

Abstract
The immune response to HIV-1 in patients who carry human histocompatibility leukocyte antigen (HLA)-B27 is characterized by an immunodominant response to an epitope in p24 gag (amino acids 263–272, KRWIILGLNK). Substitution of lysine (K) or glycine (G) for arginine (R) at HIV-1 gag residue 264 (R264K and R264G) results in epitopes that bind to HLA-B27 poorly. We have detected a R264K mutation in four patients carrying HLA-B27. In three of these patients the mutation occurred late, coinciding with disease progression. In another it occurred within 1 yr of infection and was associated with a virus of syncytium-inducing phenotype. In each case, R264K was tightly associated with a leucine to methionine change at residue 268. After the loss of the cytotoxic T lymphocyte (CTL) response to this epitope and in the presence of high viral load, reversion to wild-type sequence was observed. In a fifth patient, a R264G mutation was detected when HIV-1 disease progressed. Its occurrence was associated with a glutamic acid to aspartic acid mutation at residue 260. Phylogenetic analyses indicated that these substitutions emerged under natural selection rather than by genetic drift or linkage. Outgrowth of CTL escape viruses required high viral loads and additional, possibly compensatory, mutations in the gag protein.
Keywords: human immunodeficiency virus, immune escape, selection, CD8+ T lymphocyte, phylogenetic analysis
Introduction
The ability of HIV to adapt to changes in its environment through mutation of its genome is well described. Evidence from the simian immunodeficiency virus (SIV)-macaque model of immunodeficiency virus pathogenesis indicates that CD8+ T cells can control retrovirus replication 1 2 and that SIV evolves to escape the CD8+ CTL response 3 4. These data add substantial support to earlier studies in humans which demonstrated that HIV-1 mutants can escape CTL responses 5 6 7 8. Viral escape from CTLs may occur rapidly during primary infection, or late in the disease as features of AIDS appear, but the constraints on the evolution of escape mutants are unclear.
In patients who carry HLA-B2705, the HIV-specific CTL response, during the chronic phase of infection, is usually characterized by an immunodominant response to an epitope in the core protein, p24 (amino acids 263–272, KRWIILGLNK; references 7 and 9). The interaction of the arginine (R) residue at position 2 of this and other HLA-B2705–restricted epitopes with the B pocket of the HLA-B2705 plays a crucial role in stabilizing the MHC–peptide complexes 9 10 11 12 13. Substitution of either lysine (K) or glycine (G) for arginine (R) at gag residue 264 (R264K and R264G) results in an epitope that binds poorly to HLA-B2705, thus forming unstable complexes 7 14.
In two previously described patients, the mutation K (AAA) for R (AGA) at residue 264 (R264K) occurred late in the infection and coincided with disease progression 7. This mutation could have enabled viral escape from CTLs. Another nonsynonymous mutation, AGA to GGA (R264G), has not been detected in patients, but was engineered into the LAI strain of HIV. This synthetic mutant virus was replication competent in vitro 14. As viral turnover in untreated HIV-infected patients is high 15 16 and HIV reverse transcriptase has an error rate of 10−4 base incorporations 17 18 19, Nietfield et al. argued that R264G should be preferentially selected if CTLs exerted a significant selection pressure on the virus 14. However, this sequence is absent from the database 20, arguing against CTLs being important in control of virus replication 14. The epitope is within a structurally important region of p24 which is involved in the conformational multimerization of p24 during capsid formation 21. As this protein structure must be maintained, few amino acid substitutions within or near the HLA-B2705–restricted epitope are likely to be tolerated.
Here we describe five HIV-infected patients with HLA-B2705 in whom the appearance of mutations at R264 was observed. This selection occurred both after primary infection and during late HIV disease. When CTL selection pressure was lost, partial reversion to wild-type (w/t) virus sequence occurred.
Materials and Methods
Patients.
HIV-1–infected patients attending clinics for the first time were HLA typed by sequence-specific primer methods 22. Patients with HLA-B2705 had PBMCs taken whenever the patient attended clinic. Patients without HLA-B27 who were recruited to other studies had samples collected on at least one occasion. All studies were approved by the appropriate local Research Ethics committees.
Isolation of PBMCs.
PBMCs were isolated from heparinized blood by standard density centrifugation on Lymphoprep (Nycomed). Cells were washed twice in RPMI 1640 (Sigma-Aldrich) and then either cryopreserved or set up in culture.
Sequencing of Cell-associated Viral DNA.
2–3 × 106 PBMCs were resuspended in RPMI 1640 supplemented with 10% FCS, penicillin/streptomycin (GIBCO BRL), and glutamine (GIBCO BRL), and stimulated for 36–48 h with a 1:200 dilution of PHA (Murex). DNA was isolated from PBMCs using the Puregene kit (Gentra Systems) and stored at −20°C. A nested PCR was performed as described which resulted in the amplification of a 335-bp sequence from p24 7. The products were ligated into a thymidine/adenosine (T/A) vector and used to transform competent cells (Invitrogen). Positive colonies were identified by blue/white selection and grown up over night in Luria-Bertani (LB) media in the presence of ampicillin. Plasmid DNA was isolated using either QIAGEN or Hybaid miniprep kits and cycle sequencing reactions were performed using 7-deaza-dGTP (Amersham Pharmacia Biotech) and Cy5-labeled T7 primer (Amersham Pharmacia Biotech). Reactions were loaded onto a 5.75% acryl amide gel and run out on an automated sequencer (Amersham Pharmacia Biotech) and analyzed using the ALF/WIN Express software. A minimum of 20 clones were sequenced at each time point in the HLA-B27–positive patients.
Viral subtype was determined using “HIV subtyping using BLAST” software available on the Los Alamos HIV sequence data base web site (http://hiv-web.lanl.gov/). p24 sequence data from each patient were compared with several reference sequences of each subtype, scored for similarity to each, and a subtype allocated.
Phylogenetic Analysis.
Phylogenetic trees were reconstructed using the maximum likelihood (ML) available in PAUP* (v4) provided by D.L. Swofford (Sinauer Associates, Sunderland, MA). The HKY85 model of DNA substitution was used in all cases with the maximum likelihood transition/transversion ratio (Ts/Tv) and alpha (α), the shape parameter of a discrete approximation to a gamma distribution of rate heterogeneity among sites (here assumed to contain eight rate categories), determined using an iterative procedure in which these parameters were continually adjusted until the tree of highest likelihood was found. These parameter values are available from the authors on request.
Population Genetic Analysis.
The effective population size (Ne) of HIV-1 within patients was estimated by rearranging the relation θ = 2Neμ, where θ, a measure of genetic diversity assuming neutral evolution, was estimated using a Metropolis-Hastings sampling method (program Fluctuate, v1.3; reference 23), and the mutation rate, μ, was set to 3 × 10−5 per site, per generation 24.
Viral Loads and CD4+ Cell Counts.
Viral loads were determined on sterile plasma separated within 6 h of collection into EDTA, and stored at −70°C by the Amplicor kit (Roche). CD4+ counts were determined by standard methodology.
HLA Class I Tetramers.
HLA-B27, HLA-A2, HLA-A11, HLA-B35, HLA-B7, and HLA-B8 tetramers were synthesized as reported previously 22. The following HLA class I–peptide complexes were synthesized: B2705 (C67S) gag 263–272 (KRWIILGLNK), B2705 gag 263–272 (KRWIIMGLNK), A201 gag 77–85 (SLYNTVATL), A201 pol 476–484 (ILKEPVHGV), B35 nef 78–85 (VPLRPMTY), B35 gag 260–268 (PPIPVGDIY), B35 gp120 42–52 (vpvwkeatttl), B7 nef 128–137 (TPGPGVRYPL), B7 gag 148–156 (SPRTLNAWV), B8 gag 24–31 (GGKKKYKL), B8 nef 89–97(FLKEKGGL), B8 nef 13–20 (WPTVRERM), A11 pol 325–333 (AIFQSSMTK), and A11 nef 75–86 (QVPLRPMTYK). For staining, PBMCs were resuspended in PBS and 1% BSA, washed, and then incubated for 60 min at 4°C in the presence of the tetramer. The cells were then washed twice in PBS plus 1% BSA and then incubated for another 20 min at 4°C in the presence of an FITC-conjugated CD8 antibody (Dako). Cells were washed twice as above and then resuspended in PBS plus 1% BSA plus 4% formaldehyde and stored at 4°C for up to 48 h before analysis on a FACScan™ (Becton Dickinson) using CELLQuest™ (Becton Dickinson) software. Lymphocyte gates were set on forward versus side scatter. CD8+ gates were set by isotype control staining and cutoffs for positive staining were set by staining with an irrelevant tetramer.
Bulk Culture and Cell Lines.
Bulk culture and cell lines were set up as detailed previously. Chromium release assays were performed as detailed previously 7 9.
Online Supplemental Material.
For each patient described, the date of identification, viral load, CD4+ cell count, and the percentage of total sequences at each time point with substitutions are shown in Table SI. CTL escape mutations are shown in bold. Available at http://www.jem/org/cgi/content/full/193/3/375/DC1.
Results
HLA-B27–positive Patients
12 HIV-infected patients with HLA-B27 were studied. All were male. Eight had acquired HIV through sexual contact. The others were hemophiliacs who acquired HIV from blood products (Table ). Three were studied during primary infection, five during the asymptomatic phase of the disease, and four during late stage disease. p24 sequences from each patient were subtype B.
Table 1.
Summary of Patient Characteristics
| Risk group | HLA | Disease state when identified | |||
|---|---|---|---|---|---|
| Identifier | Sex | A | B | ||
| 007 | M | Hph | 1, 32 | 27, 44 | Asymptomatic |
| 049 | M | Hph | 3, 3 | 27, 39 | Asymptomatic |
| 422 | M | Hs | 24, 31 | 27, 57 | Asymptomatic |
| 868 | M | Hs | 2, 22 | 27, 35 | Asymptomatic |
| 025 | M | Hph | 3, 10 | 7, 27 | Asymptomatic |
| CW | M | Hph | 1, 2 | 8, 27 | AIDS |
| MH | M | Hph | 2, 32 | 7, 27 | AIDS |
| SW | M | Hs | 11, 32 | 27, 35 | AIDS |
| RT | M | Hs | 1, 2 | 8, 27 | AIDS |
| 777 | M | Hs | 1, 3 | 27, 35 | Primary infection |
| SC8 | M | Hs | 2, 3 | 27, 39 | Primary infection |
| SC40 | M | Hs | 1, 30/31 | 27, 35 | Primary infection |
Hs, homosexual contact; Hph, hemophiliac exposed to infected blood products.
Selection of R264K and R264G Mutants
Acute Primary Infection.
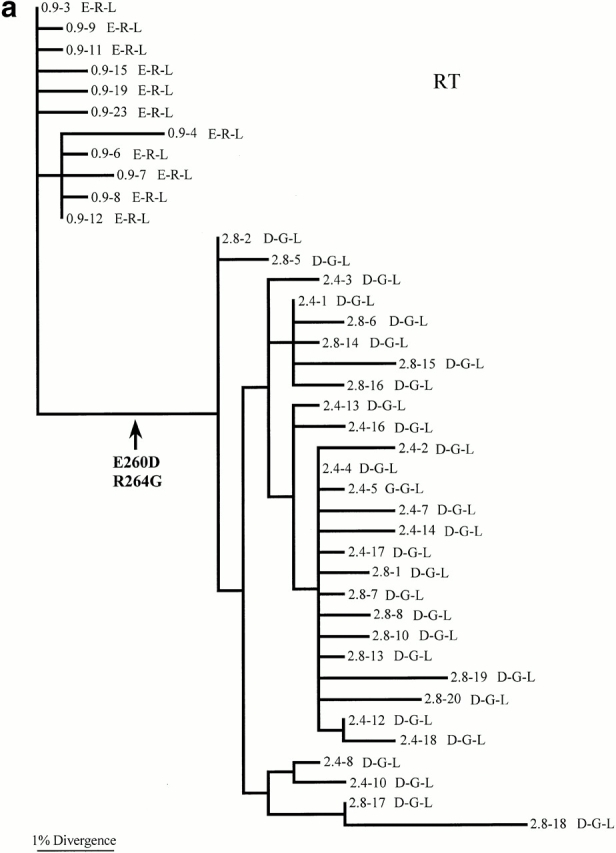
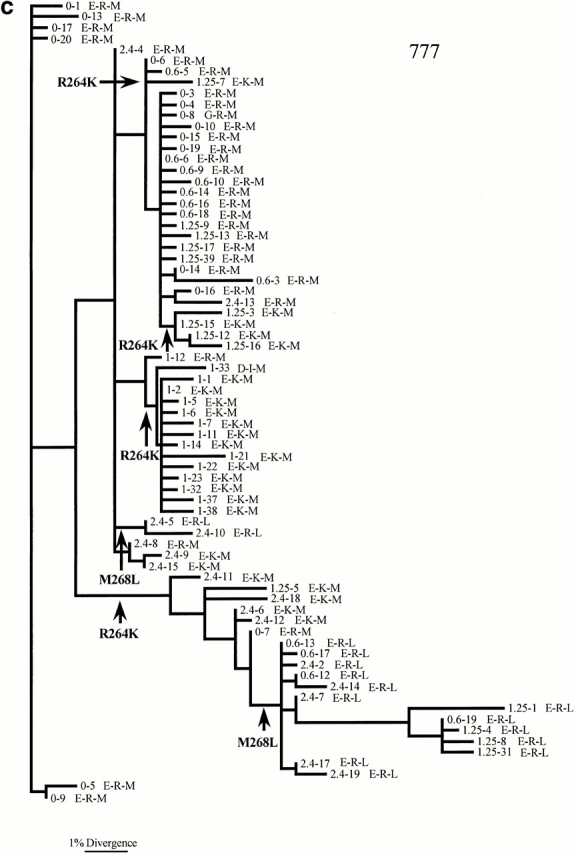
Patient 777 was identified 14 d after a high risk exposure with fever, headache and esophageal candidiasis. His HIV RNA was >106 copies/ml of plasma. He declined antiretroviral therapy. Viral loads and CD4+ counts are shown in Fig. 1. During symptomatic infection viral sequences were w/t at position 264 (R264) and showed methionine (M), rather than leucine (L) at position 268 (Fig. 1 a, and online supplemental Table SI). 28 wk later, his virus contained a mixture of both M268 and L268, but retained R264 in all sequences. 52 wk after acquiring infection, 96% of his viral sequences carried the R264K sequence (1/25 had R264I) and all had M268. Combination antiretroviral chemotherapy (CAC) was instituted at this visit because of persistent viremia and the development of a virus with a syncytium-inducing phenotype (SI). Viremia was better controlled after therapy and a mixed population of viral sequences was detected (Fig. 1 a, and online supplemental Table SI). Throughout the period of observation, this patient had a robust HLA-B27–restricted immunodominant CD8+ T cell response to p24 gag (263–272) as detected by tetramer staining (Fig. 1 a).
Figure 1.
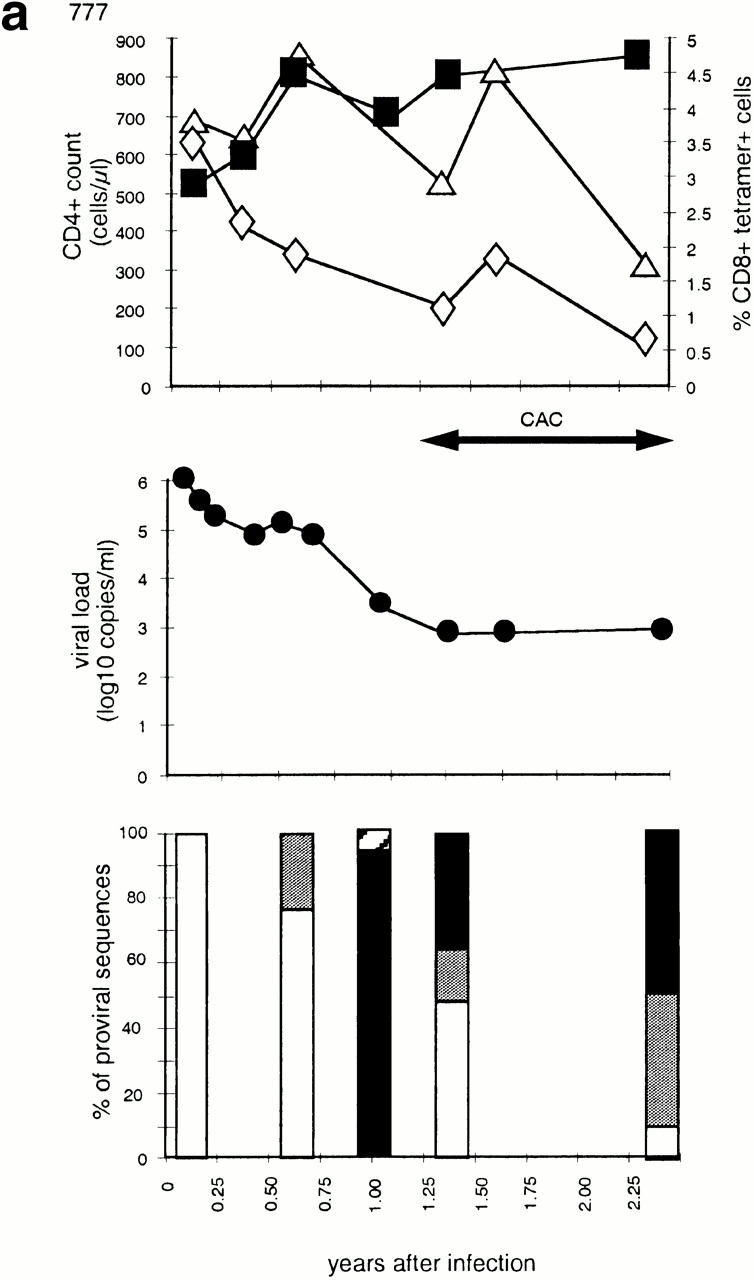
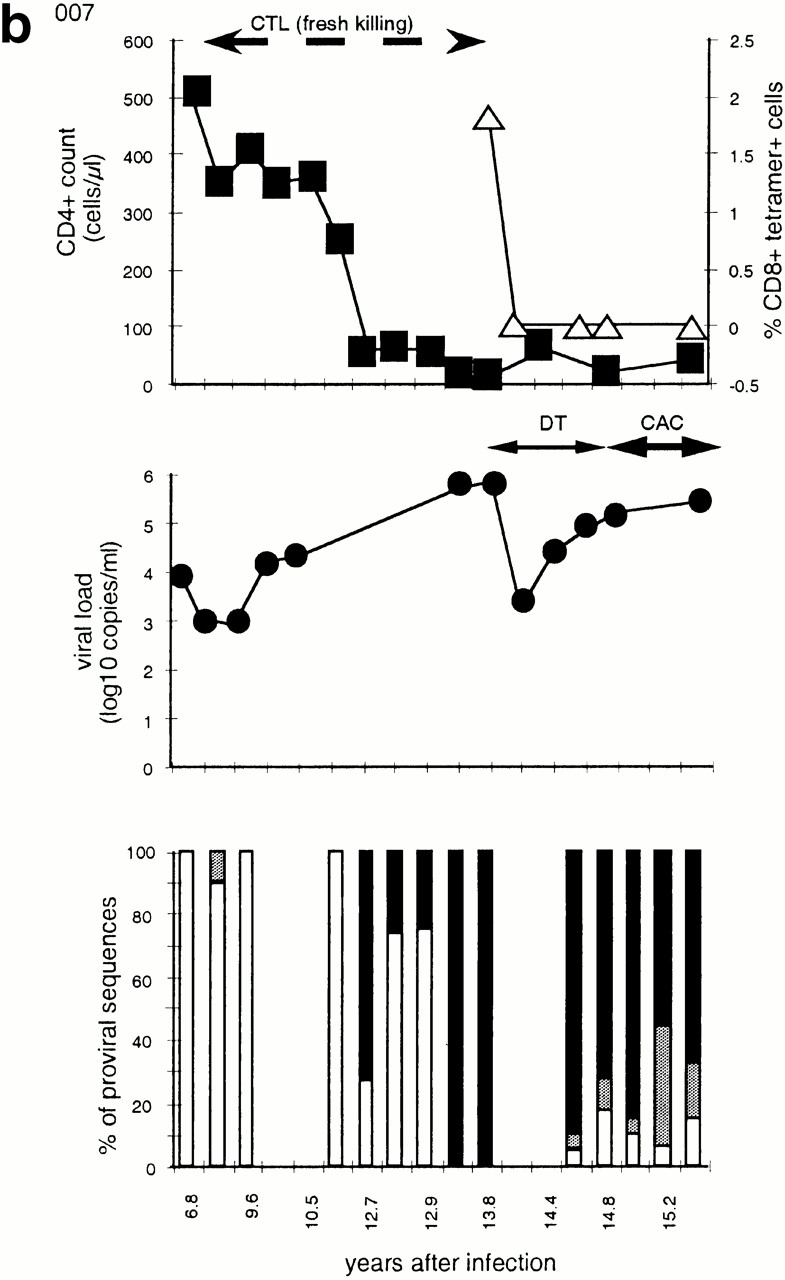
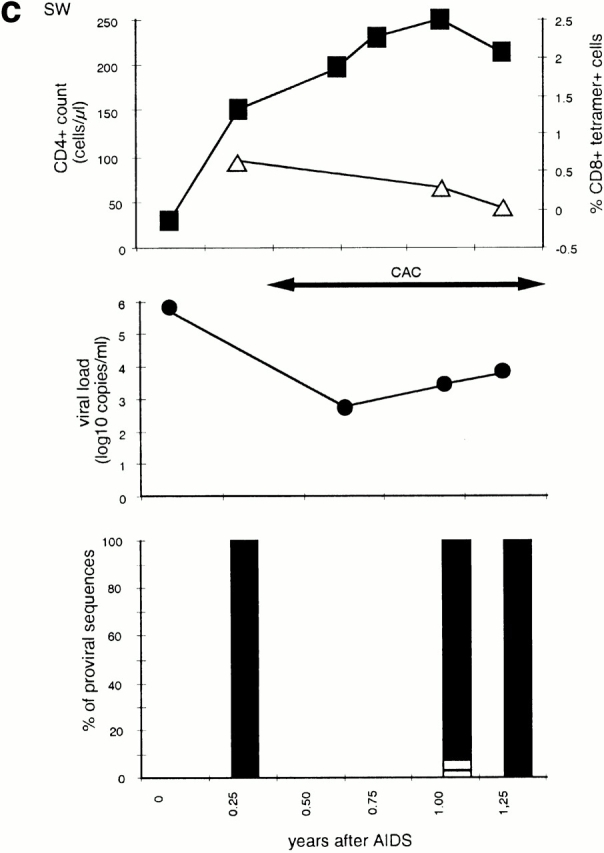
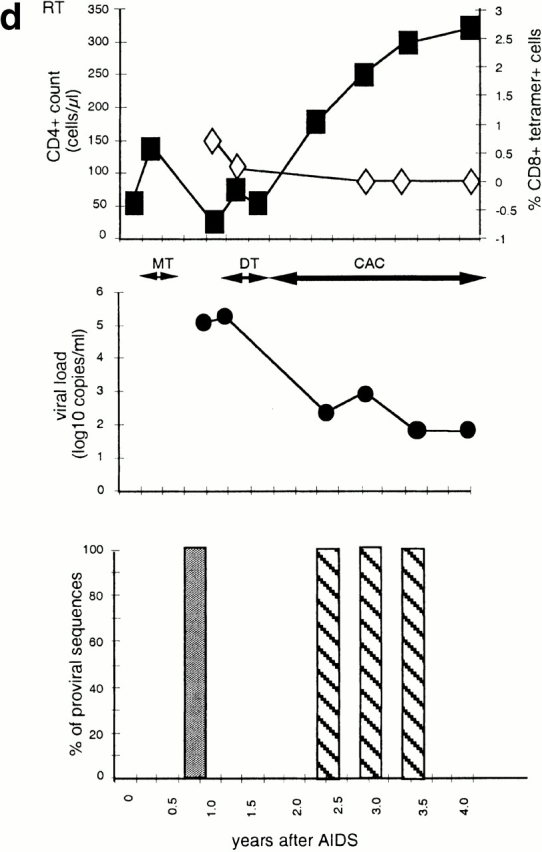
Time course of four subjects developing nonbinding mutations: (a) 777, (b) SW, (c) 007, and (d) RT. In each, the top panel shows CD4+ count (left axis) and HLA B-27 tetramer staining (right axis); the middle panel shows viral load (log RNA copies/ml); and the bottom panel shows the percentage of clones with each variant epitope sequence. Top panels: filled boxes, CD4+ T cell count; open triangles, percentage of CD8+ cells staining with M268 variant peptide B27 tetramer; open diamonds, percentage of CD8+ cells staining with L268 variant peptide B27 tetramer. Double-headed arrows indicate period during which fresh killing was detected of peptide-pulsed autologous targets. Middle panels: closed circles, viral load log10 copies of HIV RNA/ml plasma. Periods of mononucleoside analogue therapy (MT), dual nucleoside analogue therapy (DT), and combination therapy with dual nucleoside analogues and a protease inhibitor (CAC) are indicated by double arrow headed lines. Bottom panels: white bars, percentage of viral DNA sequences with R264 M268; gray bars, percentage of viral DNA sequences with E260 R264 L268 (w/t); black bars, percentage of viral DNA sequences with K264 M268; horizontal stripe, percentage of viral DNA sequences with K264 L268; left to right downward hatch, percentage of viral DNA sequences with D260 G264 L268; and right to left downward hatch, percentage of viral DNA sequences with another mutation at 264.
Patients SC8 and SC40 were also identified during primary infection with seroconversion illnesses and both were treated with CAC very early in their infections (online supplemental Table SI). Both have HLA-B2705, but did not select R264K during the study period. SC8 had a reduction of viral load to <400 copies/ml by day 50. This was maintained for 2.1 yr. Viral sequences remained w/t with L268 (online supplemental Table SI). In SC40, viral load was reduced to undetectable levels 80 d after initial presentation and remained <1,000 copies/ml thereafter. In this patient, virus sequences were w/t with L268. Immunodominant HLA-B27–restricted CD8+ T cell responses to p24 gag (263–272), as defined by enzyme-linked immunospot assay and tetramer staining, were documented in both these patients 25.
Late Stage Infection.
Patient SW presented with Pneumocystis carinii pneumonia, and was subsequently found to be HIV seropositive with a high viral load and a low CD4+ cell count. In the first sample available for sequencing, collected 12 wk after initial presentation, all proviral sequences showed the R264K mutation and M268 (Fig. 1 c). A HLA-B27–restricted T cell response to the M268 variant peptide was detectable by tetrameric staining (0.68% of CD8+ cells) at this and the next time point (0.28% of CD8+ cells) but not later (Fig. 1 c). No HLA-A11– or HLA-B35–restricted responses were detected in this patient at any time by tetramer staining or chromium release assays performed on bulk cultures. When effective antiretroviral therapy was started 16 wk after presentation, the viral load fell and the R264K mutation persisted.
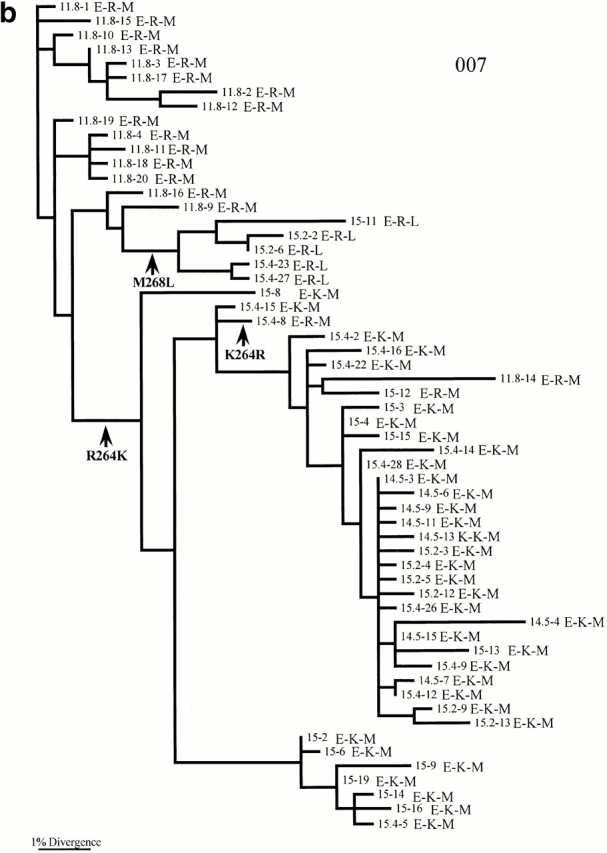
As described previously 7, the hemophiliac patients 007 and 025 had w/t epitope sequences with R264 and L268; however, L268M occurred relatively early in the course of infection (online supplemental Table SI). Subsequently, each developed the R264K mutation as disease progressed with increasing viral loads and falling CD4+ counts (7; online supplemental Table SI, and Fig. 1). Patient 007 had exhibited a sustained immunodominant CTL response to the HLA-B27–restricted gag 263–272 epitope 7 9 26. This was confirmed by tetramer staining but responses became undetectable as measured by tetrameric complexes, bulk culture, and peptide generated cell lines 1 yr after the rise to fixation of the R264K mutation (Fig. 1 b). Patient 025 also had detectable HLA-B27 gag-specific CTLs before the appearance of the R264K mutation.
Because of disease progression, patient 007 was commenced on dual nucleoside therapy, the only drug therapy available at the time. This had a transient effect on viral load and little effect on his low CD4+ count. Later, CAC was instituted but this had no effect on viral load or CD4+ cell count (Fig. 1 b). When the viral load was high and the CTL response was undetectable, the epitope sequences partially reverted from K264 to w/t (K264R). In the sequences where this occurred, but not in the escape sequences, there was an increasing frequency of the other reversion mutation M268L (online supplemental Table SI, and Fig. 1 b).
Patient RT presented with Pneumocystis carinii pneumonia and was then found to be infected with HIV. He received 8 wk zidovudine monotherapy, which resulted in a transient increase in CD4+ cell number, but this drug was stopped because of side effects. HLA-B27 gag epitope sequences 48 wk after AIDS was diagnosed were uniformly R264 and L268. 58 wk after diagnosis, zidovudine and didanosine, introduced because of falling CD4+ cell counts, resulted in a transient rise in CD4+ cell numbers. 6 mo later, a protease inhibitor was added resulting in a sustained rise in CD4+ cells and reduced viral loads. Approximately 20 mo after presentation the R264G mutation was found in all sequences (Fig. 1 d). This mutation was always associated with L268, and a E260D mutation in the NH2-terminal flanking region of the epitope, at this and at all subsequent time points. CD8+ T cell responses to the w/t epitope were detected by tetrameric staining at weeks 28 and 48, but disappeared when the mutant was present (Fig. 1 d). Tetramer staining for HIV-specific CD8+ T cells restricted through HLA-A2 and HLA-B8 was not found at any time.
Three other patients studied had progressive disease but no mutation at R264 was found during the course of the study. Patients CW and MH were recruited after an AIDS diagnosis. Patient CW has never received antiretroviral therapy but has had relatively stable clinical disease with an intermediate viremia (online supplemental Table SI). Patient MH commenced CAC shortly after being identified and since then has had a stable CD4+ cell count and undetectable viral load (online supplemental Table SI). Neither had HIV-specific CD8+ T cells detected either by traditional CTL assays or by tetramer staining with HLA-A2, HLA-B8, HLA-B7, or HLA-B27 constructs at any time point studied (data not shown). Both have proviral sequences that are w/t at the HLA-B27 gag epitope with L268. Patient 868 has been followed for over 10 yr. Over the last 2–3 yr he has had progressive disease and transiently effective dual nucleoside therapy, followed by CAC with a sustained virological response. Initially, he had robust CTL responses restricted through: HLA-A2 to gag (77–85) but not pol epitopes, HLA-B35 to nef (75–82), and HLA-B27 to gag (263–272; dominant response). After CAC, these responses were initially maintained but by 6 mo became undetectable by any technique. All sequences had w/t virus with M268. Despite high viral turnover before initiation of dual nucleosides and when therapy failed, mutation at position 264 was not detected (online supplemental Table SI).
Long-term Nonprogressors
Two patients (049 and 422) fulfil criteria for long-term nonprogression (at least 10 yr after diagnosis with CD4+ counts >500 cells/μl; online supplemental Table SI). Both have CD8+ T cells that recognize gag 263–272 restricted through HLA-B27 (data not shown), but there is no evidence for escape. Both have L268 and have been shown to have responses to HIV mediated through other HLA class I alleles, although in each case the HLA-B27 response appears to be dominant (reference 7, and Kelleher, A.D., unpublished results).
Context of Mutations at Codon 264
In each patient when the R264K mutant was first detected, a majority of the preceding viruses sampled had M268. R264 can be found with either M268 or L268, whereas K264 is strongly associated with M268. In only 2 out of 329 sequences were K264 and L268 found together. In neither case was this sequence detected in subsequent samples. Even in populations containing mixtures of R264 and K264, the latter always segregated with M268, whereas R264 associated with both M268 and L268 (Table ). Similarly, the R264G mutation appears only in the context of the amino acid residues L268 and E260D.
Table 2.
2 × 2 Table Demonstrating the Association R264 or K264 with either M268 or L268 in All Sequences Derived from Time Points Where a Mixture of Clones Expressing either R264 or K264 Were Found
| Clones | R264 | K264 | Total |
|---|---|---|---|
| L268 | 27 | 0 | 27 |
| M268 | 32 | 81 | 113 |
| Total clones | 59 | 81 | 140 |
Fisher's exact P value < 0.0001. Time points used in this analysis were: 777, 1.0, 1.3, and 2.4 yr; 007, 12.7, 12.8, 12.9,14.5, 15.0, 15.2, and 15.4 yr; SW, 1.1 yr.
Mutations in This Region of p24 in HLA-B27–negative Patients
All gag sequences derived from HLA-B27–negative patients studied in our laboratories, which included this region of p24, were analyzed. 24 HIV-infected patients had subtype B p24 sequence data available for analysis from at least one time point. 15 of these were patients with recently acquired infection and 2 of these were sequenced on two occasions 18–20 mo apart. Nine others had chronic disease, with infections of 4–11 yr duration (median duration: 6 yr, median CD4 count 300 cells/μl, median viral load 3.5 × 104 copies of viral RNA/ml) and were sequenced on 2–5 occasions during that time. Two of these patients had commenced therapy with CAC for high viral loads and low CD4 cell counts at the time of sequencing. An average of 15 clones was assessed at each time point. 1 of these 24 patients, a patient with primary infection, had K264 in 7/7 clones sequenced at a single time point before seroconversion. In this subject, K264 occurred in association with T280S and R286K in all sequences. These mutations at codons 280 and 286 are not seen in any of the four HLA-B27–positive patients with R264K and L268M, but are commonly seen in subtype A viruses 20. In all the remaining 483 clones derived from the other HLA-B27–negative patients, R264 was uniformly detected. One of the HLA-B27 negative patients had M268 in all sequences at each of two time points and two of these patients had D260 in a majority of sequences from several time points. These data, along with a review of the Los Alamos database (Table ), indicate that R264K mutation occurs rarely in subtype B viruses.
Table 3.
Summary of All Examples of R264K or L268M and Their Associated Amino Acid Changes in the Immediate Region of the gag HLA-B2705–restricted Epitope Seen in 529 gag Sequences from the Los Alamos Database (Reference 20)
| Virus | Clade | Origin | 260 | 264 | 268 | 272 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Consensus | B | G | E | I | Y | K | R | W | I | I | L | G | L | N | K | I | V | R | |
| WEAU 160 | |||||||||||||||||||
| AF069140a | B | USA | − | − | − | − | − | − | − | − | − | M | − | − | − | − | − | − | − |
| GA18 | B | Spain | − | − | − | − | − | − | − | − | − | M | − | − | − | − | − | − | K |
| CI51 | A | Ivory Coast | − | − | − | − | − | K | − | − | − | − | − | − | − | − | − | − | − |
| LBV2-3 | A | Gabon | − | D | − | − | R | K | − | − | − | − | − | − | |||||
| AF110979b, | C | Botswana | − | D | − | − | − | − | − | − | S | M | − | − | − | − | − | − | − |
| G109 | D | Gabon | − | − | − | − | − | K | − | − | − | M | − | − | − | − | − | − | − |
| Consensus | O | − | D | − | − | R | − | − | − | V | − | − | − | − | − | M | − | K | |
| ANT70Cc | O | Cameroon | − | D | − | − | R | K | − | − | V | − | − | − | − | − | M | − | K |
| CA9 | O | Cameroon | − | D | − | − | R | K | − | − | V | F | − | − | − | − | L | − | K |
| BCF02B | O | Cameroon | − | − | − | − | W | K | − | − | V | − | − | − | − | − | L | − | K |
| AF009015d, | U | Cameroon | − | D | − | − | R | K | − | − | V | − | − | − | M | − | |||
| AF009019 | U | Cameroon | − | D | − | − | S | K | − | − | V | − | − | − | Q | M | − | N | |
| AF009016e, | U | Cameroon | − | D | − | − | R | K | − | − | V | − | − | − | − | L | − | K | |
| AF006857 | U | Uganda | − | ? | − | − | R | K | − | − | − | − | − | ? | − | − | − | − | − |
| AF006924 | U | Uganda | − | D | − | − | − | K | − | − | − | − | − | − | − | − | − | − | − |
| HIV90CF402 | AE | Thailand | − | − | − | − | − | K | − | − | − | − | − | − | − | − | − | − | − |
| VI354 | AG | Gabon | − | K | − | − | − | K | * | − | − | − | − | − | − | − | − | − | − |
| HIV224190 | B/E | Thailand | − | D | − | − | − | Q | − | − | − | M | − | − | − | − | − | − | − |
| HIV224194 | B/E | Thailand | − | − | − | − | − | − | − | − | M | − | − | − | − | − | − | − | |
| HIV224196 | B/E | Thailand | − | D | − | − | − | − | − | − | − | M | − | − | − | − | − | − | − |
*, stop codon; other sequences have stop codon at 793 (e.g., accession no. L11970/U43172) and other pseudogenes have been excluded from the analysis. U, untyped. Identical sequences, in this region, are seen in the following viruses: aPH136, from the Philippines and BZ190 from Brazil, bAF110980 and 110981 from Botswana, cMVP5180, BCF01B, BCF03B, BCF07B, BCF08B, BCF06B,BCF11B, AF001966, AF001967, and AF001969 from Cameroon, AF001970, AF001971, AF001972, and AF001973, from Equatorial Guinea, and VI686 from Gabon, dAF009018 from Cameroon, and eAF009024 from Cameroon.
Phylogenetic Analysis
Phylogenetic trees were constructed for the viral sequences obtained from three of the patients: RT, 007, and 777 (Fig. 2, a–c) to study the evolutionary processes contributing to the accumulation of mutations clustered around codon 264. In the tree constructed for patient RT, there is a long branch separating the sequences sampled 0.9 yr after enrolment from those collected at later time points (2.4–2.8 yr after enrolment). Two amino acid changes have occurred on this branch: E260D and R264G. It is striking that these two changes are in such close proximity in the amino acid sequence and that they are fixed in all later samples (Fig. 1 d, and online supplemental Table SI). This suggests that either both represent escape mutants, or that one is favored by selection and that the other has been pulled to fixation by genetic linkage.
Figure 2.
Maximum likelihood phylogenetic trees depicting the evolutionary relationships of viruses from patients (a) RT, (b) 007, and (c) 777. The tree is unrooted but the direction of mutations can be inferred by assuming that the oldest sampled sequences are ancestral. All branch lengths are drawn to scale. The labels on each branch correspond to the time point at which the viral sample was taken (years after diagnosis), followed by the amino acid (single letter code) found at positions 260, 264, and 268, respectively. The positions of all putative changes involving amino acids E to D at position 260, R and K at position 264, and M to L at position 268 are indicated with arrows. Note that designation of amino acid changes is difficult inpatients 007 and 777 because viruses from the first sampling time point do not form a single group and are dispersed across the phylogeny. Genbank accession numbers for sequences used to construct phylogenetic trees are available from Genbank/EMBL/DDBJ under accession nos. RT, AF319258, AF319259, AF319260, AF319261, AF319262, AF319263, AF319264, AF319265, AF319266, AF319267, AF319268, AF319269, AF319270, AF319271, AF319272, AF319273, AF319274, AF319275, AF319276, AF319277, AF319278, AF319279, AF319280, AF319281, AF319282, AF319283, AF319284, AF319285, AF319286, AF319287, AF319288, AF319289, AF319290, AF319291, AF319292, AF319293, AF319294, AF319295, AF319296, AF319297, AF319298, AF319299, AF319300, AF319301, AF319302, AF319303, AF319304, AF319305, AF319306, AF319307, AF319308, AF319309, AF319310; 007, AF319174, AF319175, AF319176, AF319177, AF319178, AF319179, AF319180, AF319181, AF319182, AF319183, AF319184, AF319185, AF319186, AF319187, AF319188, AF319189, AF319190, AF319191, AF319192, AF319193, AF319194, AF319195, AF319196, AF319197, AF319198, AF319199, AF319200, AF319201, AF319202, AF319203, AF319204, AF319205, AF319206, AF319207, AF319208, AF319209, AF319210, AF319211, AF319212, AF319213, AF319214, AF319215, AF319216, AF319217, AF319218, AF319219, AF319220, AF319221, AF319222, AF319223, AF319224, AF319225, AF319226, AF319227, AF319228, AF319229, AF319230, AF319231, AF319232, AF319233, AF319234, AF319235, AF319236, AF319237, AF319238, AF319239, AF319240, AF319241, AF319242, AF319243, AF319244, AF319245, AF319246, AF319247, AF319248, AF319249, AF319250, AF319251, AF319252, AF319253, AF319254, AF319255, AF319256, AF319257; and 777, AF319311, AF319312, AF319313, AF319314, AF319315, AF319316, AF319317, AF319318, AF319319, AF319320, AF319321, AF319322, AF319323, AF319324, AF319325, AF319326, AF319327, AF319328, AF319329, AF319330, AF319331, AF319332, AF319333, AF319334, AF319335, AF319336, AF319337, AF319338, AF319339, AF319340, AF319341, AF319342, AF319343, AF319344, AF319345, AF319346, AF319347, AF319348, AF319349, AF319350, AF319351, AF319352, AF319353, AF319354, AF319355, AF319356, AF319357, AF319358, AF319359, AF319360, AF319361, AF319362, AF319363, AF319364, AF319365, AF319366, AF319367, AF319368, AF319369, AF319370, AF319371, AF319372, AF319373, AF319374, AF319375, AF319376, AF319377, AF319378, AF319379, AF319380, AF319381, AF319382, AF319383, AF319384, AF319385, AF319386, AF319387, AF319388, AF319389, AF319390, AF319391, AF319392, AF319393, AF319394, AF319395, AF319396.
The action of positive selection at codon 264 is supported by an analysis of the numbers of synonymous (dS) and nonsynonymous (dN) substitutions per site in this codon. In patients RT, 007, and 777 combined, 11 nonsynonymous and 0 synonymous substitutions are observed at codon 264. This bias toward nonsynonymous substitutions is significantly more than expected by chance alone (P = 0.021 in a χ2 test weighted for the different rates of substitution among bases) showing that dN > dS for this codon as expected under positive selection. In other words, codon 264 does not evolve quickly simply because it is subject to weaker selective constraints.
Support for positive selection also comes from a population genetic analysis of diversity. If, for example, the fixation of R264G in patient RT occurred between 0.9 and 2.4 yr after enrolment, as is compatible with the phylogenetic analysis, then this fixation event took no more than ∼18 mo. For this process to occur by genetic drift alone would require an Neof only ∼100, assuming that the HIV generation time is 2.6 d 16 and that fixation of neutral mutations takes on average 2Ne generations 27. However, if Ne is estimated directly from the sequence data of patient RT using a neutral measure of genetic diversity (θ), then the average value of Ne across time points is ∼800 (θ values from 0.025 to 0.076). Consequently, although the effective population size in patient RT is small, as might be expected to be the case after a selective sweep, it is still not small enough to allow fixation of a neutral mutation by genetic drift in such a short time period.
The phylogenetic analysis also supports the selection of R264K in patients 007 and 777. In patient 007, all sequences present at 11.8 yr after diagnosis were R264, but by 33 mo later (14.5 yr after diagnosis) all sequences were K264 (Fig. 2 b). Interestingly, R264 then reappeared after antiviral therapy, remained in the population for the last year of sampling, and reached a frequency of 25%. The phylogenetic tree reveals that the main source of this virus is persistence of a variant bearing R264, as those sequences which bore R264 later are clustered with those earlier samples which carry R264. However, it is also apparent from the phylogenetic tree that at least one K264R reversion mutation has occurred (Fig. 2 b).
A similarly complex evolutionary picture is seen in patient 777. In this patient, the R264K mutation may have arisen four times, but never reaches fixation (i.e., 100% frequency), perhaps because antiviral therapy was given after that time which altered the selection pressure on viral evolution. R264 then rises in frequency later on in infection seemingly because of the persistence of lineages with R264 across several time points.
The phylogenetic analysis also provides strong support for the compensatory relationship between the mutations at positions 264 and 268. In both patients 007 and 777, L268 only occurs in association with R264 (Fig. 2b and Fig. c). Moreover, the phylogenetic analysis reveals that changes at amino acid positions 264 and 268 occur on different branches of the tree, indicating that they are independent. Such observations suggest that although L268 and R264 are intimately related, they are not in direct linkage; the rise of M268L is not simply due to it being linked to a selectively favored K264R virus. For patient 777, the M to L change has actually occurred twice independently.
Discussion
This study defines some of the complex requirements for the evolution of CTL escape mutants at a locus in a highly conserved HIV protein. We have described five HLA-B27 carrying HIV-infected patients, four of whom have acquired the same (R264K) escape mutation in gag. In a fifth patient an alternative mutation (R264G) was seen. We have shown that the R264K mutation is strongly associated with a second change L268M and that the acquisition of this doubly modified epitope does not appear to result from simple linkage between the mutations at sites 264 and 268. Escape occurred both early and late in HIV disease but required high viral replication and compensatory mutations nearby in HIV-1 p24. Frequent selection of the same mutations, which are uncommon in the database and HLA-B27–negative subjects, and the rapidity with which they are fixed within patients, strongly implies selection by CTLs specific for the epitope.
The mutability of HIV and its ability to adapt to environmental pressures has been well demonstrated by the rapid emergence of drug resistance mutations in reverse transcriptase and protease (for reviews, see references 28 and 29). In the case of resistance to protease inhibitors, mutations that appear to have no direct effect on the active site of the enzyme have been described. These appear to compensate for critical mutations within the active site itself 30 31 32.
Evidence supporting a role for CTLs in the control of HIV includes: depletion of CD8+ cells results in more rapid progression of SIV-related disease in macaques 1 2, the appearance of HIV-specific CTLs at primary infection coincides with control of viremia 25 33 34, and CTL escape mutations allow immune evasion by HIV early in disease 5 6. These data indicate that CTLs can exert a significant selective force upon the virus. Later in the disease, the mutability of the virus allows it to generate escape mutations rapidly after artificial perturbations of the steady state which resulted in a highly focused CTL response 8. In animals infected with identical strains of SIV, nonsynonymous mutations in nef and env accumulate exclusively within antigenic sites subject to CTL pressure 3.
These studies suggest that several conditions need to be met to permit the evolution of CTL escape mutants at the HLA-B27 gag epitope. High viral turnover appears to be a requirement whether escape occurs early in the disease as with patient 777, or late in the disease as with patients 007, RT, SW, and 025. In those treated effectively and early in primary infection, escape did not occur for as long as 2.1 yr. Long-term nonprogressors and patient CW, whose viral load was constant at 4.5 logs, did not develop very high levels of viral turnover and did not generate escape mutants.
Why do these mutations, which arise in an epitope that is the target of a highly focused CTL response, not occur earlier in the disease? There are several possible explanations. These mutations may only arise in certain viral strains. SI phenotypes of HIV-1 are more pathogenic and usually occur later in the disease 35 36 37. The only patient who had early escape developed an SI virus early in the infection. Those whose virus escaped late in infection are likely to have had SI virus. SI virus is not susceptible to inhibition by β-chemokines released from CD8+ T cells 38. In the absence of soluble factor inhibition, SI virus faces an immune selection pressure more clearly restricted to the CTL response than non-SI virus, and thus generation of escape mutants to lytic activity of CD8+ T cells may be favored.
A second explanation for constraint on evolution within gag involves the structure and function of this protein. The HLA-B27 epitope lies embedded in the NH2-terminal domain of the p24 capsid protein. Evidence for the need to conserve structure in this region of the protein comes from several sources. First of all, most mutations in this area result in nonviable virus 14, and deletion mutants indicate that this region is necessary for the conformational changes that allow p24 to form functional capsid after p24 multimerisation 21 39. Second, the crystal structure of the NH2-terminal domain of p24 reveals that the epitope and its NH2-terminal flanking regions lie within helix 7 (amino acid residues 258–276), one of five helices within the NH2-terminal domain that form a coiled coil structure 40 41 42. Residues 260, 264, and 268 all lie on the same aspect of helix 7 (Fig. 3 a). Furthermore, the packing of molecules into crystals of the NH2 terminus of p24 is dependent on the formation of two interfaces, and one of these interfaces is formed by parallel packing of helix 7 from adjacent molecules. Residues 264 and 268, along with 271 and 275, play a role in the formation of this interface, and R264 and L268 lose significant accessible surface area (Fig. 3 b). In this structure, the aliphatic portions of the two R264 side chains are in van der Waals contact with each other and guanidium groups participate in a charged interaction with the side chains of E260 41. Moreover, a model of core particle assembly predicts that antiparallel interactions between helix 7 and helix 2 are the basis for NH2-terminal dimerization and that helix 7 plays a role in the association of these dimers to form arrays of capsid molecules before the formation of the mature capsid 43. Taken together, these findings indicate that this region plays an important role in capsid self-association and conformational assembly. Furthermore, although this area is not the primary site of p55 association with cyclophilin 44, deletions in this area can abrogate cyclophilin binding to p24 45. Therefore, mutations in this area are likely to interfere with viral assembly and thus impair fitness. Accommodation of a new mutation in one part of this structure may require at least one compensatory mutation.
Figure 3.
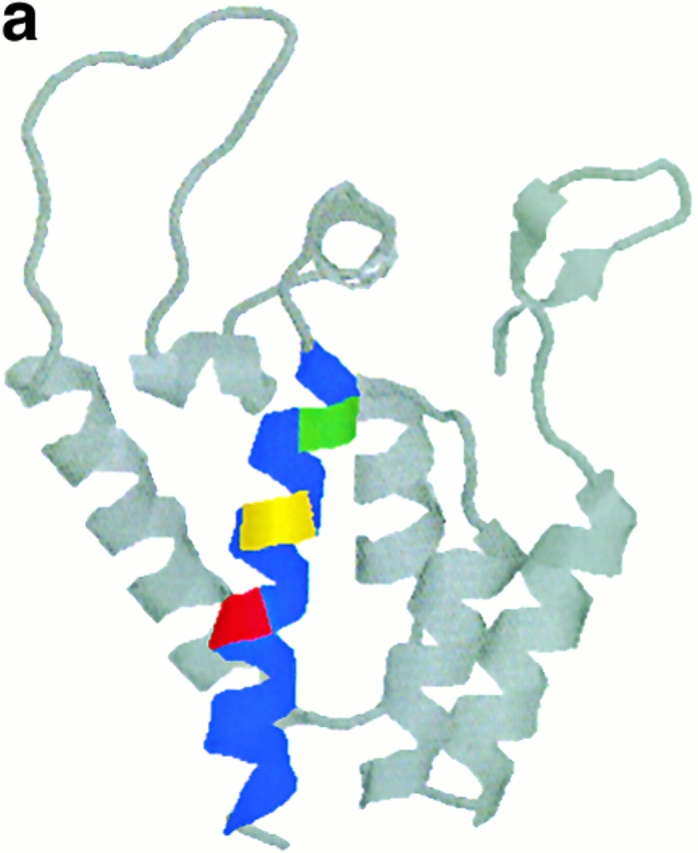
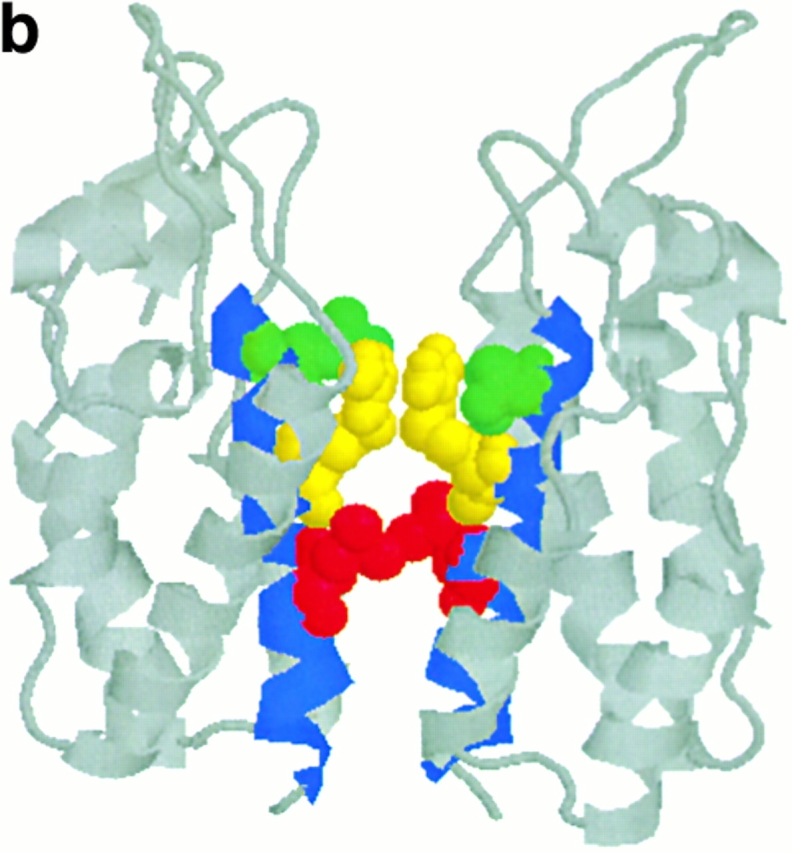
Ribbon diagrams of structure of NH2-terminal domain of p24 adapted from reference 41. Helix 7 is shown in blue, residues 260, 264, and 268 are highlighted in green, yellow, and red, respectively. (a) The structure of the monomer. (b) The structure of the dimer in the crystal structure, highlighting the role that residues 260, 264, and 268 play in one interface; in this diagram these residues are shown in the “space fill” convention.
The results presented here indicate that in subtype B viruses, if the R264K mutation is to be tolerated, a L268M mutation is preferred. Furthermore, after development of the escape mutation, the reversion of methionine to w/t leucine at position 268 (M268L) only occurs after the reappearance of arginine at position 264. In subtype B viruses M268 is uncommon, reported in only 3 out of 57 subtype B p24 sequences in the Los Alamos data base (20; Table ). The K264 mutation is not reported in subtype B viruses. In subtype D viruses, where K264 is present in a minority of sequences, there is an accompanying M268 mutation (Table ). However, there are three p24 sequences (CI51, HIV90CF402, and AF006924) in the database where K264 occurs without any mutations in the immediate vicinity of the epitope (Table ). In each case the p24 sequence, or the available segment of p24 sequence, is subtype A. CI51 and HIV90CF402 viruses both differ by >20 amino acids across p24 from the consensus B subtype sequence. The AF006924 sequence differs from consensus B subtype sequence by 14 amino acids out of the 131 amino acids of sequence available for analysis. It may be that some of these amino acid substitutions in subtype A p24 allow K264 to be accommodated more easily than in subtype B p24. Furthermore, one of these viruses (HIV90CF402) had been extensively passaged in vitro including through chimpanzee cells, before sequencing. This prolonged period of laboratory adaptation may allow mutations that may not be advantageous in vivo to persist. In subtype O viruses, and in some untyped viruses, R264K is a common variant; however, it is associated with other mutations such as V267I, K263R, and usually with D260E (Table ). Taken together, these observations support the concept that sequence constraints, imposed by the necessity to maintain the structure of p24 require several changes to support an escape virus. Thus, escape from the immunodominant HLA-B27–restricted CTL response may require a particular array of mutations. This could account for the delayed appearance of HIV-1 escape mutations in most cases.
R264G does not appear in the Los Alamos database. This mutation is probably disadvantageous in isolation. It may also indicate that in vivo the mutation is only tolerable in subtype B viruses, all of which on the database have E260, when E260D and R264G mutations occur in the presence of L268. Such requirements may explain why this mutation occurs infrequently. The observation that the R264G mutation can be tolerated by the laboratory-adapted LAI strain in vitro in the presence of E260 14 would argue that E260D is not an absolute requirement for the viability of R264G. However, the growth requirements of a mutant laboratory adapted strain of virus grown in isolation in vitro are likely to be different from in vivo requirements where a mutant must out compete all other strains to become dominant. The advantage of this dual mutation may only need to be small, as even minor enhancements of viral fitness can quickly lead to replacement of the less fit variants in a population 46. Standard in vitro culture techniques do not permit the definition of small differences in viral fitness that are revealed by viral competition and selective pressure 47 48.
Further evidence that CTLs exert selective pressure on these escape mutations comes from the observation that when CTLs are absent and there is escalating viral replication, as in patient 007, the detectable virus partially reverts to w/t. Phylogenetic analysis suggests that although some of this recrudescence of w/t virus is derived from sequences laid down as provirus, other reversions arise de novo and do so in a manner where mutations occur in a certain order; that is, L268M appears to occur after reversion of K264R. This phenomenon of reversion on removal of selective pressure is reminiscent of the changes seen in drug resistant strains on drug cessation 49 50 51 52.
When antiretroviral therapy fails to control HIV replication well, evolution of the virus can continue. When therapy is effective proviral evolution appears to be slowed substantially 53, and sequences tend to reflect the virus population that was present when therapy started. The fact that an escape mutant can persist for many months after the initiation of therapy has therapeutic implications; any cessation or failure of therapy will result inevitably in recrudescence of a virus containing immune escape variants.
The exact causes of disease progression in HIV infection are still poorly understood. However, the analysis presented here and accumulating evidence from other work 3 4 5 6 7 8 strengthens the early suggestion 54 that evasion of the CTL response through mutation of the virus is a significant mechanism of viral persistence.
Acknowledgments
The authors would like to thank Tim Rostron for the HLA typing and viral loads, the clinical staff for the collection of samples, David Price and Charles Bangham for their critical review of the manuscript, and Eric Rosenberg for provision of clinical data.
A.D. Kelleher was supported by grants from Royal College of Physicians of Australasia, the National Health and Medical Research Council (Australia), and Medical Research Council (UK). R. Phillips, C. Long, and K. Olson were supported by the Wellcome Trust. P. Goulder was supported by Medical Research Council (UK) and the Elizabeth Glaser Pediatric AIDS Foundation.
Footnotes
Abbreviations used in this paper: CAC, combination antiretroviral chemotherapy; SI, syncytium-inducing; SIV, simian immunodeficiency virus; w/t, wild-type.
The online version of this article contains supplemental material.
References
- Jin X., Bauer D.E., Tuttleton S.E., Lewin S., Gettie A., Blanchard J., Irwin C.E., Safrit J.T., Mittler J., Weinberger L. Dramatic rise in plasma viremia after CD8+ T cell depletion in simian immunodeficiency virus–infected macaques. J. Exp. Med. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz J.E., Kuroda M.J., Santra S., Sasseville V.G., Simon M.A., Lifton M.A., Racz P., Tenner-Racz K., Delesandro M., Scallon B.J. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- Evans D.T., O'Connor D.H., Jing P., Dzuris J.L., Sidney J., da Silva J., Allen T.M., Horton H., Venham J.E., Rudersdorf R.A. Virus-specific cytotoxic T-lymphocyte responses select for amino-acid variation in simian immunodeficiency virus Env and Nef. Nat. Med. 1999;5:1270–1276. doi: 10.1038/15224. [DOI] [PubMed] [Google Scholar]
- Allen T.M., O'Connor D.H., Jing P., Dzuris J.L., Mothe B.R., Vogel T.U., Dunphy E., Liebl M.E., Emerson C., Wilson N. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- Price D.A., Goulder P.J.R., Klenerman P., Sewell A.K., Easterbrook P.J., Troop M., Bangham C.R.M., Phillips R.E. Positive selection of cytotoxic T lymphocyte escape variants during primary infection. Proc. Natl. Acad. Sci. USA. 1997;94:1890–1895. doi: 10.1073/pnas.94.5.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P., Lewicki H., Wei X., Horwitz M.S., Peffer N., Meyers H., Nelson J.A., Gairin J.E., Hahn B.H., Oldstone M.B.A. Antiviral pressure exerted by HIV-1 specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 1997;3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- Goulder P.J.R., Phillips R.E., Colbert R.A., McAdam S., Ogg G., Nowak M.A., Giangrande P., Luzzi G., Morgan B., Edwards A. Late escape from an immunodominant cytotoxic T lymphocyte response associated with progression to AIDS. Nat. Med. 1997;3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- Koenig S., Conley A.J., Brewah Y.A., Jones G.M., Leath S., Boots L.J., Davey V., Panteleo G., Demarest J.F., Carter C. Transfer of HIV-1-specific cytotoxic T lymphocytes to an AIDS patient leads to selection for mutant HIV variants and subsequent disease progression. Nat. Med. 1995;1:330–336. doi: 10.1038/nm0495-330. [DOI] [PubMed] [Google Scholar]
- Nixon D.F., Townsend A.R.M., Elvin J.G., Rizza C.R., Gallwey J., McMichael A.J. HIV-1 gag specific cytotoxic T lymphocytes defined with recombinant vaccinia virus and synthetic peptides. Nature. 1988;336:484–487. doi: 10.1038/336484a0. [DOI] [PubMed] [Google Scholar]
- Rammensee H.G., Friede T., Stevanoviic S. MHC ligands and peptide motifsfirst listing. Immunogenetics. 1995;41:178–228. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- Goulder P.J.R., Edwards A., Phillips R.E., McMichael A.J. Identification of a novel HLAB*2705-restricted cytotoxic T lymphocyte epitope within a conserved region of HIV-1 nef. AIDS. 1997;11:536–538. [PubMed] [Google Scholar]
- Jardetsky T.S., Lane W.S., Robinson R.A., Madden D.R., Wiley D.C. Identification of self peptides bound to purified HLA-B27. Nature. 1991;353:326–329. doi: 10.1038/353326a0. [DOI] [PubMed] [Google Scholar]
- Madden D.R., Gorga J.C., Strominger J.L., Wiley D.C. The three dimensional structure of HLA-B27 at 2.1A resolution suggests a general mechanism for tight peptide binding to MHC. Cell. 1992;70:1035–1058. doi: 10.1016/0092-8674(92)90252-8. [DOI] [PubMed] [Google Scholar]
- Nietfield W., Bauer M., Fevier M., Maier R., Holzwarth B., Frank R., Maier B., Riviere Y., Meyerhans A. Sequence constraints and recognition by CTL of an HLA-B27-restricted HIV-1 gag epitope. J. Immunol. 1995;154:2189–2197. [PubMed] [Google Scholar]
- Ho D.D., Neumann A.U., Perelson A.S., Chen W., Leonard J.M., Markovitz M. Rapid turnover of plasma virions and CD4+ lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Perelson A.S., Neumann A.U., Markowitz M., Leonard J.M., Ho D.D. HIV-1 dynamics in vivovirion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- Temin H.M. Retrovirus variation and reverse transcriptionabnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA. 1993;90:6900–6904. doi: 10.1073/pnas.90.15.6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts J.D., Bebenek K., Kunkel T.A. The accuracy of reverse transcriptase from HIV-1. Science. 1988;242:1171–1173. doi: 10.1126/science.2460925. [DOI] [PubMed] [Google Scholar]
- Preston D.B., Poiesz B.J., Loeb L.A. Fidelity of HIV-1 reverse transcriptase. Science. 1988;242:1168–1171. doi: 10.1126/science.2460924. [DOI] [PubMed] [Google Scholar]
- Human Retroviruses and AIDS. A Compilation and Analysis of Nucleic Acid and Amino Acid Sequences Korber B., Hahn B., Foley J.W., Mellors T., Leitner G., Myers F., McCutchan, Kuiken C.L. B 1997. Theoretical Biology and Biophysics Group; Los Alamos National Laboratory: 367Los Alamos, NM [Google Scholar]
- Zhang W.H., Hockley D., Nermut M.V., Morikawa Y., Jones I.M. Gag-Gag interactions in the C-terminal domain of human immunodeficiency virus type 1 p24 capsid antigen are essential for Gag particle assembly. J. Gen. Virol. 1996;1996:743–751. doi: 10.1099/0022-1317-77-4-743. [DOI] [PubMed] [Google Scholar]
- Bunce M., O'Neill C.M., Barnardo M.C., Krausa P., Browning M.I., Morris P.J., Welsh K.I. Phototypingcomprehensive DNA typing for HLA-A, B, C, DRB1, DRB3, DRB4, DRB5 and DQB1, by PCR with 144 primer mixes utilizing sequence specific primers. Tissue Antigens. 1995;46:355–367. doi: 10.1111/j.1399-0039.1995.tb03127.x. [DOI] [PubMed] [Google Scholar]
- Kuhner M.K., Yamato J., Felsenstein J. Estimating effective population size and mutation rate from sequence data using Metropolis-Hastings sampling. Genetics. 1995;140:1421–1430. doi: 10.1093/genetics/140.4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin J. Molecular biology of HIV. In: Crandell K.A., editor. The Evolution of HIV. Johns Hopkins University Press; Baltimore, MD: 1999. pp. 3–40. [Google Scholar]
- Wilson J.D.K., Ogg G.S., Allen R.L., Davis C., Shaunak S., Workman C., Downie J., Dyer W., Sullivan J., McMichael A.J., Rowland-Jones S.L. Direct visualisation of the HIV-1-specific cytotoxic T lymphocyte (CTL) response during primary HIV infection. AIDS. 2000;14:225–233. doi: 10.1097/00002030-200002180-00003. [DOI] [PubMed] [Google Scholar]
- Moss P.A.H., Rowland-Jones S.L., Frodsham P.M., McAdam S., Giangrande P., McMichael A.J., Bell J.I. Persistent high frequency of human immunodeficiency virus-specific cytotoxic T cells in peripheral blood of infected donors. Proc. Natl. Acad. Sci. USA. 1995;92:5773–5777. doi: 10.1073/pnas.92.13.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M. Neutral Theory of Molecular Evolution 1983. Cambridge University Press, ; Cambridge, UK: pp. 789 pp [Google Scholar]
- Mayers D. Rational approach to resistancenucleoside analogues AIDS. 10Suppl. 11996. S9 S13 [PubMed] [Google Scholar]
- Boucher C. Rational approach to resistanceusing saquinavir AIDS. 10Suppl. 11996. S15 S19 [PubMed] [Google Scholar]
- Nijhuis M., Schuurman R., de Jong D., Erickson J., Gustchina E., Albert J., Schipper P., Gulnick S., Boucher C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS. 1999;13:2349–2359. doi: 10.1097/00002030-199912030-00006. [DOI] [PubMed] [Google Scholar]
- Schock H.B., Garsky V.M., Kuo L.C. Mutational anatomy of an HIV-1 protease variant conferring cross-resistance to protease inhibitors in clinical trials. Compensatory modulations of binding and activity. J. Biol. Chem. 1996;271:31957–31963. doi: 10.1074/jbc.271.50.31957. [DOI] [PubMed] [Google Scholar]
- Ala P.J., Huston E.E., Klabe R.M., McCabe D.D., Duke J.L., Rizzo C.J., Korant B.D., DeLoskey R.J., Lam P.Y., Hodge C.N., Chang C.H. Molecular basis of HIV-1 protease drug resistancestructural analysis of mutant proteases complexed with cyclic urea inhibitors. Biochemistry. 1997;36:1573–1580. doi: 10.1021/bi962234u. [DOI] [PubMed] [Google Scholar]
- Koup R.A., Safrit J.T., Cao Y., Andrews C.A., McLeod G., Borkowsky W., Farthing C., Ho D.D. Temporal association of cellular immune responses with initial control of viraemia in primary human immunodeficiency type 1 syndrome. J. Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P., Lewicki H., Hahn B.H., Shaw G.M., Oldstone M.B.A. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo E.M., Mordfeldt-Mason L., Chiodi F., Lind B., von Gegerfelt A., Albert J., Olausson E., Asjo B. Distinctive replicative and cytopathic characteristics of human immunodeficiency virus isolates. J. Virol. 1988;62:4414–4419. doi: 10.1128/jvi.62.11.4414-4419.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersmette M, de Goede R.E., Al B.J., Winkel I.N., Gruters R.A., Cuypers H.T., Huisman H.G., Miedema F. Differential syncytium-inducing capacity of human immunodeficiency virus isolatesfrequent detection of syncytium-inducing isolates in patients with acquired immunodeficiency syndrome (AIDS) and AIDS-related complex. J. Virol. 1988;62:2026–2032. doi: 10.1128/jvi.62.6.2026-2032.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tersmette M., Lange J.M.A., deGoede R.E.Y., deWold F., Eeftink-Schattankerk J.K.M., Schellekens P.T.A., Coutinho R.A., Huisman J.G, Goudsmit J., Miedema F. Association between biological properties of human immunodeficiency virus variants and risk for AIDS and AIDS mortality. Lancet. 1989;1:983–985. doi: 10.1016/s0140-6736(89)92628-7. [DOI] [PubMed] [Google Scholar]
- Cocchi F., DeVico A.L., Garzino-Demo A., Arya S.K., Gallo R.C., Lusso P. Identification of RANTES, MIP-1α and MIP-1β the major HIV-suppressive factors produced by CD8+ T cells. Science. 1995;270:1811–1815. doi: 10.1126/science.270.5243.1811. [DOI] [PubMed] [Google Scholar]
- Dorfman T., Burkovsky A., Ohagen A., Hoglund S., Gottlinger H.C. Functional domains of the capsid protein of human immunodeficiency virus type 1. J. Virol. 1994;68:8180–8187. doi: 10.1128/jvi.68.12.8180-8187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momany C., Kovari L.C., Prongay A.J., Keller W., Gitti R.K., Lee B.M., Gorbalenya A.E., Tong L., McClure J., Ehrlich L.S. Crystal structure of dimeric HIV-1 capsid protein. Nat. Struct. Biol. 1996;3:763–770. doi: 10.1038/nsb0996-763. [DOI] [PubMed] [Google Scholar]
- Gamble T.R., Vajdos F.F., Yoo S., Worthylake D.K., Houseweart M., Sundquist W.I., Hill C.P. Crystal structure of human cyclophilin A bound to the amino terminal domain of HIV-1 capsid. Cell. 1996;87:1285–1294. doi: 10.1016/s0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- Gitti R.K., Lee B.M., Walker J., Summers M.F., Yoo S., Sundquist W.I. Structure of the amino acid terminal core domain of the HIV-1 capsid protein. Science. 1996;273:231–235. doi: 10.1126/science.273.5272.231. [DOI] [PubMed] [Google Scholar]
- Jin Z., Jin L., Peterson D.L., Lawson C.L. Model for lentivirus capsid core assembly based on crystal dimers of EIAV p26. J. Mol. Biol. 1999;286:83–93. doi: 10.1006/jmbi.1998.2443. [DOI] [PubMed] [Google Scholar]
- Yoo S., Myska D.G, Yeh C., McMurray M., Hill C.P., Sundquist W.I. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 1997;269:780–785. doi: 10.1006/jmbi.1997.1051. [DOI] [PubMed] [Google Scholar]
- Thali M., Bukovsky A., Kundo I., Rosenwirth B., Walsh C.T., Sodroski J., Gottlinger H.G. Functional association of cyclophilin A with HIV-1 virions. Nature. 1994;372:363–365. doi: 10.1038/372363a0. [DOI] [PubMed] [Google Scholar]
- Coffin J.M. HIV population dynamics in vivoimplications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- Kellam P., Boucher C.A., Tijnagel J.M., Larder B.A. Zidovudine treatment results in selection of human immunodeficiency virus type 1 variants whose genotypes confer increasing levels of drug resistance. J. Gen. Virol. 1994;75:341–351. doi: 10.1099/0022-1317-75-2-341. [DOI] [PubMed] [Google Scholar]
- Harrigan P.R., Bloor S., Larder B.A. Relative replicative fitness of zidovudine-resistant human immunodeficiency virus type 1 isolates in vitro . J. Virol. 1998;72:3773–3778. doi: 10.1128/jvi.72.5.3773-3778.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucher C.A., van Leeuween R., Kellam P., Schipper P., Tijnagel J., Lange J.M., Larder B.A. Effects of discontinuation of zidovudine treatment on zidovudine sensitivity of human immunodeficiency virus type 1 isolates. Antimicrob. Agents Chemother. 1993;37:1525–1530. doi: 10.1128/aac.37.7.1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudsmit J., de Ronde A., de Rooij E., de Boer R. Broad spectrum of in vivo fitness of human immunodeficiency virus type 1 subpopulations differing at reverse transcriptase codons 41 and 215. J. Virol. 1997;71:4479–4484. doi: 10.1128/jvi.71.6.4479-4484.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudsmit J., de Ronde A., Ho D.D., Perelson A.S. Human immunodeficiency virus fitness in vivocalculations based on single zidovudine resistance mutation at codon 215 of reverse transcriptase. J. Virol. 1996;70:5662–5666. doi: 10.1128/jvi.70.8.5662-5664.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurusinghe A.D., Land S.A., Birch C., McGavin C., Hooker D.J., Tachedijian G., Doherty R., Deacon N.J. Reverse transcriptase mutations in sequential HIV-isolates in patients with AIDS. J. Med. Virol. 1995;46:238–243. doi: 10.1002/jmv.1890460312. [DOI] [PubMed] [Google Scholar]
- Furtado M.R., Callaway D.S., Phair J.P., Kunstman K.J., Stanton J.L., Macken C.A., Perelson A.S., Wolinsky S.M. Persistence of HIV-1 transcription in peripheral-blood mononuclear cells in patients receiving potent antiretroviral therapy. N. Engl. J. Med. 1999;340:1614–1622. doi: 10.1056/NEJM199905273402102. [DOI] [PubMed] [Google Scholar]
- Phillips R.E., Rowland-Jones S., Nixon D.F., Gotch F.M., Edwards J.P., Ogunlesi A.O., Elvin J.G., Rothbard J.A., Bangham C.R.M., Rizza C.R., McMichael A.J. Human immunodeficiency virus genetic variation can escape cytotoxic T cell recognition. Nature. 1991;354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]