, William Roswit, Theresa Tolley, and Donghui Xia for excellent technical assistance.
This research was supported by grants from the National Institutes of Health, American Lung Association, Martin Schaeffer Fund, and Alan A. and Edith L. Wolff Charitable Trust.
Footnotes
Abbreviations used in this paper: ANOVA, analysis of variance; BAL, bronchoalveolar lavage; EID50, 50% egg infectious dose; ICAM, intercellular adhesion molecule; mTE, mouse tracheal epithelial; NF, nuclear factor; RANTES, regulated upon activation, normal T cell expressed and secreted; SdV, Sendai virus.
References
- Medzhitov R., Janeway C.A., Jr. An ancient system of host defense. Curr. Opin. Immunol. 1998;10:12–15. doi: 10.1016/s0952-7915(98)80024-1. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Zhang V., Hussain H., Roswit W.T., Wilson J.D. Prostaglandin H synthase and lipoxygenase gene families in the epithelial cell barrier. In: Goetzl E.J., Lewis R.A., Rola-Pleszczynski M., editors. Cellular Generation, Transport and Effects of EicosanoidsBiological Roles and Pharmacological Intervention. Ann. NY. Acad. Sci; New York: 1995. pp. 58–77. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Castro M., Look D.C., O'Sullivan M., Walter M.J. Regulation of epithelial-leukocyte interaction and epithelial immune-response genes. In: Busse W., Holgate S., editors. Asthma and Rhinitis. Blackwell Scientific; Cambridge, MA: 2000. pp. 784–800. [Google Scholar]
- Look D.C., Keller B.T., Rapp S.R., Holtzman M.J. Selective induction of intercellular adhesion molecule-1 by interferon-γ in human airway epithelial cells. Am. J. Physiol. 1992;263:L79–L87. doi: 10.1152/ajplung.1992.263.1.L79. [DOI] [PubMed] [Google Scholar]
- Nakajima S., Look D.C., Roswit W.T., Bragdon M.J., Holtzman M.J. Selective differences in vascular endothelial- vs. airway epithelial-T cell adhesion mechanisms. Am. J. Physiol. 1994;267:L422–L432. doi: 10.1152/ajplung.1994.267.4.L422. [DOI] [PubMed] [Google Scholar]
- Nakajima S., Roswit W.T., Look D.C., Holtzman M.J. A hierarchy for integrin expression and adhesiveness among T cell subsets that is linked to TCR gene usage and emphasizes Vδ1+ γδ T cell adherence and tissue retention. J. Immunol. 1995;155:1117–1131. [PubMed] [Google Scholar]
- Taguchi M., Sampath D., Koga T., Castro M., Look D.C., Nakajima S., Holtzman M.J. Patterns for RANTES secretion and intercellular adhesion molecule-1 expression mediate transepithelial T cell traffic based on analyses in vitro and in vivo. J. Exp. Med. 1998;187:1927–1940. doi: 10.1084/jem.187.12.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M.J., Kajiwara N., Xia D., Holtzman M.J. Early-phase innate immunity and late-phase remodeling of the epithelium during primary viral bronchitis and hyperreactivity. J. Invest. Med. 1999;47:256A. [Google Scholar]
- Sampath D., Castro M., Look D.C., Holtzman M.J. Constitutive activation of an epithelial signal transducer and activator of transcription (Stat1) pathway in asthma. J. Clin. Invest. 1999;103:1353–1361. doi: 10.1172/JCI6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look D.C., Pelletier M.R., Holtzman M.J. Selective interaction of a subset of interferon-γ response element binding proteins with the intercellular adhesion molecule-1 (ICAM-1) gene promoter controls the pattern of expression on epithelial cells. J. Biol. Chem. 1994;269:8952–8958. [PubMed] [Google Scholar]
- Look D.C., Pelletier M.R., Tidwell R.M., Roswit W.T., Holtzman M.J. Stat1 depends on transcriptional synergy with Sp1. J. Biol. Chem. 1995;270:30264–30267. doi: 10.1074/jbc.270.51.30264. [DOI] [PubMed] [Google Scholar]
- Look D.C., Roswit W.T., Frick A.G., Gris-Alevy Y., Dickhaus D.M., Walter M.J., Holtzman M.J. Direct suppression of Stat1 function during adenoviral infection. Immunity. 1998;9:871–880. doi: 10.1016/s1074-7613(00)80652-4. [DOI] [PubMed] [Google Scholar]
- Stern A.S., Podlaski F.J., Hulmes J.D., Pan Y.C., Quinn P.M., Wolitzky A.G., Familletti P.C., Stremlo D.L., Truitt T., Chizzonite R., Gately M.K. Purification to homogeneity and partial characterization of cytotoxic lymphocyte maturation factor from human B-lymphoblastoid cells. Proc. Natl. Acad. Sci. USA. 1990;87:6808–6812. doi: 10.1073/pnas.87.17.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea A., Rengaraju M., Valiante N.M., Chehimi J., Kubin M., Aste M., Chan S.H., Kobayashi M., Young D., Nickbarg E. Production of natural killer cell stimulatory factor (interleukin 12) by peripheral blood mononuclear cells. J. Exp. Med. 1992;176:1387–1398. doi: 10.1084/jem.176.5.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macatonia S.E., Hosken N.A., Litton M., Vieira P., Hsieh C.S., Culpepper J.A., Wysocka M., Trinchieri G., Murphy K.M., O'Garra A. Dendritic cells produce IL-12 and direct the development of Th1 cells from naive CD4+ T cells. J. Immunol. 1995;154:5071–5079. [PubMed] [Google Scholar]
- Trinchieri G., Scott P. Interleukin-12basic principles and clinical applications. Curr. Top. Microbiol. Immunol. 1999;238:57–78. doi: 10.1007/978-3-662-09709-0_4. [DOI] [PubMed] [Google Scholar]
- Holtzman M.J., Look D.C., Iademarco M.F., Dean D.C., Sampath D., Castro M. Asthma. In: Jameson J.L., editor. Principles of Molecular Medicine. Humana Press; Totawa, NJ: 1998. pp. 319–327. [Google Scholar]
- Ling P., Gately M.K., Gubler U., Stern A.S., Lin P., Hollfelder K., Su C., Pan Y.-C. E., Hakimi J. Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J. Immunol. 1995;154:116–127. [PubMed] [Google Scholar]
- Kato K., Shimozato O., Hoshi K., Wakimoto H., Hamada H., Yagita H., Okumura K. Local production of the p40 subunit of interleukin-12 suppresses T-helper 1-mediated immune responses and prevents allogeneic myoblast rejection. Proc. Natl. Acad. Sci. USA. 1996;93:9085–9089. doi: 10.1073/pnas.93.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzel F.P., Hujer A.M., Ahmed F.N., Rerko R.M. In vivo production and function of IL-12 p40 homodimers. J. Immunol. 1997;158:4381–4388. [PubMed] [Google Scholar]
- Yoshimoto T., Wang C.-R., Yoneto T., Waki S., Sunaga S., Komagata Y., Mitsuyama M., Miyazaki J., Nariuchi H. Reduced T helper 1 responses in IL-12 p40 transgenic mice. J. Immunol. 1998;160:588–594. [PubMed] [Google Scholar]
- Gazzinelli R.T., Wysocka M., Hieny S., Scharton-Kersten T., Cheever A., Kuhn R., Muller W., Trinchieri G., Sher A. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-γ, and TNF-α. J. Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- Reis e Sousa C., Yap G., Schulz O., Rogers N., Schito M., Aliberti J., Hieny S., Sher A. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 1999;11:637–647. doi: 10.1016/s1074-7613(00)80138-7. [DOI] [PubMed] [Google Scholar]
- Dalton D.K., Pitts-Meek S., Keshav S., Figari I.S., Bradley A., Stewart T.A. Multiple defects of immune cell function in mice with disrupted interferon-γ genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- Magram J., Connaughton S.E., Warrier R.R., Carvajal D.M., Wu C., Ferrante J., Stewart C., Sarmiento U., Faherty D.A., Gately M.K. IL-12-deficient mice are defective in IFNγ production and type 1 cytokine responses. Immunity. 1996;4:471–481. doi: 10.1016/s1074-7613(00)80413-6. [DOI] [PubMed] [Google Scholar]
- Mattner F., Magram J., Ferrante J., Launois P., Di Padova K., Behin R., Gately M.K., Louis J.A., Alber G. Genetically resistant mice lacking interleukin-12 are susceptible to infection with Leishmania major and mount a polarized Th2 cell response. Eur. J. Immunol. 1996;26:1553–1559. doi: 10.1002/eji.1830260722. [DOI] [PubMed] [Google Scholar]
- Hunter J.A., Finkbeiner W.E., Nadel J.A., Goetzl E.J., Holtzman M.J. Predominant generation of 15-lipoxygenase metabolites of arachidonic acid by epithelial cells from human trachea. Proc. Natl. Acad. Sci. USA. 1985;82:4633–4637. doi: 10.1073/pnas.82.14.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meneely G.R., Renzetti A.D., Jr., Steele J.D., Wyatt J.P., Harris H.W. Definitions and classification of chronic bronchitis, asthma, and pulmonary emphysema. Am. Rev. Respir. Dis. 1962;85:762–768. [Google Scholar]
- Hsieh C.S., Macatonia S.E., Tripp C.S., Wolf S.F., O'Garra A., Murphy K.M. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science. 1993;260:547–549. doi: 10.1126/science.8097338. [DOI] [PubMed] [Google Scholar]
- Carter L.L., Murphy K.M. Lineage-specific requirement for signal transducer and activator of transcription (Stat)4 in interferon γ production from CD4+ versus CD8+ T cells. J. Exp. Med. 1999;189:1355–1360. doi: 10.1084/jem.189.8.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babik J.M., Adams E., Tone Y., Fairchild P.J., Tone M., Waldmann H. Expression of murine IL-12 is regulated by translational control of the p35 subunit. J. Immunol. 1999;162:4069–4078. [PubMed] [Google Scholar]
- Cousens L.P., Peterson R., Hsu S., Dorner A., Altman J.D., Ahmed R., Biron C.A. Two roads divergedinterferon α/β- and interleukin 12-mediated pathways in promoting T cell interferon γ responses during viral infection. J. Exp. Med. 1999;189:1315–1327. doi: 10.1084/jem.189.8.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter M.J., Kajiwara N., Sampath D., Rucker J., Xia D., Holtzman M.J. Epithelial immune-response gene expression during viral bronchitis and hyperreactivity in wild-type and IFN-γ-deficient mice. Am. J. Respir. Crit. Care Med. 1999;159:A437. [Google Scholar]
- Ha S.J., Lee C.H., Lee S.B., Kim C.M., Jang K.L., Shin H.S., Sung Y.C. A novel function of IL-12p40 as a chemotactic molecule for macrophages. J. Immunol. 1999;163:2902–2908. [PubMed] [Google Scholar]
- Schoenhaut D.S., Chua A.O., Wolitzky A.G., Quinn P.M., Dwyer C.M., McComas W., Familletti P.C., Gately M.K., Gubler U. Cloning and expression of murine IL-12. J. Immunol. 1992;148:3433–3440. [PubMed] [Google Scholar]
- Flesch I.E.A., Hess J.H., Huang S., Aguet M., Rothe J., Bluethmann H., Kaufman S.H.E. Early interleukin 12 production by macrophages in response to mycobacterial infection depends on interferon γ and tumor necrosis factor α. J. Exp. Med. 1995;181:1615–1621. doi: 10.1084/jem.181.5.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y., Cheers C. Control of IL-12 and IFN-γ production in response to live or dead bacteria by TNF and other factors. J. Immunol. 1998;161:1447–1453. [PubMed] [Google Scholar]
- Ma X., Aste-Amezaga M., Trinchieri G. Regulation of interleukin-12 production. Ann. NY Acad. Sci. 1996;795:13–25. doi: 10.1111/j.1749-6632.1996.tb52651.x. [DOI] [PubMed] [Google Scholar]
- Murphy T.L., Cleveland M.G., Kulesza P., Magram J., Murphy K.M. Regulation of interleukin-12 p40 expression through an NF-κB half-site. Mol. Cell. Biol. 1995;15:5258–5267. doi: 10.1128/mcb.15.10.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plevy S.E., Gemberling H.M., Hsu S., Dorner A.J., Smale S.T. Multiple control elements mediate activation of the murine and human interleukin 12 p40 promotersevidence of functional synergy between C/EBP and Rel proteins. Mol. Cell. Biol. 1997;17:4572–4588. doi: 10.1128/mcb.17.8.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbulescu J., Becker C., Schlaak J.F., Schmitt E., Buschenfelde K.M., Neurath M.F. IL-12 and IL-18 differentially regulate the transcriptional activity of the human IFN-γ promoter in primary CD4+ T lymphocytes. J. Immunol. 1998;160:3642–3647. [PubMed] [Google Scholar]
- Alzona M., Jack H.M., Simms P.E., Ellis T.M. Interleukin-12 activates interferon-γ production by targeted activation of CD30+ T cells. Ann. NY Acad. Sci. 1996;795:127–136. doi: 10.1111/j.1749-6632.1996.tb52661.x. [DOI] [PubMed] [Google Scholar]
- Chace J.H., Hooker N.A., Mildenstein K.L., Krieg A.M., Cowdery J.S. Bacterial DNA-induced NK cell IFN-γ production is dependent on macrophage secretion of IL-12. Clin. Immunol. Immunopathol. 1997;84:185–193. doi: 10.1006/clin.1997.4380. [DOI] [PubMed] [Google Scholar]
- Giese N.A., Gazzinelli R.T., Actor J.K., Morawetz R.A., Sarzotti M., Morse H.C. Retrovirus-elicited interleukin-12 and tumor necrosis factor-α as inducers of interferon-γ-mediated pathology in mouse AIDS. Immunol. 1996;87:467–474. doi: 10.1046/j.1365-2567.1996.492569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpern M.D., Kurlander R.J., Pisetsky D.S. Bacterial DNA induces murine interferon-γ production by stimulation of interleukin-12 and tumor necrosis factor-α. Cell. Immunol. 1996;167:72–78. doi: 10.1006/cimm.1996.0009. [DOI] [PubMed] [Google Scholar]
- Tripp C.S., Wolf S.F., Unanue E.R. Interleukin 12 and tumor necrosis factor α are costimulators of interferon γ production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc. Natl. Acad. Sci. USA. 1993;90:3725–3729. doi: 10.1073/pnas.90.8.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris S.C., Madden K.B., Adamovicz J.J., Gause W.C., Hubbard B.R., Gately M.K., Finkelman F.D. Effects of IL-12 on in vivo cytokine gene expression and Ig isotype selection. J. Immunol. 1994;152:1047–1056. [PubMed] [Google Scholar]
- Car B.D., Eng V.M., Schnyder B., LeHir M., Shakhov A.N., Woerly G., Huang S., Aguet M., Anderson T.D., Ryffel B. Role of interferon-γ in interleukin 12-induced pathology in mice. Am. J. Pathol. 1995;147:1693–1707. [PMC free article] [PubMed] [Google Scholar]
- van den Broek M., Bachmann M.F., Kohler G., Barner M., Escher R., Zinkernagel R., Kopf M. IL-4 and IL-10 antagonize IL-12-mediated protection against acute vaccinia virus infection with a limited role of IFN-γ and nitric oxide synthetase 2. J. Immunol. 2000;164:371–378. doi: 10.4049/jimmunol.164.1.371. [DOI] [PubMed] [Google Scholar]
- Newport M.J., Huxley C.M., Huston S., Hawrylowicz C.M., Oostra B.A., Williamson R., Levin M. A mutation in the interferon-γ-receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 1996;335:1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- Altare F., Durandy A., Lammas D., Emile J., Lamhamedi S., Le Deist F., Drysdale P., Jouanguy E., Doffinger R., Bernaudin F. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 1998;280:1432–1435. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- de Jong R.D., Altare F., Haagen I., Elferink D.G., de Boer T., van Breda Vriesman P.J.C., Kabel P.J., Draaisma J.M.T., van Dissel J.T., Kroon F.P. Severe mycobacterial and Salmonella infections in interleukin-12 receptor deficient patients. Science. 1998;280:1435–1438. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- Mattner F., Fischer S., Guckes S., Jin S., Kaulen H., Rude E., Germann T. The interleukin-12 subunit p40 specifically inhibits effects of the interleukin-12 heterodimer. Eur. J. Immunol. 1993;23:2202–2208. doi: 10.1002/eji.1830230923. [DOI] [PubMed] [Google Scholar]
- Gillessen S., Carvajal D., Ling P., Podlaski F.J., Stremlo D.L., Familletti P.C., Gubler U., Presky D.H., Stern A.S., Gately M.K. Mouse interleukin-12 (IL-12) p40 homodimera potent IL-12 antagonist. Eur. J. Immunol. 1995;25:200–206. doi: 10.1002/eji.1830250133. [DOI] [PubMed] [Google Scholar]
- Abdi K., Herrmann S.H. CTL generation in the presence of IL-4 is inhibited by free p40. J. Immunol. 1997;159:3148–3155. [PubMed] [Google Scholar]
- Chen L., Chen D., Block E., O'Donnell M., Kufe D.W., Clinton S.K. Eradication of murine bladder carcinoma by intratumor injection of a bicistronic adenoviral vector carrying cDNAs for the IL-12 heterodimer and its inhibition by the IL-12 p40 subunit homodimer. J. Immunol. 1997;159:351–359. [PubMed] [Google Scholar]
- Piccotti J.R., Chan S.Y., Li K., Eichwald E.J., Bishop D.K. Differential effects of IL-12 receptor blockade with IL-12 p40 homodimer on the induction of CD4+ and CD8+ IFN-γ-producing cells. J. Immunol. 1997;158:643–648. [PubMed] [Google Scholar]
- Hou S., Doherty P.C., Zijlstra M., Jaenisch R., Katz J.M. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J. Immunol. 1992;149:1319–1325. [PubMed] [Google Scholar]
- Wang X., Wilkinson V.I., Podlaski F.J., Wu C., Stern A.S., Presky D.H., Magram J. Characterization of mouse interleukin-12 p40 homodimer binding to the interleukin-12 receptor subunits. Eur. J. Immunol. 1999;29:2007–2013. doi: 10.1002/(SICI)1521-4141(199906)29:06<2007::AID-IMMU2007>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Burastero S., Magnani Z., Confetti C., Abbruzzese L., Oddera S., Balbo P., Rossi G., Crimi E. Increased expression of the CD80 accessory molecule by alveolar macrophages in asthmatic subjects and its functional involvement in allergen presentation to autologous TH2 lymphocytes. J. Allergy Clin. Immunol. 1999;103:1136–1142. doi: 10.1016/s0091-6749(99)70189-2. [DOI] [PubMed] [Google Scholar]
- Coutelier J.-P., van Broeck J., Wolf S.F. Interleukin-12 gene expression after viral infection in the mouse. J. Virol. 1995;69:1955–1958. doi: 10.1128/jvi.69.3.1955-1958.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Chow J.M., Gri G., Carra G., Gerosa F., Wolf S.F., Dzialo R., Trinchieri G. The interleukin-12 p40 gene promoter is primed by interferon-γ in monocytic cells. J. Exp. Med. 1996;183:147–157. doi: 10.1084/jem.183.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge-Dufour J., Marino M.W., Horton M.R., Jungbluth A., Burdick M.D., Strieter R.M., Noble P.W., Hunter C.A., Pure E. Inhibition of interferon γ induced interleukin 12 productiona potential mechanism for anti-inflammatory activities of tumor necrosis factor. Proc. Natl. Acad. Sci. USA. 1998;95:13806–13811. doi: 10.1073/pnas.95.23.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X., Sun J., Papasavvas E., Riemann H., Robertson S., Marshall J., Bailer R.T., Moore A., Donnelly R.P., Trinchieri G., Montaner L.J. Inhibition of IL-12 production in human monocyte-derived macrophages by TNF. J. Immunol. 2000;164:1722–1729. doi: 10.4049/jimmunol.164.4.1722. [DOI] [PubMed] [Google Scholar]
- Marino M.W., Dunn A., Grail D., Inglese M., Noguchi Y., Richards E., Jungbluth A., Wada H., Moore M., Williamson B., Basu S., Old L.J. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. USA. 1997;94:8093–8098. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga T., Sardina E., Tidwell R.M., Pelletier M.R., Look D.C., Holtzman M.J. Virus-inducible expression of a host chemokine gene relies on replication-linked mRNA stabilization. Proc. Natl. Acad. Sci. USA. 1999;96:5680–5685. doi: 10.1073/pnas.96.10.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller G., Saloga J., Germann T., Bellinghausen I., Mohamadzadeh M., Knop J., Enk A.H. Identification and induction of human keratinocyte-derived IL-12. J. Clin. Invest. 1994;94:1799–1805. doi: 10.1172/JCI117528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragane Y., Rieman H., Bhardwaj R.S., Schwarz A., Sawada Y., Yamada H., Luger T.A., Kubin M., Trinchieri G., Schwarz T. IL-12 is expressed and released by human keratinocytes and epidermoid carcinoma cell lines. J. Immunol. 1994;153:5366–5372. [PubMed] [Google Scholar]
- Kondo S., Jimbow K. Dose-dependent induction of IL-12 but not IL-10 from human keratinocytes after exposure to ultraviolet light A. J. Cell. Physiol. 1998;177:493–498. doi: 10.1002/(SICI)1097-4652(199812)177:3<493::AID-JCP12>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Miklosa Z., Danis V.A., Adams S., Lloyd A.R., Adrian D.L., Cuningham A.L. In vivo production of cytokines and β (C-C) chemokines in human recurrent herpes simplex lesions—do herpes simplex virus-infected keratinocytes contribute to their production? J. Infect. Dis. 1997;177:827–838. doi: 10.1086/515236. [DOI] [PubMed] [Google Scholar]
- Neurath M.F., Fuss I., Kelsall B.L., Stuber E., Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poston R.N., Chanez P., Lacoste J.Y., Litchfield T., Lee T.H., Bousquet J. Immunohistochemical characterization of the cellular infiltration in asthmatic bronchi. Am. Rev. Respir. Dis. 1992;145:918–921. doi: 10.1164/ajrccm/145.4_Pt_1.918. [DOI] [PubMed] [Google Scholar]
- Ollerenshaw S., Woolcock A.J. Characteristics of the inflammation in biopsies from large airways of subjects with asthma and subjects with chronic airflow limitaton. Am. Rev. Respir. Dis. 1992;145:922–927. doi: 10.1164/ajrccm/145.4_Pt_1.922. [DOI] [PubMed] [Google Scholar]
- Cluzel M., Damon M., Chanez P., Bousquet J., Crastes de Paulet A., Godard P. Enhanced alveolar cell luminol-dependent chemiluminescence in asthma. J. Allergy Clin. Immunol. 1987;80:195–201. doi: 10.1016/0091-6749(87)90129-1. [DOI] [PubMed] [Google Scholar]
- Metzger W.J., Zavala D., Richerson H.B., Moseley P., Iwamota P., Monick M., Sjoerdsma K., Hunninghake G.W. Local allergen challenge and bronchoalveolar lavage of allergic asthmatic lungs. Am. Rev. Respir. Dis. 1987;135:433–440. doi: 10.1164/arrd.1987.135.2.433. [DOI] [PubMed] [Google Scholar]